

Advanced Molecular Characterisation in Relapsed and Refractory Paediatric Acute Leukaemia, the Key for Personalised Medicine

 , , , ,
, , , ,  and
and
Abstract
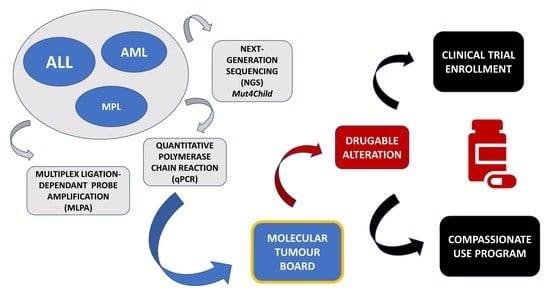
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Samples
2.3. Multiplex Ligand-Probe Amplification (MLPA)
2.4. Quantitative Polymerase Chain Reaction (qPCR)
2.5. Next-Generation Sequencing (NGS)
2.6. Molecular Tumour Board
2.7. Statistical Analysis
3. Results
3.1. Clinical Data
3.2. Molecular Findings
3.2.1. CRLF2 and WT1 Overexpression
3.2.2. Copy Number Alterations (CNAs)
3.2.3. Genetic Alterations Identified by NGS
3.3. Directed Therapy According to Genetic Characterisation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pui, C.-H.; Gajjar, A.J.; Kane, J.R.; Qaddoumi, I.A.; Pappo, A.S. Challenging Issues in Pediatric Oncology. Nat. Rev. Clin. Oncol. 2011, 8, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Downing, J.R.; Wilson, R.K.; Zhang, J.; Mardis, E.R.; Pui, C.-H.; Ding, L.; Ley, T.J.; Evans, W.E. The Pediatric Cancer Genome Project. Nat. Genet. 2012, 44, 619–622. [Google Scholar] [CrossRef]
- Onciu, M.; Pui, C.-H. Diagnosis and Classification. In Childhood Leukemias; Pui, C.-H., Ed.; Cambridge University Press: Cambridge, UK, 2012; pp. 21–48. ISBN 978-0-511-97763-3. [Google Scholar]
- Inaba, H.; Mullighan, C.G. Pediatric Acute Lymphoblastic Leukemia. Haematology 2020, 105, 2524–2539. [Google Scholar] [CrossRef] [PubMed]
- de Morais, R.V.; de Souza, M.V.; de Silva, K.A.S.; Santiago, P.; Lorenzoni, M.C.; Lorea, C.F.; de Junior, C.G.C.; Taniguchi, A.N.R.; Scherer, F.F.; Michalowski, M.B.; et al. Epidemiological Evaluation and Survival of Children with Acute Myeloid Leukemia. J. Pediatr. 2021, 97, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Hudson, M.M.; Neglia, J.P.; Woods, W.G.; Sandlund, J.T.; Pui, C.-H.; Kun, L.E.; Robison, L.L.; Green, D.M. Lessons from the Past: Opportunities to Improve Childhood Cancer Survivor Care through Outcomes Investigations of Historical Therapeutic Approaches for Pediatric Hematological Malignancies. Pediatr. Blood Cancer 2012, 58, 334–343. [Google Scholar] [CrossRef]
- Hucks, G.; Rheingold, S.R. The Journey to CAR T Cell Therapy: The Pediatric and Young Adult Experience with Relapsed or Refractory B-ALL. Blood Cancer J. 2019, 9, 10. [Google Scholar] [CrossRef]
- von Stackelberg, A.; Völzke, E.; Kühl, J.-S.; Seeger, K.; Schrauder, A.; Escherich, G.; Henze, G.; Tallen, G. ALL-REZ BFM Study Group Outcome of Children and Adolescents with Relapsed Acute Lymphoblastic Leukaemia and Non-Response to Salvage Protocol Therapy: A Retrospective Analysis of the ALL-REZ BFM Study Group. Eur. J. Cancer 2011, 47, 90–97. [Google Scholar] [CrossRef]
- Kopp, L.M.; Gupta, P.; Pelayo-Katsanis, L.; Wittman, B.; Katsanis, E. Late Effects in Adult Survivors of Pediatric Cancer: A Guide for the Primary Care Physician. Am. J. Med. 2012, 125, 636–641. [Google Scholar] [CrossRef]
- Tran, T.H.; Shah, A.T.; Loh, M.L. Precision Medicine in Pediatric Oncology: Translating Genomic Discoveries into Optimized Therapies. Clin. Cancer Res. 2017, 23, 5329–5338. [Google Scholar] [CrossRef][Green Version]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing-Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef]
