
Prioritization of Candidate Biomarkers for Degenerative Aortic Stenosis through a Systems Biology-Based In-Silico Approach

 , ,
, , 
Abstract
1. Introduction
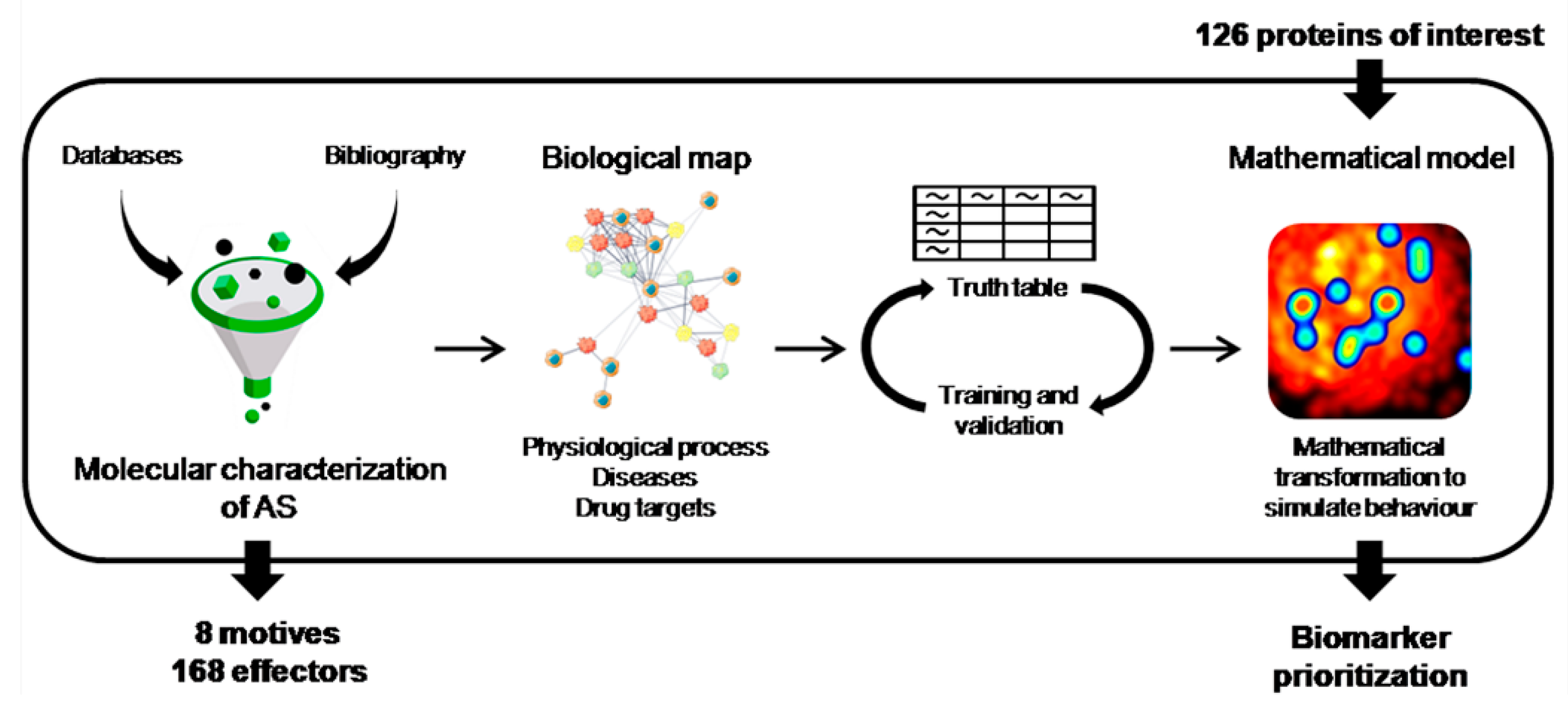
2. Materials and Methods
2.1. Molecular Characterization of AS
2.2. Generation of the Mathematical Models
2.3. Candidate Prioritization
- “strong relationship” with the processes under study, with very high, high or, medium-high predicted relationships with any of the sub-processes used in the characterization, and considered to be good candidates;
- “medium relationship” with the processes under study, with at least a medium predicted relationship with any of the sub-processes used in the characterization;
- “low or no relationship” with the processes under study and with a weak predicted relationship with all the sub-processes used in the characterization.
2.4. Cell Culture and Differentiation
2.5. Alizarin Red Staining
2.6. Patient Selection and Plasma Extraction
2.7. Western Blotting
2.8. Statistics
3. Results
3.1. Molecular Motives of AS
3.2. Candidate Prioritization
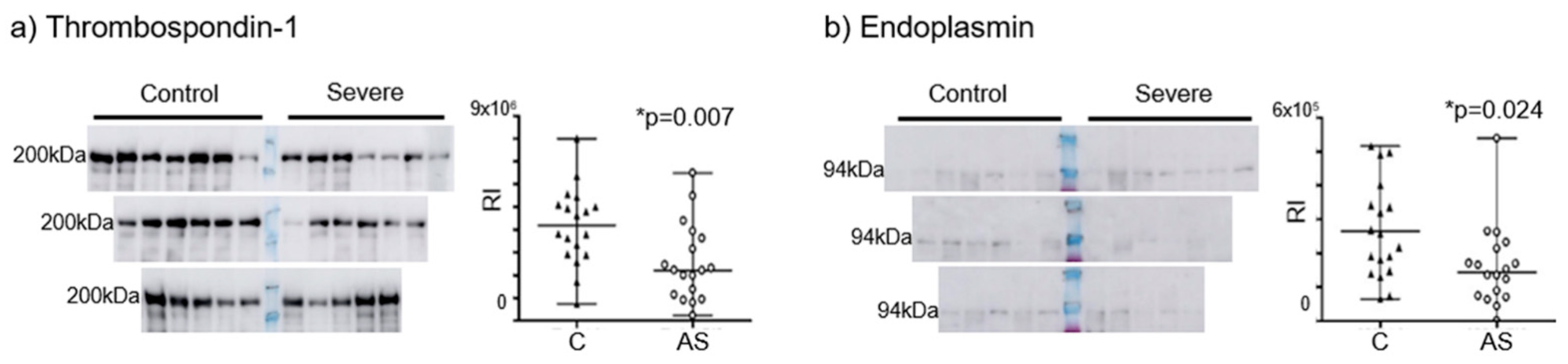
3.3. Confirmation of the Prioritized Candidates in a Cell Model and Plasma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goldbarg, S.H.; Elmariah, S.; Miller, M.A.; Fuster, V. Insights into degenerative aortic valve disease. J. Am. Coll. Cardiol. 2007, 50, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Helske, S.; Kupari, M.; Lindstedt, K.A.; Kovanen, P.T. Aortic valve stenosis: An active atheroinflammatory process. Curr. Opin. Lipidol. 2007, 18, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Freeman, R.V.; Otto, C.M. Spectrum of calcific aortic valve disease: Pathogenesis, disease progression, and treatment strategies. Circulation 2005, 111, 3316–3326. [Google Scholar] [CrossRef] [PubMed]
- Leon, M.B.; Smith, C.R.; Mack, M.J.; Makkar, R.R.; Svensson, L.G.; Kodali, S.K.; Thourani, V.H.; Tuzcu, E.M.; Miller, D.C.; Herrmann, H.C.; et al. Transcatheter or surgical aortic-valve replacement in intermediate-risk patients. N. Engl. J. Med. 2016, 374, 1609–1620. [Google Scholar] [CrossRef]
- Mourino-Alvarez, L.; Martin-Rojas, T.; Corros-Vicente, C.; Corbacho-Alonso, N.; Padial, L.R.; Solis, J.; Barderas, M.G. Patient management in aortic stenosis: Towards precision medicine through protein analysis, imaging and diagnostic tests. J. Clin. Med. 2020, 9, 2421. [Google Scholar] [CrossRef]
- Small, A.; Kiss, D.; Giri, J.; Anwaruddin, S.; Siddiqi, H.; Guerraty, M.; Chirinos, J.A.; Ferrari, G.; Rader, D.J. Biomarkers of calcific aortic valve disease. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 623–632. [Google Scholar] [CrossRef]
- Everett, R.J.; Clavel, M.-A.; Pibarot, P.; Dweck, M.R. Timing of intervention in aortic stenosis: A review of current and future strategies. Heart 2018, 104, 2067–2076. [Google Scholar] [CrossRef]
- Ky, B.; French, B.; Levy, W.C.; Sweitzer, N.K.; Fang, J.C.; Wu, A.H.B.; Goldberg, L.R.; Jessup, M.; Cappola, T.P. Multiple biomarkers for risk prediction in chronic heart failure. Circ. Heart Fail. 2012, 5, 183–190. [Google Scholar] [CrossRef]
- Wang, T.J.; Wollert, K.C.; Larson, M.G.; Coglianese, E.; McCabe, E.L.; Cheng, S.; Ho, J.E.; Fradley, M.G.; Ghorbani, A.; Xanthakis, V.; et al. Prognostic utility of novel biomarkers of cardiovascular stress: The Framingham Heart Study. Circulation 2012, 126, 1596–1604. [Google Scholar] [CrossRef]
- Alvarez-Llamas, G.; Martin-Rojas, T.; de la Cuesta, F.; Calvo, E.; Gil-Dones, F.; Darde, V.M.; Lopez-Almodovar, L.F.; Padial, L.R.; Lopez, J.-A.; Vivanco, F.; et al. Modification of the secretion pattern of proteases, inflammatory mediators, and extracellular matrix proteins by human aortic valve is key in severe aortic stenosis. Mol. Cell. Proteom. 2013, 12, 2426–2439. [Google Scholar] [CrossRef]
- Martin-Rojas, T.; Mourino-Alvarez, L.; Alonso-Orgaz, S.; Rosello-Lleti, E.; Calvo, E.; Lopez-Almodovar, L.F.; Rivera, M.; Padial, L.R.; Lopez, J.A.; de la Cuesta, F.; et al. iTRAQ proteomic analysis of extracellular matrix remodeling in aortic valve disease. Sci. Rep. 2015, 5, 17290. [Google Scholar] [CrossRef] [PubMed]
- Gil-Dones, F.; Darde, V.; Orgaz, S.A.; Lopez-Almodovar, L.F.; Mourino-Alvarez, L.; Padial, L.R.; Vivanco, F.; Barderas, M.G. Inside human aortic stenosis: A proteomic analysis of plasma. J. Proteom. 2012, 75, 1639–1653. [Google Scholar] [CrossRef] [PubMed]
- Martin-Rojas, T.; Gil-Dones, F.; Lopez-Almodovar, L.F.; Padial, L.R.; Vivanco, F.; Barderas, M.G. Proteomic profile of human aortic stenosis: Insights into the degenerative process. J. Proteome Res. 2012, 11, 1537–1550. [Google Scholar] [CrossRef] [PubMed]
- Mourino-Alvarez, L.; Iloro, I.; de la Cuesta, F.; Azkargorta, M.; Sastre-Oliva, T.; Escobes, I.; Lopez-Almodovar, L.F.; Sanchez, P.L.; Urreta, H.; Fernandez-Aviles, F.; et al. MALDI-Imaging Mass Spectrometry: A step forward in the anatomopathological characterization of stenotic aortic valve tissue. Sci. Rep. 2016, 6, 27106. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Chatr-Aryamontri, A.; Oughtred, R.; Boucher, L.; Rust, J.; Chang, C.; Kolas, N.K.; O’Donnell, L.; Oster, S.; Theesfeld, C.; Sellam, A.; et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017, 45, D369–D379. [Google Scholar] [CrossRef]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; McKay, S.; et al. The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016, 44, D481–D487. [Google Scholar] [CrossRef]