- Hiemenz, M.C.; Ostrow, D.G.; Busse, T.M.; Buckley, J.; Maglinte, D.T.; Bootwalla, M.; Done, J.; Ji, J.; Raca, G.; Ryutov, A.; et al. OncoKids: A Comprehensive Next-Generation Sequencing Panel for Pediatric Malignancies. J. Mol. Diagn. 2018, 20, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Beau, M.M.L.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Cilloni, D.; Renneville, A.; Hermitte, F.; Hills, R.K.; Daly, S.; Jovanovic, J.V.; Gottardi, E.; Fava, M.; Schnittger, S.; Weiss, T.; et al. Real-Time Quantitative Polymerase Chain Reaction Detection of Minimal Residual Disease by Standardized WT1 Assay to Enhance Risk Stratification in Acute Myeloid Leukemia: A European LeukemiaNet Study. J. Clin. Oncol. 2009, 27, 5195–5201. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Picard Tools—By Broad Institute. Available online: https://broadinstitute.github.io/picard/ (accessed on 13 March 2022).
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Dunn, T.; Berry, G.; Emig-Agius, D.; Jiang, Y.; Lei, S.; Iyer, A.; Udar, N.; Chuang, H.-Y.; Hegarty, J.; Dickover, M.; et al. Pisces: An Accurate and Versatile Variant Caller for Somatic and Germline next-Generation Sequencing Data. Bioinformatics 2019, 35, 1579–1581. [Google Scholar] [CrossRef]
- Benjamin, D.; Sato, T.; Cibulskis, K.; Getz, G.; Stewart, C.; Lichtenstein, L. Calling Somatic SNVs and Indels with Mutect2. BioRxiv 2019, 861054. [Google Scholar] [CrossRef]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A Sequence-Quality Aware, Ultra-Sensitive Variant Caller for Uncovering Cell-Population Heterogeneity from High-Throughput Sequencing Datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid Detection of Structural Variants and Indels for Germline and Cancer Sequencing Applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.-L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.-H.; Kim, H.-J.; Shin, S.-H.; Yahng, S.-A.; Lee, S.-E.; Cho, B.-S.; Eom, K.-S.; Kim, Y.-J.; Lee, S.; Min, C.-K.; et al. Serial Measurement of WT1 Expression and Decrement Ratio until Hematopoietic Cell Transplantation as a Marker of Residual Disease in Patients with Cytogenetically Normal Acute Myelogenous Leukemia. Biol. Blood Marrow. Transplant. 2013, 19, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Cilloni, D.; Gottardi, E.; Saglio, G. WT1 Overexpression in Acute Myeloid Leukemia and Myelodysplastic Syndromes. Methods Mol. Med. 2006, 125, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.-H.; Yang, J.J.; Hunger, S.P.; Pieters, R.; Schrappe, M.; Biondi, A.; Vora, A.; Baruchel, A.; Silverman, L.B.; Schmiegelow, K.; et al. Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. J. Clin. Oncol. 2015, 33, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Söderhäll, S.; Arvidson, J.; Forestier, E.; Montgomery, S.; Bottai, M.; Lausen, B.; Carlsen, N.; Hellebostad, M.; Lähteenmäki, P.; et al. Relapsed Childhood Acute Lymphoblastic Leukemia in the Nordic Countries: Prognostic Factors, Treatment and Outcome. Haematologica 2016, 101, 68–76. [Google Scholar] [CrossRef]