- Han, H.; Cho, J.W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database—2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef]
- Bishop, C.M. Pattern Recognition and Machine Learning; Information Science and Statistics; Springer: New York, NY, USA, 2006; p. 738. [Google Scholar]
- Latif, N.; Quillon, A.; Sarathchandra, P.; McCormack, A.; Lozanoski, A.; Yacoub, M.H.; Chester, A.H. Modulation of human valve interstitial cell phenotype and function using a Fibroblast Growth Factor 2 formulation. PLoS ONE 2015, 10, e0127844. [Google Scholar] [CrossRef] [PubMed]
- van Engeland, N.C.A.; Bertazzo, S.; Sarathchandra, P.; McCormack, A.; Bouten, C.V.C.; Yacoub, M.H.; Chester, A.H.; Latif, N. Aortic calcified particles modulate valvular endothelial and interstitial cells. Cardiovasc. Pathol. 2017, 28, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Gregory, C.A.; Gunn, W.G.; Peister, A.; Prockop, D.J. An Alizarin red-based assay of mineralization by adherent cells in culture: Comparison with cetylpyridinium chloride extraction. Anal. Biochem. 2004, 329, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Gil-Dones, F.; Martin-Rojas, T.; Lopez-Almodovar, L.; De la Cuesta, F.; Darde, V.; Alvarez-Llamas, G.; Juarez-Tosina, T.; Barroso, G.; Vivanco, F.; Padial, L.; et al. Valvular aortic stenosis: A proteomic insight. Clin. Med. Insights Cardiol. 2010, 4, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Gilda, J.E.; Gomes, A.V. Western blotting using in-gel protein labeling as a normalization control: Stain-free technology. Methods Mol. Biol. 2015, 1295, 381–391. [Google Scholar] [CrossRef]
- Chung, K.F.; Adcock, I.M. Precision medicine for the discovery of treatable mechanisms in severe asthma. Allergy 2019, 74, 1649–1659. [Google Scholar] [CrossRef]
- Sathyamurthy, I.; Alex, S.; Kirubakaran, K.; Sengottuvelu, G.; Srinivasan, K.N. Risk factor profile of calcific aortic stenosis. Indian Heart J. 2016, 68, 828–831. [Google Scholar] [CrossRef][Green Version]
- Hofmanis, J.; Hofmane, D.; Svirskis, S.; Mackevics, V.; Tretjakovs, P.; Lejnieks, A.; Signorelli, S.S. HDL-C role in acquired aortic valve stenosis patients and its relationship with oxidative stress. Medicina 2019, 55, 416. [Google Scholar] [CrossRef]
- Lerman, D.A.; Prasad, S.; Alotti, N. Calcific aortic valve disease: Molecular mechanisms and therapeutic approaches. Eur. Cardiol. 2015, 10, 108–112. [Google Scholar] [CrossRef]
- Fielitz, J.; Hein, S.; Mitrovic, V.; Pregla, R.; Zurbrügg, H.R.; Warnecke, C.; Schaper, J.; Fleck, E.; Regitz-Zagrosek, V. Activation of the cardiac renin-angiotensin system and increased myocardial collagen expression in human aortic valve disease. J. Am. Coll. Cardiol. 2001, 37, 1443–1449. [Google Scholar] [CrossRef]
- Lindman, B.R.; Clavel, M.A.; Mathieu, P.; Iung, B.; Lancellotti, P.; Otto, C.M.; Pibarot, P. Calcific aortic stenosis. Nat. Rev.Dis. Primers 2016, 2, 16006. [Google Scholar] [CrossRef] [PubMed]
- Pibarot, P.; Dumesnil, J.G. Improving assessment of aortic stenosis. J. Am. Coll. Cardiol. 2012, 60, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Antonini-Canterin, F.; Huang, G.; Cervesato, E.; Faggiano, P.; Pavan, D.; Piazza, R.; Nicolosi, G.L. Symptomatic aortic stenosis: Does systemic hypertension play an additional role? Hypertension 2003, 41, 1268–1272. [Google Scholar] [CrossRef] [PubMed]
- Sanson, M.; Augé, N.; Vindis, C.; Muller, C.; Bando, Y.; Thiers, J.C.; Marachet, M.A.; Zarkovic, K.; Sawa, Y.; Salvayre, R.; et al. Oxidized low-density lipoproteins trigger endoplasmic reticulum stress in vascular cells: Prevention by oxygen-regulated protein 150 expression. Circ. Res. 2009, 104, 328–336. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, M.; Wang, S.; Liang, B.; Zhao, Z.; Liu, C.; Wu, M.; Choi, H.C.; Lyons, T.J.; Zou, M.H. Activation of AMP-activated protein kinase inhibits oxidized LDL-triggered endoplasmic reticulum stress in vivo. Diabetes 2010, 59, 1386–1396. [Google Scholar] [CrossRef]
- Deng, J.; Lu, P.D.; Zhang, Y.; Scheuner, D.; Kaufman, R.J.; Sonenberg, N.; Harding, H.P.; Ron, D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell. Biol. 2004, 24, 10161–10168. [Google Scholar] [CrossRef]
- Cai, Z.; Li, F.; Gong, W.; Liu, W.; Duan, Q.; Chen, C.; Ni, L.; Xia, Y.; Cianflone, K.; Dong, N.; et al. Endoplasmic reticulum stress participates in aortic valve calcification in hypercholesterolemic animals. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2345–2354. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Luo, S.; Baumeister, P.; Yang, S.; Abcouwer, S.F.; Lee, A.S. Induction of Grp78/BiP by translational block: Activation of the Grp78 promoter by ATF4 through and upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J. Biol. Chem. 2003, 278, 37375–37385. [Google Scholar] [CrossRef]
- Clauss, I.M.; Gravallese, E.M.; Darling, J.M.; Shapiro, F.; Glimcher, M.J.; Glimcher, L.H. In situ hybridization studies suggest a role for the basic region-leucine zipper protein hXBP-1 in exocrine gland and skeletal development during mouse embryogenesis. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 1993, 197, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Tohmonda, T.; Miyauchi, Y.; Ghosh, R.; Yoda, M.; Uchikawa, S.; Takito, J.; Morioka, H.; Nakamura, M.; Iwawaki, T.; Chiba, K.; et al. The IRE1α-XBP1 pathway is essential for osteoblast differentiation through promoting transcription of Osterix. EMBO Rep. 2011, 12, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhou, J.; Zhang, P.; Song, F.; Jiang, R.; Li, M.; Xia, F.; Guo, F.J. IRE1α dissociates with BiP and inhibits ER stress-mediated apoptosis in cartilage development. Cell Signal. 2013, 25, 2136–2146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; McGrath, B.; Li, S.; Frank, A.; Zambito, F.; Reinert, J.; Gannon, M.; Ma, K.; McNaughton, K.; Cavener, D.R. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol. Cell. Biol. 2002, 22, 3864–3874. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Matsuda, K.; Bialek, P.; Jacquot, S.; Masuoka, H.C.; Schinke, T.; Li, L.; Brancorsini, S.; Sassone-Corsi, P.; Townes, T.M.; et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell 2004, 117, 387–398. [Google Scholar] [CrossRef]
- Furmanik, M.; van Gorp, R.; Whitehead, M.; Ahmad, S.; Bordoloi, J.; Kapustin, A.; Schurgers, L.J.; Shanahan, C.M. Endoplasmic reticulum stress mediates vascular smooth muscle cell calcification via increased release of Grp78 (Glucose-Regulated Protein, 78 kDa)-loaded extracellular vesicles. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 898–914. [Google Scholar] [CrossRef]
- Wang, Q.; Lin, P.; Feng, L.; Ren, Q.; Xie, X.; Zhang, B. Ameliorative effect of allicin on vascular calcification via inhibiting endoplasmic reticulum stress. Vascular 2021, 17085381211035291. [Google Scholar] [CrossRef]
- Song, X.; Li, J.; Jiao, M.; Chen, Y.; Pan, K. Effect of endoplasmic reticulum stress-induced apoptosis in the role of periodontitis on vascular calcification in a rat model. J. Mol. Histol. 2021, 52, 1097–1104. [Google Scholar] [CrossRef]
- Veliceasa, D.; Ivanovic, M.; Hoepfner, F.T.; Thumbikat, P.; Volpert, O.V.; Smith, N.D. Transient potential receptor channel 4 controls thrombospondin-1 secretion and angiogenesis in renal cell carcinoma. FEBS J. 2007, 274, 6365–6377. [Google Scholar] [CrossRef]
- Kuznetsov, G.; Chen, L.B.; Nigam, S.K. Multiple molecular chaperones complex with misfolded large oligomeric glycoproteins in the endoplasmic reticulum. J. Biol. Chem. 1997, 272, 3057–3063. [Google Scholar] [CrossRef]
- Lynch, J.M.; Maillet, M.; Vanhoutte, D.; Schloemer, A.; Sargent, M.A.; Blair, N.S.; Lynch, K.A.; Okada, T.; Aronow, B.J.; Osinska, H.; et al. A thrombospondin-dependent pathway for a protective ER stress response. Cell 2012, 149, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Dobaczewski, M.; Gonzalez-Quesada, C.; Chen, W.; Biernacka, A.; Li, N.; Lee, D.W.; Frangogiannis, N.G. Endogenous thrombospondin 1 protects the pressure-overloaded myocardium by modulating fibroblast phenotype and matrix metabolism. Hypertension 2011, 58, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, D.; Schips, T.G.; Vo, A.; Grimes, K.M.; Baldwin, T.A.; Brody, M.J.; Accornero, F.; Sargent, M.A.; Molkentin, J.D. Thbs1 induces lethal cardiac atrophy through PERK-ATF4 regulated autophagy. Nat. Commun. 2021, 12, 3928. [Google Scholar] [CrossRef] [PubMed]
- Calzada, M.J.; Sipes, J.M.; Krutzsch, H.C.; Yurchenco, P.D.; Annis, D.S.; Mosher, D.F.; Roberts, D.D. Recognition of the N-terminal modules of thrombospondin-1 and thrombospondin-2 by alpha6beta1 integrin. J. Biol. Chem. 2003, 278, 40679–40687. [Google Scholar] [CrossRef]
- Lopez-Dee, Z.; Pidcock, K.; Gutierrez, L.S. Thrombospondin-1: Multiple paths to inflammation. Mediat. Inflamm. 2011, 2011, 296069. [Google Scholar] [CrossRef]
- Lawler, J.; Hynes, R.O. The structure of human thrombospondin, an adhesive glycoprotein with multiple calcium-binding sites and homologies with several different proteins. J. Cell Biol. 1986, 103, 1635–1648. [Google Scholar] [CrossRef]
- Good, D.J.; Polverini, P.J.; Rastinejad, F.; Le Beau, M.M.; Lemons, R.S.; Frazier, W.A.; Bouck, N.P. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc. Natl. Acad. Sci. USA 1990, 87, 6624–6628. [Google Scholar] [CrossRef]
- Mumby, S.M.; Raugi, G.J.; Bornstein, P. Interactions of thrombospondin with extracellular matrix proteins: Selective binding to type V collagen. J. Cell Biol. 1984, 98, 646–652. [Google Scholar] [CrossRef]
- Li, Z.; He, L.; Wilson, K.; Roberts, D. Thrombospondin-1 inhibits TCR-mediated T lymphocyte early activation. J. Immunol. 2001, 166, 2427–2436. [Google Scholar] [CrossRef]
- Iruela-Arispe, M.L. Regulation of thrombospondin1 by extracellular proteases. Curr. Drug Targets 2008, 9, 863–868. [Google Scholar] [CrossRef]