- Rau, R.E.; Loh, M.L. Using Genomics to Define Pediatric Blood Cancers and Inform Practice. Hematol. Am. Soc. Hematol. Educ. Program 2018, 2018, 286–300. [Google Scholar] [CrossRef]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-Cancer Genome and Transcriptome Analyses of 1,699 Paediatric Leukaemias and Solid Tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef]
- Liu, Y.-F.; Wang, B.-Y.; Zhang, W.-N.; Huang, J.-Y.; Li, B.-S.; Zhang, M.; Jiang, L.; Li, J.-F.; Wang, M.-J.; Dai, Y.-J.; et al. Genomic Profiling of Adult and Pediatric B-Cell Acute Lymphoblastic Leukemia. EBioMedicine 2016, 8, 173–183. [Google Scholar] [CrossRef]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The Genomic Landscape of Pediatric and Young Adult T-Lineage Acute Lymphoblastic Leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef]
- Mercher, T.; Schwaller, J. Pediatric Acute Myeloid Leukemia (AML): From Genes to Models Toward Targeted Therapeutic Intervention. Front. Pediatr. 2019, 7, 401. [Google Scholar] [CrossRef]
- Inaba, H.; Greaves, M.; Mullighan, C.G. Acute Lymphoblastic Leukaemia. Lancet 2013, 381, 1943–1955. [Google Scholar] [CrossRef]
- Zhang, W.; Kuang, P.; Liu, T. Prognostic Significance of CDKN2A/B Deletions in Acute Lymphoblastic Leukaemia: A Meta-Analysis. Ann. Med. 2019, 51, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Bortolozzi, R.; Mattiuzzo, E.; Trentin, L.; Accordi, B.; Basso, G.; Viola, G. Ribociclib, a Cdk4/Cdk6 Kinase Inhibitor, Enhances Glucocorticoid Sensitivity in B-Acute Lymphoblastic Leukemia (B-All). Biochem. Pharmacol. 2018, 153, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Bride, K.L.; Hu, H.; Tikhonova, A.; Fuller, T.J.; Vincent, T.L.; Shraim, R.; Li, M.M.; Carroll, W.L.; Raetz, E.A.; Aifantis, I.; et al. Rational Drug Combinations with CDK4/6 Inhibitors in Acute Lymphoblastic Leukemia. Haematologica 2021. [Google Scholar] [CrossRef]
- Bautista, F.; Paoletti, X.; Rubino, J.; Brard, C.; Rezai, K.; Nebchi, S.; Andre, N.; Aerts, I.; De Carli, E.; van Eijkelenburg, N.; et al. Phase I or II Study of Ribociclib in Combination With Topotecan-Temozolomide or Everolimus in Children With Advanced Malignancies: Arms A and B of the AcSé-ESMART Trial. J. Clin. Oncol. 2021, 39, 3546–3560. [Google Scholar] [CrossRef]
- Pikman, Y.; Alexe, G.; Roti, G.; Conway, A.S.; Furman, A.; Lee, E.S.; Place, A.E.; Kim, S.; Saran, C.; Modiste, R.; et al. Synergistic Drug Combinations with a CDK4/6 Inhibitor in T-Cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2017, 23, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Konecny, G.E.; Winterhoff, B.; Kolarova, T.; Qi, J.; Manivong, K.; Dering, J.; Yang, G.; Chalukya, M.; Wang, H.-J.; Anderson, L.; et al. Expression of P16 and Retinoblastoma Determines Response to CDK4/6 Inhibition in Ovarian Cancer. Clin. Cancer Res. 2011, 17, 1591–1602. [Google Scholar] [CrossRef]
- Hamilton, E.; Infante, J.R. Targeting CDK4/6 in Patients with Cancer. Cancer Treat. Rev. 2016, 45, 129–138. [Google Scholar] [CrossRef]
- Roskoski, R. Cyclin-Dependent Protein Serine/Threonine Kinase Inhibitors as Anticancer Drugs. Pharmacol. Res. 2019, 139, 471–488. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Knudsen, K.E.; Dicker, A.P.; Knudsen, E.S. The Meaning of P16(Ink4a) Expression in Tumors: Functional Significance, Clinical Associations and Future Developments. Cell Cycle 2011, 10, 2497–2503. [Google Scholar] [CrossRef]
- Du, Q.; Guo, X.; Wang, M.; Li, Y.; Sun, X.; Li, Q. The Application and Prospect of CDK4/6 Inhibitors in Malignant Solid Tumors. J. Hematol. Oncol. 2020, 13, 41. [Google Scholar] [CrossRef] [PubMed]
- Antić, Ž.; Yu, J.; Van Reijmersdal, S.V.; Van Dijk, A.; Dekker, L.; Segerink, W.H.; Sonneveld, E.; Fiocco, M.; Pieters, R.; Hoogerbrugge, P.M.; et al. Multiclonal Complexity of Pediatric Acute Lymphoblastic Leukemia and the Prognostic Relevance of Subclonal Mutations. Haematologica 2021, 106, 3046–3055. [Google Scholar] [CrossRef] [PubMed]
- Malinowska-Ozdowy, K.; Frech, C.; Schönegger, A.; Eckert, C.; Cazzaniga, G.; Stanulla, M.; zur Stadt, U.; Mecklenbräuker, A.; Schuster, M.; Kneidinger, D.; et al. KRAS and CREBBP Mutations: A Relapse-Linked Malicious Liaison in Childhood High Hyperdiploid Acute Lymphoblastic Leukemia. Leukemia 2015, 29, 1656–1667. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-H.; Wang, H.-S.; Qian, X.-W.; Zhu, X.-H.; Miao, H.; Yu, Y.; Meng, J.-H.; Le, J.; Jiang, J.-Y.; Cao, P.; et al. Ras Pathway Mutation Feature in the Same Individuals at Diagnosis and Relapse of Childhood Acute Lymphoblastic Leukemia. Transl. Pediatr. 2020, 9, 4–12. [Google Scholar] [CrossRef]
- Jerchel, I.S.; Hoogkamer, A.Q.; Ariës, I.M.; Steeghs, E.M.P.; Boer, J.M.; Besselink, N.J.M.; Boeree, A.; van de Ven, C.; de Groot-Kruseman, H.A.; de Haas, V.; et al. RAS Pathway Mutations as a Predictive Biomarker for Treatment Adaptation in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. Leukemia 2018, 32, 931–940. [Google Scholar] [CrossRef]
- Kerstjens, M.; Driessen, E.M.C.; Willekes, M.; Pinhanços, S.S.; Schneider, P.; Pieters, R.; Stam, R.W. MEK Inhibition Is a Promising Therapeutic Strategy for MLL-Rearranged Infant Acute Lymphoblastic Leukemia Patients Carrying RAS Mutations. Oncotarget 2017, 8, 14835–14846. [Google Scholar] [CrossRef]
- Kennedy, V.E.; Smith, C.C. FLT3 Mutations in Acute Myeloid Leukemia: Key Concepts and Emerging Controversies. Front. Oncol. 2020, 10, 612880. [Google Scholar] [CrossRef]
- Harvey, R.C.; Tasian, S.K. Clinical Diagnostics and Treatment Strategies for Philadelphia Chromosome-like Acute Lymphoblastic Leukemia. Blood Adv. 2020, 4, 218–228. [Google Scholar] [CrossRef]
- Palmi, C.; Vendramini, E.; Silvestri, D.; Longinotti, G.; Frison, D.; Cario, G.; Shochat, C.; Stanulla, M.; Rossi, V.; Di Meglio, A.M.; et al. Poor Prognosis for P2RY8-CRLF2 Fusion but Not for CRLF2 over-Expression in Children with Intermediate Risk B-Cell Precursor Acute Lymphoblastic Leukemia. Leukemia 2012, 26, 2245–2253. [Google Scholar] [CrossRef]
- Yamashita, Y.; Shimada, A.; Yamada, T.; Yamaji, K.; Hori, T.; Tsurusawa, M.; Watanabe, A.; Kikuta, A.; Asami, K.; Saito, A.M.; et al. IKZF1 and CRLF2 Gene Alterations Correlate with Poor Prognosis in Japanese BCR-ABL1-Negative High-Risk B-Cell Precursor Acute Lymphoblastic Leukemia. Pediatr. Blood Cancer 2013, 60, 1587–1592. [Google Scholar] [CrossRef]