- Starlinger, P.; Moll, H.P.; Assinger, A.; Nemeth, C.; Hoetzenecker, K.; Gruenberger, B.; Gruenberger, T.; Kuehrer, I.; Schoppmann, S.F.; Gnant, M.; et al. Thrombospondin-1: A unique marker to identify in vitro platelet activation when monitoring in vivo processes. J. Thromb. Haemost. 2010, 8, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Mikhailenko, I.; Krylov, D.; Argraves, K.M.; Roberts, D.D.; Liau, G.; Strickland, D.K. Cellular internalization and degradation of thrombospondin-1 is mediated by the amino-terminal heparin binding domain (HBD). High affinity interaction of dimeric HBD with the low density lipoprotein receptor-related protein. J. Biol. Chem. 1997, 272, 6784–6791. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, L.S.; Gutierrez, J. Thrombospondin 1 in metabolic diseases. Front. Endocrinol. 2021, 12, 638536. [Google Scholar] [CrossRef] [PubMed]
- Stenina, O.I.; Plow, E.F. Counterbalancing forces: What is thrombospondin-1 doing in atherosclerotic lesions? Circ. Res. 2008, 103, 1053–1055. [Google Scholar] [CrossRef] [PubMed]
- Adil, S.O.; Ibran, E.A.; Nisar, N.; Shafique, K. Pattern of unintentional burns: A hospital based study from Pakistan. Burns 2016, 42, 1345–1349. [Google Scholar] [CrossRef]
- Capoulade, R.; Mahmut, A.; Tastet, L.; Arsenault, M.; Bédard, É.; Dumesnil, J.G.; Després, J.P.; Larose, É.; Arsenault, B.J.; Bossé, Y.; et al. Impact of plasma Lp-PLA2 activity on the progression of aortic stenosis: The PROGRESSA study. JACC Cardiovasc. Imaging 2015, 8, 26–33. [Google Scholar] [CrossRef]
- Cho, K.I.; Sakuma, I.; Sohn, I.S.; Jo, S.H.; Koh, K.K. Inflammatory and metabolic mechanisms underlying the calcific aortic valve disease. Atherosclerosis 2018, 277, 60–65. [Google Scholar] [CrossRef]
- Cowell, S.J.; Newby, D.E.; Boon, N.A.; Elder, A.T. Calcific aortic stenosis: Same old story? Age Ageing 2004, 33, 538–544. [Google Scholar] [CrossRef]
- Doris, M.K.; Everett, R.J.; Shun-Shin, M.; Clavel, M.A.; Dweck, M.R. The Role of imaging in measuring disease progression and assessing novel therapies in aortic stenosis. JACC Cardiovasc. Imaging 2019, 12, 185–197. [Google Scholar] [CrossRef]
- Elmariah, S.; Mohler, E.R., 3rd. The Pathogenesis and treatment of the valvulopathy of aortic stenosis: Beyond the SEAS. Curr. Cardiol. Rep. 2010, 12, 125–132. [Google Scholar] [CrossRef]
- Gallo, G.; Presta, V.; Volpe, M.; Rubattu, S. Molecular and clinical implications of natriuretic peptides in aortic valve stenosis. J. Mol. Cell. Cardiol. 2019, 129, 266–271. [Google Scholar] [CrossRef] [PubMed]
- García-Rodríguez, C.; Parra-Izquierdo, I.; Castaños-Mollor, I.; López, J.; San Román, J.A.; Sánchez Crespo, M. Toll-Like receptors, inflammation, and calcific aortic valve disease. Front. Physiol. 2018, 9, 201. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Gómez, M.M.; Hernández-Betancor, I.; García-Niebla, J.; Marí-López, B.; Laynez-Cerdeña, I.; Lacalzada-Almeida, J. Valve calcification in aortic stenosis: Etiology and diagnostic imaging techniques. BioMed Res. Int. 2017, 2017, 5178631. [Google Scholar] [CrossRef] [PubMed]
- Kaden, J.J.; Bickelhaupt, S.; Grobholz, R.; Vahl, C.F.; Hagl, S.; Brueckmann, M.; Haase, K.K.; Dempfle, C.E.; Borggrefe, M. Expression of bone sialoprotein and bone morphogenetic protein-2 in calcific aortic stenosis. J. Heart Valve Dis. 2004, 13, 560–566. [Google Scholar]
- Kapelouzou, A.; Tsourelis, L.; Kaklamanis, L.; Degiannis, D.; Kogerakis, N.; Cokkinos, D.V. Serum and tissue biomarkers in aortic stenosis. Glob. Cardiol. Sci. Pract. 2015, 2015, 49. [Google Scholar] [CrossRef]
- Katholi, R.E.; Couri, D.M. Left ventricular hypertrophy: Major risk factor in patients with hypertension: Update and practical clinical applications. Int. J. Hypertens. 2011, 2011, 495349. [Google Scholar] [CrossRef]
- Kleinauskienė, R.; Jonkaitienė, R. Degenerative aortic stenosis, dyslipidemia and possibilities of medical treatment. Medicina 2018, 54, 24. [Google Scholar] [CrossRef]
- Kolasa-Trela, R.; Konieczynska, M.; Bazanek, M.; Undas, A. Specific changes in circulating cytokines and growth factors induced by exercise stress testing in asymptomatic aortic valve stenosis. PLoS ONE 2017, 12, e0173787. [Google Scholar] [CrossRef]
- Lee, S.H.; Choi, J.H. Involvement of inflammatory responses in the early development of calcific aortic valve disease: Lessons from statin therapy. Anim. Cells Syst. 2018, 22, 390–399. [Google Scholar] [CrossRef]
- Legere, S.A.; Haidl, I.D.; Légaré, J.F.; Marshall, J.S. Mast cells in cardiac fibrosis: New insights suggest opportunities for intervention. Front. Immunol. 2019, 10, 580. [Google Scholar] [CrossRef]
- Liu, T.; Song, D.; Dong, J.; Zhu, P.; Liu, J.; Liu, W.; Ma, X.; Zhao, L.; Ling, S. Current understanding of the pathophysiology of myocardial fibrosis and its quantitative assessment in heart failure. Front. Physiol. 2017, 8, 238. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.G.; Yuan, Y.P.; Wu, H.M.; Zhang, X.; Tang, Q.Z. Cardiac fibrosis: New insights into the pathogenesis. Int. J. Biol. Sci. 2018, 14, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, P.; Bouchareb, R.; Boulanger, M.C. Innate and adaptive immunity in calcific aortic valve disease. J. Immunol. Res. 2015, 2015, 851945. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, P.; Boulanger, M.C. Basic mechanisms of calcific aortic valve disease. Can. J. Cardiol. 2014, 30, 982–993. [Google Scholar] [CrossRef]