- Joseph, P.D.; Craig, J.C.; Caldwell, P.H.Y. Clinical Trials in Children. Br. J. Clin. Pharmacol. 2015, 79, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Kyr, M.; Svobodnik, A.; Stepanova, R.; Hejnova, R. N-of-1 Trials in Pediatric Oncology: From a Population-Based Approach to Personalized Medicine—A Review. Cancers 2021, 13, 5428. [Google Scholar] [CrossRef] [PubMed]
- Moerdler, S.; Zhang, L.; Gerasimov, E.; Zhu, C.; Wolinsky, T.; Roth, M.; Goodman, N.; Weiser, D.A. Physician Perspectives on Compassionate Use in Pediatric Oncology. Pediatr. Blood Cancer 2019, 66, e27545. [Google Scholar] [CrossRef] [PubMed]
- Dieck, C.L.; Ferrando, A. Genetics and Mechanisms of NT5C2-Driven Chemotherapy Resistance in Relapsed ALL. Blood 2019, 133, 2263–2268. [Google Scholar] [CrossRef]
- Barz, M.J.; Hof, J.; Groeneveld-Krentz, S.; Loh, J.W.; Szymansky, A.; Astrahantseff, K.; von Stackelberg, A.; Khiabanian, H.; Ferrando, A.A.; Eckert, C.; et al. Subclonal NT5C2 Mutations Are Associated with Poor Outcomes after Relapse of Pediatric Acute Lymphoblastic Leukemia. Blood 2020, 135, 921–933. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Hu, J.; Ren, Y.; Wang, H. Clinical Features and Prognosis of Normal Karyotype Acute Myeloid Leukemia Pediatric Patients with WT1 Mutations: An Analysis Based on TCGA Database. Hematology 2020, 25, 79–84. [Google Scholar] [CrossRef]
- Zidan, M.A.A.; Kamal Shaaban, H.M.; Elghannam, D.M. Prognostic Impact of Wilms Tumor Gene Mutations in Egyptian Patients with Acute Myeloid Leukemia with Normal Karyotype. Hematology 2014, 19, 267–274. [Google Scholar] [CrossRef]
- Owen, C.; Fitzgibbon, J.; Paschka, P. The Clinical Relevance of Wilms Tumour 1 (WT1) Gene Mutations in Acute Leukaemia. Hematol. Oncol. 2010, 28, 13–19. [Google Scholar] [CrossRef]
- Valliyammai, N.; Nancy, N.K.; Sagar, T.G.; Rajkumar, T. Study of NOTCH1 and FBXW7 Mutations and Its Prognostic Significance in South Indian T-Cell Acute Lymphoblastic Leukemia. J. Pediatr. Hematol. Oncol. 2018, 40, e1–e8. [Google Scholar] [CrossRef]
- Fogelstrand, L.; Staffas, A.; Wasslavik, C.; Sjögren, H.; Söderhäll, S.; Frost, B.-M.; Forestier, E.; Degerman, S.; Behrendtz, M.; Heldrup, J.; et al. Prognostic Implications of Mutations in NOTCH1 and FBXW7 in Childhood T-ALL Treated According to the NOPHO ALL-1992 and ALL-2000 Protocols. Pediatr. Blood Cancer 2014, 61, 424–430. [Google Scholar] [CrossRef]
- Park, M.-J.; Taki, T.; Oda, M.; Watanabe, T.; Yumura-Yagi, K.; Kobayashi, R.; Suzuki, N.; Hara, J.; Horibe, K.; Hayashi, Y. FBXW7 and NOTCH1 Mutations in Childhood T Cell Acute Lymphoblastic Leukaemia and T Cell Non-Hodgkin Lymphoma. Br. J. Haematol. 2009, 145, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Azzato, E.M.; Mullighan, C.G. Integration of Next-Generation Sequencing to Treat Acute Lymphoblastic Leukemia with Targetable Lesions: The St. Jude Children’s Research Hospital Approach. Front. Pediatr. 2017, 5, 258. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.-H. Precision Medicine in Acute Lymphoblastic Leukemia. Front. Med. 2020, 14, 689–700. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| N (%) | |
|---|---|
| SEX | |
| Male | 17 (65.4%) |
| Female | 9 (34.6%) |
| DISEASE | |
| B-ALL | 18 (69.2%) |
| T-ALL | 4 (15.4%) |