- Mathieu, P.; Boulanger, M.C.; Bouchareb, R. Molecular biology of calcific aortic valve disease: Towards new pharmacological therapies. Expert Rev. Cardiovasc. Ther. 2014, 12, 851–862. [Google Scholar] [CrossRef]
- Miller, J.D.; Chu, Y.; Brooks, R.M.; Richenbacher, W.E.; Peña-Silva, R.; Heistad, D.D. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J. Am. Coll. Cardiol. 2008, 52, 843–850. [Google Scholar] [CrossRef]
- Musa, T.A.; Treibel, T.A.; Vassiliou, V.S.; Captur, G.; Singh, A.; Chin, C.; Dobson, L.E.; Pica, S.; Loudon, M.; Malley, T.; et al. Myocardial Scar and Mortality in Severe Aortic Stenosis. Circulation 2018, 138, 1935–1947. [Google Scholar] [CrossRef]
- Myasoedova, V.A.; Ravani, A.L.; Frigerio, B.; Valerio, V.; Moschetta, D.; Songia, P.; Poggio, P. Novel pharmacological targets for calcific aortic valve disease: Prevention and treatments. Pharmacol. Res. 2018, 136, 74–82. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- O’Brien, K.D.; Reichenbach, D.D.; Marcovina, S.M.; Kuusisto, J.; Alpers, C.E.; Otto, C.M. Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of ’degenerative’ valvular aortic stenosis. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 523–532. [Google Scholar] [CrossRef]
- Osman, L.; Yacoub, M.H.; Latif, N.; Amrani, M.; Chester, A.H. Role of human valve interstitial cells in valve calcification and their response to atorvastatin. Circulation 2006, 114, I547–I552. [Google Scholar] [CrossRef] [PubMed]
- Pasipoularides, A. Calcific aortic valve disease: Part 2-morphomechanical abnormalities, gene reexpression, and gender effects on ventricular hypertrophy and its reversibility. J. Cardiovasc. Transl. Res. 2016, 9, 374–399. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, T.; Taskinen, P.; Näpänkangas, J.; Leskinen, H.; Ohtonen, P.; Soini, Y.; Juvonen, T.; Satta, J.; Vuolteenaho, O.; Ruskoaho, H. Increase in tissue endothelin-1 and ETA receptor levels in human aortic valve stenosis. Eur. Heart J. 2009, 30, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Perrucci, G.L.; Zanobini, M.; Gripari, P.; Songia, P.; Alshaikh, B.; Tremoli, E.; Poggio, P. Pathophysiology of aortic stenosis and mitral regurgitation. Compr. Physiol. 2017, 7, 799–818. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, A.L.; Ngo, D.T.; Chapman, M.J.; Ali, O.A.; Chirkov, Y.Y.; Horowitz, J.D. Pathogenesis of aortic stenosis: Not just a matter of wear and tear. Am. J. Cardiovasc. Dis. 2011, 1, 185–199. [Google Scholar]
- Syväranta, S.; Alanne-Kinnunen, M.; Oörni, K.; Oksjoki, R.; Kupari, M.; Kovanen, P.T.; Helske-Suihko, S. Potential pathological roles for oxidized low-density lipoprotein and scavenger receptors SR-AI, CD36, and LOX-1 in aortic valve stenosis. Atherosclerosis 2014, 235, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Towler, D.A. Oxidation, inflammation, and aortic valve calcification peroxide paves an osteogenic path. J. Am. Coll. Cardiol. 2008, 52, 851–854. [Google Scholar] [CrossRef]
- Venardos, N.; Nadlonek, N.A.; Zhan, Q.; Weyant, M.J.; Reece, T.B.; Meng, X.; Fullerton, D.A. Aortic valve calcification is mediated by a differential response of aortic valve interstitial cells to inflammation. J. Surg. Res. 2014, 190, 1–8. [Google Scholar] [CrossRef][Green Version]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef]
- Yetkin, E.; Waltenberger, J. Molecular and cellular mechanisms of aortic stenosis. Int. J. Cardiol. 2009, 135, 4–13. [Google Scholar] [CrossRef]
- Yip, C.Y.; Simmons, C.A. The aortic valve microenvironment and its role in calcific aortic valve disease. Cardiovasc. Pathol. 2011, 20, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Q.; Zeng, Q.; Song, R.; Zhai, Y.; Xu, D.; Fullerton, D.A.; Dinarello, C.A.; Meng, X. IL-37 suppresses MyD88-mediated inflammatory responses in human aortic valve interstitial cells. Mol. Med. 2017, 23, 83–91. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Protein Name | Uniprot ID | Reference | Protein Name | Uniprot ID | Reference |
|---|---|---|---|---|---|
| 72 kDa type IV collagenase | P08253 | Alvarez-Llamas G et al., 2013 [10] | Glutathione S-transferase P | P09211 | Martin-Rojas T et al., 2012 [13] |
| Alcohol dehydrogenase 1B | P00325 | Martin-Rojas T et al., 2015 [11] | Glycogen phosphorylase, liver form | P06737 | Alvarez-Llamas G et al., 2013 [10] |
| Alpha-1-acid glycoprotein 1 | P02763 | Martin-Rojas T et al., 2015 [11] | Haptoglobin | P00738 | Martin-Rojas T et al., 2012 [13]; Martin-Rojas T et al., 2015 [11] |
| Alpha-1-antichymotrypsin | P01011 | Gil-Dones F et al., 2012 [12]; Alvarez-Llamas G et al., 2013 [10] | Hemoglobin subunit beta | P68871 | Gil-Dones F et al., 2012 [12] |
| Alpha-1-antitrypsin | P01009 | Martin-Rojas T et al., 2012 [13]; Martin-Rojas T et al., 2015 [11]; Gil-Dones F et al., 2012 [12] | Hemopexin | P02790 | Martin-Rojas T et al., 2015 [11]; Gil-Dones F et al., 2012 [12] |
| Alpha-1B-glycoprotein | P04217 | Martin-Rojas T et al., 2012 [13] | Histone H2A type 1-H | Q96KK5 | Martin-Rojas T et al., 2015 [11] |
| Alpha-2-HS-glycoprotein | P02765 | Martin-Rojas T et al., 2015 [11]; Gil-Dones F et al., 2012 [12] | Ig gamma-1 chain C region | P01857 | Martin-Rojas T et al., 2015 [11]; Alvarez-Llamas G et al., 2013 [10] |
| Alpha-2-macroglobulin | P01023 | Alvarez-Llamas G et al., 2013 [10] | Ig kappa chain C region | P01834 | Gil-Dones F et al., 2012 [12] |
| Alpha-enolase | P06733 | Martin-Rojas T et al., 2015 [11] | Ig lambda-1 chain C regions | P0CG04 | Gil-Dones F et al., 2012 [12] |
| Angiotensinogen | P01019 | Alvarez-Llamas G et al., 2013 [10] | Ig mu chain C region | P01871 | Gil-Dones F et al., 2012 [12] |
| Annexin A1 | P04083 | Martin-Rojas T et al., 2015 [11] | Insulin-like growth factor-binding protein 5 | P24593 | Alvarez-Llamas G et al., 2013 [10] |
| Annexin A2 | P07355 | Martin-Rojas T et al., 2015 [11] | Insulin-like growth factor-binding protein 7 | Q16270 | Alvarez-Llamas G et al., 2013 [10] |
| Antithrombin-III | P01008 | Gil-Dones F et al., 2012 [12] | Inter-alpha-trypsin inhibitor heavy chain H4 | Q14624 | Gil-Dones F et al., 2012 [12] |
| Apolipoprotein A-I | P02647 | Martin-Rojas T et al., 2012 [13]; Gil-Dones F et al., 2012 [12] | Interleukin-6 | P05231 | Alvarez-Llamas G et al., 2013 [10] |
| Apolipoprotein A-IV | P06727 | Gil-Dones F et al., 2012 [12] | Killer cell immunoglobulin-like receptor 3DL3 | Q8N743 | Alvarez-Llamas G et al., 2013 [10] |
| Apolipoprotein B-100 | P04114 | Alvarez-Llamas G et al., 2013 [10] | Kininogen-1 | P01042 | Gil-Dones F et al., 2012 [12] |
| Apolipoprotein C-II | P02655 | Martin-Rojas T et al., 2015 [11] | Leucine-rich alpha-2-glycoprotein | P02750 | Gil-Dones F et al., 2012 [12] |
| Apolipoprotein C-III | P02656 | Gil-Dones F et al., 2012 [12] | Leukocyte receptor cluster member 9 | Q96B70 | Alvarez-Llamas G et al., 2013 [10] |
| Apolipoprotein E | P02649 | Gil-Dones F et al., 2012 [12] | L-lactate dehydrogenase A chain | P00338 | Martin-Rojas T et al., 2015 [11] |
| Beta-1,4-galactosyl- transferase 2 | O60909 | Alvarez-Llamas G et al., 2013 [10] | Lumican | P51884 | Martin-Rojas T et al., 2012 [13]; Martin-Rojas T et al., 2015 [11]; Alvarez-Llamas G et al., 2013 [10] |
| Biglycan | P21810 | Martin-Rojas T et al., 2015 [11]; Alvarez-Llamas G et al., 2013 [10] | Mannose-binding protein C | P11226 | Gil-Dones F et al., 2012 [12] |
| Biogenesis of lysosome-related organelles complex 1 subunit 5 | Q8TDH9 | Martin-Rojas T et al., 2015 [11] | Metalloproteinase inhibitor 1 | P01033 | Alvarez-Llamas G et al., 2013 [10] |
| Calcineurin-binding protein cabin-1 | Q9Y6J0 | Martin-Rojas T et al., 2015 [11] | Metalloproteinase inhibitor 3 | P35625 | Alvarez-Llamas G et al., 2013 [10] |
| Calreticulin | P27797 | Martin-Rojas T et al., 2012 [13] | Moesin | P26038 | Martin-Rojas T et al., 2015 [11] |
| Cartilage oligomeric matrix protein | P49747 | Alvarez-Llamas G et al., 2013 [10] | Nuclear factor NF-kappa-B p100 subunit | Q00653 | Alvarez-Llamas G et al., 2013 [10] |
| Cathepsin B | P07858 | Alvarez-Llamas G et al., 2013 [10] | Pentraxin-related protein PTX3 | P26022 | Alvarez-Llamas G et al., 2013 [10] |
| Cathepsin D | P07339 | Alvarez-Llamas G et al., 2013 [10] | Peptidyl-prolyl cis-trans isomerase A | P62937 | Martin-Rojas T et al., 2015 [11] |
| CD5 antigen-like | O43866 | Gil-Dones F et al., 2012 [12] | Periostin | Q15063 | Martin-Rojas T et al., 2015 [11] |
| CD9 antigen | P21926 | Alvarez-Llamas G et al., 2013 [10] | Peroxiredoxin-1 | Q06830 | Martin-Rojas T et al., 2015 [11] |
| Ceruloplasmin | P00450 | Gil-Dones F et al., 2012 [12]; Alvarez-Llamas G et al., 2013 [10] | Phosphoglycerate kinase 1 | P00558 | Martin-Rojas T et al., 2015 [11] |
| Chitinase-3-like protein 1 | P36222 | Alvarez-Llamas G et al., 2013 [10] | Phospholipid transfer protein | P55058 | Alvarez-Llamas G et al., 2013 [10] |
| Chitinase-3-like protein 2 | Q15782 | Alvarez-Llamas G et al., 2013 [10] | Pigment epithelium-derived factor | P36955 | Alvarez-Llamas G et al., 2013 [10] |
| Clusterin | P10909 | Gil-Dones F et al., 2012 [12]; Alvarez-Llamas G et al., 2013 [10] | Plasma protease C1 inhibitor | P05155 | Gil-Dones F et al., 2012 [12]; Alvarez-Llamas G et al., 2013 [10] |
| Coagulation factor XII | P00748 | Gil-Dones F et al., 2012 [12] | Plasminogen activator inhibitor 1 | P05121 | Alvarez-Llamas G et al., 2013 [10] |
| Collagen alpha-1(III) chain | P02461 | Alvarez-Llamas G et al., 2013 [10] | Pre-B-cell leukemia transcription factor-interacting protein 1 | Q96AQ6 | Alvarez-Llamas G et al., 2013 [10] |
| Collagen alpha-1(VI) chain | P12109 | Martin-Rojas T et al., 2015 [11]; Alvarez-Llamas G et al., 2013 [10] | Procollagen C-endopeptidase enhancer 2 | Q9UKZ9 | Alvarez-Llamas G et al., 2013 [10] |
| Collagen alpha-1(XIV) chain | Q05707 | Alvarez-Llamas G et al., 2013 [10] | Prolargin | P51888 | Martin-Rojas T et al., 2015 [11] |
| Collagen alpha-2(I) chain | P08123 | Alvarez-Llamas G et al., 2013 [10] | Prosaposin | P07602 | Alvarez-Llamas G et al., 2013 [10] |
| Collagen alpha-3(VI) chain | P12111 | Mourino-Alvarez L et al., 2016 [14] | Prostaglandin-H2 D-isomerase | P41222 | Alvarez-Llamas G et al., 2013 [10] |
| Complement C1s subcomponent | P09871 | Alvarez-Llamas G et al., 2013 [10] | Protein AMBP | P02760 | Gil-Dones F et al., 2012 [12] |