| AML | 3 (11.5%) |
| MPL | 1 (3.8%) |
| DISEASE STATUS | |
| Refractory | 5 (19.2%) |
| Relapse: | 21 (80.8%) |
| Isolated bone marrow | 14 (66.7%) |
| Isolated extramedullary | 3 (14.3%) |
| Combined relapse | 4 (19%) |
| KARYOTYPE (DIAGNOSIS) | |
| Hypodiploid | 0 |
| Hyperdiploid | 1 (3.8%) |
| Complex | 2 (7.7%) |
| Normal | 9 (34.6%) |
| CNS INVOLVEMENT (DIAGNOSIIS) | |
| Yes | 3 (11.5%) |
| No | 23 (88.5%) |
| RISK GROUP (DIAGNOSIS) | |
| Standard | 8 (30.8%) |
| Intermediate | 11 (42.3%) |
| High | 7 (26.9%) |
| HSCT | |
| Yes | 13 (50%) |
| No | 13 (50%) |
| STATUS | |
| Alive | 16 (61.5%) |
| Dead | 10 (38.5%) |
| PATIENT ID | GEN | VARIANT | CLASSIFICATION (AMP) | DRUG AND DURATION (DAYS) | PRIOR/AFTER HSCT | RESPONSE |
|---|---|---|---|---|---|---|
| HRL 2 | NRAS | c.183A>C(p.Gln61His) | Tier II | Trametinib (28) | After | NO |
| HRL 4 | CRLF2 | overexpression | Tier II | Ruxolitinib (342) | Prior | YES |
| HRL 7 | FLT3 | c.2503_2506delinsC (p.Asp835_Ile836delinsLeu) | Tier II | Sunitinib (383) | Prior | YES |
| HRL 10 | CRLF2 | overexpression | Tier II | Ruxolitinib (254) | Prior | YES |
| HRL 11 | FLT3 | FLT3-ITD | Tier I | Quizartinib (22) | After | N/A * |
| HRL 16 | CDKN2A | deletion | Tier II | Ribociclib (108) | After | YES |
| HRL 17 | KRAS | c.34G>T(p.Gly12Cys) | Tier II | Trametinib (3) | No HSCT | NO |
| HRL 24 | CDKN2A | c.319dupC(p.His107fs) | Tier II | Ribociclib (67) | After | NO |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galán-Gómez, V.; Matamala, N.; Ruz-Caracuel, B.; Valle-Simón, P.; Ochoa-Fernández, B.; Guerra-García, P.; Pernas-Sánchez, A.; Minguillón, J.; González, B.; Martínez-Romera, I.; et al. Advanced Molecular Characterisation in Relapsed and Refractory Paediatric Acute Leukaemia, the Key for Personalised Medicine. J. Pers. Med. 2022, 12, 881. https://doi.org/10.3390/jpm12060881
Galán-Gómez V, Matamala N, Ruz-Caracuel B, Valle-Simón P, Ochoa-Fernández B, Guerra-García P, Pernas-Sánchez A, Minguillón J, González B, Martínez-Romera I, et al. Advanced Molecular Characterisation in Relapsed and Refractory Paediatric Acute Leukaemia, the Key for Personalised Medicine. Journal of Personalized Medicine. 2022; 12(6):881. https://doi.org/10.3390/jpm12060881
Chicago/Turabian StyleGalán-Gómez, Víctor, Nerea Matamala, Beatriz Ruz-Caracuel, Paula Valle-Simón, Bárbara Ochoa-Fernández, Pilar Guerra-García, Alicia Pernas-Sánchez, Jordi Minguillón, Berta González, Isabel Martínez-Romera, and et al. 2022. "Advanced Molecular Characterisation in Relapsed and Refractory Paediatric Acute Leukaemia, the Key for Personalised Medicine" Journal of Personalized Medicine 12, no. 6: 881. https://doi.org/10.3390/jpm12060881
APA StyleGalán-Gómez, V., Matamala, N., Ruz-Caracuel, B., Valle-Simón, P., Ochoa-Fernández, B., Guerra-García, P., Pernas-Sánchez, A., Minguillón, J., González, B., Martínez-Romera, I., Román-Pacheco, S. S., Estival-Monteliú, P., Ibáñez-Navarro, A., Pérez-Martínez, A., & Escudero-López, A. (2022). Advanced Molecular Characterisation in Relapsed and Refractory Paediatric Acute Leukaemia, the Key for Personalised Medicine. Journal of Personalized Medicine, 12(6), 881. https://doi.org/10.3390/jpm12060881