| Complement C3 | P01024 | Gil-Dones F et al., 2012 [12]; Alvarez-Llamas G et al., 2013 [10] | Protein NDRG2 | Q9UN36 | Mourino-Alvarez L et al., 2016 [14] |
| Complement C4-A | P0C0L4 | Gil-Dones F et al., 2012 [12] | Protein phosphatase 1 regulatory subunit 3E | Q9H7J1 | Alvarez-Llamas G et al., 2013 [10] |
| Complement C4-B | P0C0L5 | Gil-Dones F et al., 2012 [12] | Protein S100-A6 | P06703 | Martin-Rojas T et al., 2015 [11] |
| Complement component C9 | P02748 | Gil-Dones F et al., 2012 [12] | Prothrombin | P00734 | Gil-Dones F et al., 2012 [12] |
| Complement factor H | P08603 | Gil-Dones F et al., 2012 [12] | Serine protease HTRA1 | Q92743 | Alvarez-Llamas G et al., 2013 [10] |
| Complement factor H-related protein 1 | Q03591 | Gil-Dones F et al., 2012 [12] | Serotransferrin | P02787 | Martin-Rojas T et al., 2015 [11] |
| Complement factor I | P05156 | Gil-Dones F et al., 2012 [12] | Serum albumin | P02768 | Martin-Rojas T et al., 2012 [13]; Martin-Rojas T et al., 2015 [11] |
| Cystatin-C | P01034 | Alvarez-Llamas G et al., 2013 [10] | Serum amyloid P-component | P02743 | Martin-Rojas T et al., 2012 [13]; Martin-Rojas T et al., 2015 [11] |
| Decorin | P07585 | Martin-Rojas T et al., 2015 [11] | Serum paraoxonase/arylesterase 1 | P27169 | Gil-Dones F et al., 2012 [12] |
| EGF-containing fibulin-like extracellular matrix protein 1 | Q12805 | Alvarez-Llamas G et al., 2013 [10] | Serum paraoxonase/lactonase 3 | Q15166 | Alvarez-Llamas G et al., 2013 [10] |
| Endoplasmin | P14625 | Martin-Rojas T et al., 2015 [11] | Spondin-1 | Q9HCB6 | Alvarez-Llamas G et al., 2013 [10] |
| Extracellular superoxide dismutase [Cu-Zn] | P08294 | Martin-Rojas T et al., 2012 [13] | Superoxide dismutase [Cu-Zn] | P00441 | Martin-Rojas T et al., 2015 [11] |
| Fatty acid-binding protein, adipocyte | P15090 | Martin-Rojas T et al., 2012 [13] | Superoxide dismutase [Mn], mitochondrial | P04179 | Martin-Rojas T et al., 2015 [11] |
| Fibrinogen alpha chain | P02671 | Gil-Dones F et al., 2012 [12] | SWI/SNF complex subunit SMARCC1 | Q92922 | Alvarez-Llamas G et al., 2013 [10] |
| Fibrinogen beta chain | P02675 | Gil-Dones F et al., 2012 [12] | Tenascin-X | P22105 | Alvarez-Llamas G et al., 2013 [10] |
| Fibrinogen gamma chain | P02679 | Martin-Rojas T et al., 2012 [13]; Gil-Dones F et al., 2012 [12] | Thrombospondin-1 | P07996 | Alvarez-Llamas G et al., 2013 [10] |
| Fibronectin | P02751 | Alvarez-Llamas G et al., 2013 [10] | Transforming growth factor-beta-induced protein ig-h3 | Q15582 | Martin-Rojas T et al., 2015 [11] |
| Ficolin-2 | Q15485 | Gil-Dones F et al., 2012 [12] | Transgelin | Q01995 | Martin-Rojas T et al., 2012 [13]; Martin-Rojas T et al., 2015 [11] |
| Follistatin-related protein 3 | O95633 | Alvarez-Llamas G et al., 2013 [10] | Transthyretin | P02766 | Martin-Rojas T et al., 2012 [13] |
| FRAS1-related extracellular matrix protein 2 | Q5SZK8 | Alvarez-Llamas G et al., 2013 [10] | Triosephosphate isomerase | P60174 | Martin-Rojas T et al., 2015 [11] |
| Galectin-1 | P09382 | Martin-Rojas T et al., 2015 [11] | Tubulin beta chain | P07437 | Martin-Rojas T et al., 2015 [11] |
| Gelsolin | P06396 | Alvarez-Llamas G et al., 2013 [10] | Vimentin | P08670 | Martin-Rojas T et al., 2012 [13]; Martin-Rojas T et al., 2015 [11] |
| Glutathione peroxidase 3 | P22352 | Martin-Rojas T et al., 2012 [13]; Martin-Rojas T et al., 2015 [11] | Vitronectin | P04004 | Gil-Dones F et al., 2012 [12] |
| ANN Category | ANN Score | Associated p-Value | |
|---|---|---|---|
| Strong relationship | Very high | >92 | <0.01 |
| High | 78–92 | 0.01–0.05 | |
| Medium-High | 63–78 | 0.05–0.15 | |
| Medium relationship | Medium | 38–63 | 0.15–0.25 |
| Low or no relationship | Low | <38 | >0.25 |
| Controls | Severe AS | p-Value | |
|---|---|---|---|
| Age | 67.76 ± 10.03 | 79.94 ± 7.21 | 0.000 |
| Gender (M/F) | 11/7 | 9/9 | 0.584 |
| BMI | 28.48 ± 4.62 | 26.92 ± 3.67 | 0.282 |
| AHT (Yes,%) | 9 (50%) | 12 (67%) | 0.406 |
| Dyslipidemia (Yes, %) | 5 (28%) | 9 (50%) | 0.265 |
| Diabetes (Yes, %) | 0 (0%) | 0 (0%) | 1.000 |
| Smokers (Yes, %) | 2 (11%) | 0 (0%) | 0.584 |
| Pneumopathy (Yes, %) | 0 (0%) | 1 (5%) | 0.791 |
| Chronic IHD (Yes, %) | 0 (0%) | 0 (0%) | 1.000 |
| Very High | High | Medium-High | |||||
|---|---|---|---|---|---|---|---|
| Effectors | No Effectors | Effectors | No Effectors | Effectors | No Effectors | TOTAL | |
| DAS general characterization | - | - | 7 | - | 13 | 15 | 35 |
| 1. Calcification | - | - | 4 | 1 | 1 | 4 | 10 |
| 2. Lipoprotein accumulation | - | - | - | - | 2 | 10 | 12 |
| 3. Inflammation | - | - | 3 | 5 | 1 | 11 | 20 |
| 4. Oxidative stress | - | - | 0 | 1 | 3 | 3 | 7 |
| 5. Endothelial dysfunction | - | - | 5 | 2 | 2 | 13 | 22 |
| 6. RAA system | - | - | - | - | 1 | 4 | 5 |
| 7. Hypertrophy | - | - | 1 | 1 | - | 8 | 10 |
| 8. Myocardial fibrosis | 1 | - | 3 | 1 | 1 | 4 | 10 |
| Uniprot ID | Gene Name | Protein Name | Motive Effector | ANN Score | Related Motive |
|---|---|---|---|---|---|
| P08123 | COL1A2 | Collagen alpha-2(I) chain | Yes | 92.52 | Myocardial fibrosis |
| No | 90.86 | Inflammation | |||
| Yes | 87.50 | Endothelial dysfunction | |||
| Yes | 69.82 | DAS General | |||
| P35625 | TIMP3 | Metalloproteinase inhibitor 3 | Yes | 91.85 | Endothelial dysfunction |
| Yes | 82.53 | DAS General | |||
| No | 69.20 | Calcification | |||
| No | 63.61 | RAA system | |||
| P02461 | COL3A1 | Collagen alpha-1(III) chain | Yes | 91.69 | Myocardial fibrosis |
| Yes | 87.29 | Endothelial dysfunction | |||
| Yes | 76.47 | DAS General | |||
| P01033 | TIMP1 | Metalloproteinase inhibitor 1 | Yes | 91.45 | Endothelial dysfunction |
| Yes | 84.60 | DAS General | |||
| No | 68.80 | Calcification | |||
| P05231 | IL6 | Interleukin-6 | Yes | 90.45 | Myocardial fibrosis |
| Yes | 88.05 | Calcification | |||
| Yes | 87.64 | Inflammation | |||
| Yes | 73.24 | DAS General | |||
| P01042 | KNG1 | Kininogen-1 | Yes | 90.45 | DAS General |
| Yes | 87.34 | Inflammation | |||
| No | 70.10 | RAA system | |||
| No | 63.40 | Calcification | |||
| P07339 | CTSD | Cathepsin D | Yes | 87.00 | DAS General |
| Yes | 84.46 | Endothelial dysfunction | |||
| No | 71.66 | Hypertrophy | |||
| P21810 | BGN | Biglycan | Yes | 86.97 | Inflammation |
| No | 84.26 | Calcification | |||
| No | 83.08 | Myocardial fibrosis | |||
| Yes | 80.74 | DAS General | |||
| No | 78.98 | Endothelial dysfunction | |||
| No | 70.50 | Lipoprotein accumulation | |||
| P14625 | HSP90B1 | Endoplasmin | No | 86.48 | Inflammation |
| No | 74.62 | Endothelial dysfunction | |||
| No | 70.58 | DAS General | |||
| No | 65.44 | Lipoprotein accumulation | |||
| P01019 | AGT | Angiotensinogen | Yes | 84.81 | DAS General |
| Yes | 82.54 | Myocardial fibrosis | |||
| Yes | 78.60 | Hypertrophy | |||
| Yes | 65.54 | RAA system | |||
| P08253 | MMP2 | 72 kDa type IV collagenase | Yes | 84.40 | Calcification |
| Yes | 75.40 | Endothelial dysfunction | |||
| Yes | 74.10 | Myocardial fibrosis | |||
| Yes | 73.98 | DAS General | |||
| No | 71.88 | Hypertrophy | |||
| No | 64.52 | Oxidative stress | |||
| P02766 | TTR | Transthyretin | No | 79.18 | Oxidative stress |
| No | 74.11 | Inflammation | |||
| No | 71.21 | DAS General | |||
| P00441 | SOD1 | Superoxide dismutase [Cu-Zn] | No | 78.56 | Inflammation |
| Yes | 74.41 | DAS General | |||
| Yes | 69.12 | Oxidative stress | |||
| P10909 | CLU | Clusterin | No | 76.50 | Inflammation |
| No | 67.68 | Endothelial dysfunction | |||
| No | 64.01 | Lipoprotein accumulation | |||
| P07996 | THBS1 | Thrombospondin-1 | No | 76.17 | Endothelial dysfunction |
| No | 71.70 | Myocardial fibrosis | |||
| No | 63.52 | RAA system | |||
| P02768 | ALB | Serum albumin | No | 76.03 | Inflammation |
| No | 75.65 | Endothelial dysfunction | |||
| No | 68.55 | Hypertrophy | |||
| No | 68.34 | Lipoprotein accumulation | |||
| Q00653 | NFKB2 | Nuclear factor NF-kappa-B p100 subunit | Yes | 75.62 | Inflammation |
| Yes | 74.83 | Calcification | |||
| Yes | 72.15 | DAS General | |||
| P04114 | APOB | Apolipoprotein B-100 | No | 75.04 | Endothelial dysfunction |
| No | 74.02 | Inflammation | |||
| Yes | 71.61 | DAS General | |||
| Yes | 64.60 | Lipoprotein accumulation | |||
| P02647 | APOA1 | Apolipoprotein A-I | Yes | 74.73 | DAS General |
| No | 72.41 | Endothelial dysfunction | |||
| Yes | 64.68 | Lipoprotein accumulation | |||
| P07585 | DCN | Decorin | No | 74.42 | Calcification |
| No | 73.11 | Endothelial dysfunction | |||
| No | 71.71 | Hypertrophy | |||
| No | 68.53 | Myocardial fibrosis | |||
| Q92743 | HTRA1 | Serine protease HTRA1 | No | 72.82 | Myocardial fibrosis |
| No | 72.73 | Endothelial dysfunction | |||
| No | 70.23 | DAS General | |||
| P01023 | A2M | Alpha-2-macroglobulin | No | 72.55 | Oxidative stress |
| No | 70.52 | Myocardial fibrosis | |||
| No | 68.56 | Inflammation | |||
| No | 68.30 | Endothelial dysfunction |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corbacho-Alonso, N.; Sastre-Oliva, T.; Corros, C.; Tejerina, T.; Solis, J.; López-Almodovar, L.F.; Padial, L.R.; Mourino-Alvarez, L.; Barderas, M.G. Prioritization of Candidate Biomarkers for Degenerative Aortic Stenosis through a Systems Biology-Based In-Silico Approach. J. Pers. Med. 2022, 12, 642. https://doi.org/10.3390/jpm12040642
Corbacho-Alonso N, Sastre-Oliva T, Corros C, Tejerina T, Solis J, López-Almodovar LF, Padial LR, Mourino-Alvarez L, Barderas MG. Prioritization of Candidate Biomarkers for Degenerative Aortic Stenosis through a Systems Biology-Based In-Silico Approach. Journal of Personalized Medicine. 2022; 12(4):642. https://doi.org/10.3390/jpm12040642
Chicago/Turabian StyleCorbacho-Alonso, Nerea, Tamara Sastre-Oliva, Cecilia Corros, Teresa Tejerina, Jorge Solis, Luis F. López-Almodovar, Luis R. Padial, Laura Mourino-Alvarez, and Maria G. Barderas. 2022. "Prioritization of Candidate Biomarkers for Degenerative Aortic Stenosis through a Systems Biology-Based In-Silico Approach" Journal of Personalized Medicine 12, no. 4: 642. https://doi.org/10.3390/jpm12040642
APA StyleCorbacho-Alonso, N., Sastre-Oliva, T., Corros, C., Tejerina, T., Solis, J., López-Almodovar, L. F., Padial, L. R., Mourino-Alvarez, L., & Barderas, M. G. (2022). Prioritization of Candidate Biomarkers for Degenerative Aortic Stenosis through a Systems Biology-Based In-Silico Approach. Journal of Personalized Medicine, 12(4), 642. https://doi.org/10.3390/jpm12040642