
Advances and Personalized Approaches in the Frontline Treatment of T-Cell Lymphomas


Abstract
:1. Introduction
2. Novel Approaches to Frontline Therapies—Successes and Failures
2.1. Brentuximab Vedotin
2.2. Histone Deacetylase Inhibitors (HDACis)
2.3. Pralatrexate
2.4. Mogamulizumab
2.5. Alemtuzumab
2.6. Lenalidomide
2.7. Azacitidine
2.8. PI3K Inhibitors (PI3Ki)
2.9. Consolidative Stem Cell Transplantation
3. Emerging Personalized Therapies
3.1. ALK Inhibitors
3.2. Cellular Therapy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vose, J.M.; Neumann, M.; Harris, M.E. International peripheral T-cell and natural killer/T-cell lymphoma study: Pathology findings and clinical outcomes international T-cell lymphoma project. J. Clin. Oncol. 2008, 26, 4124–4130. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Lee Harris, N.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Van Arnam, J.S.; Lim, M.S.; Elenitoba-Johnson, K.S.J. Novel insights into the pathogenesis of T-cell lymphomas. Blood 2018, 131, 2320–2330. [Google Scholar] [CrossRef] [PubMed]
- Falchi, L.; Ma, H.; Klein, S.; Lue, J.K.; Montanari, F.; Marchi, E.; Deng, C.; Kim, H.A.; Rada, A.; Jacob, A.T.; et al. Combined oral 5-azacytidine and romidepsin are highly effective in patients with PTCL: A multicenter phase 2 study. Blood 2021, 137, 2161–2170. [Google Scholar] [CrossRef] [PubMed]
- De Leval, L.; Gaulard, P. Cellular origin of T-cell lymphomas. Blood 2014, 123, 2909–2910. [Google Scholar] [CrossRef]
- Wang, T.; Feldman, A.L.; Wada, D.A.; Lu, Y.; Polk, A.; Briski, R.; Ristow, K.; Habermann, T.M.; Thomas, D.; Ziesmer, S.C.; et al. GATA-3 expression identifies a high-risk subset of PTCL, NOS with distinct molecular and clinical features. Blood 2014, 123, 3007–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC: Lyon, France, 2017; ISBN 9789283244943. [Google Scholar]
- Iqbal, J.; Wright, G.; Wang, C.; Rosenwald, A.; Gascoyne, R.D.; Weisenburger, D.D.; Greiner, T.C.; Smith, L.; Guo, S.; Wilcox, R.A.; et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood 2014, 123, 2915–2923. [Google Scholar] [CrossRef]
- Savage, K.J.; Chhanabhai, M.; Gascoyne, R.D.; Connors, J.M. Characterization of peripheral T-cell lymphomas in a single North American institution by the WHO classification. Ann. Oncol. 2004, 15, 1467–1475. [Google Scholar] [CrossRef]
- Gallamini, A.; Stelitano, C.; Calvi, R.; Bellei, M.; Mattei, D.; Vitolo, U.; Morabito, F.; Martelli, M.; Brusamolino, E.; Iannitto, E.; et al. Peripheral T-cell lymphoma unspecified (PTCL-U): A new prognostic model from a retrospective multicentric clinical study. Blood 2004, 103, 2474–2479. [Google Scholar] [CrossRef] [Green Version]
- Reimer, P.; Rüdiger, T.; Geissinger, E.; Weissinger, F.; Nerl, C.; Schmitz, N.; Engert, A.; Einsele, H.; Muller-Hermelink, H.K.; Wilhelm, M. Autologous stem-cell transplantation as first-line therapy in peripheral T-cell lymphomas: Results of a prospective multicenter study. J. Clin. Oncol. 2009, 27, 106–113. [Google Scholar] [CrossRef]
- D’Amore, F.; Relander, T.; Lauritzsen, G.F.; Jantunen, E.; Hagberg, H.; Anderson, H.; Holte, H.; Österborg, A.; Merup, M.; Brown, P.; et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J. Clin. Oncol. 2012, 30, 3093–3099. [Google Scholar] [CrossRef]
- Mak, V.; Hamm, J.; Chhanabhai, M.; Shenkier, T.; Klasa, R.; Sehn, L.H.; Villa, D.; Gascoyne, R.D.; Connors, J.M.; Savage, K.J. Survival of patients with peripheral T-cell lymphoma after first relapse or progression: Spectrum of disease and rare long-term survivors. J. Clin. Oncol. 2013, 31, 1970–1976. [Google Scholar] [CrossRef]
- Biasoli, I.; Casaretti, M.; Bellei, M.; Maiorana, A.; Bonacorsi, G.; Quaresima, M.; Salati, M.; Federio, M.; Luminari, S. Dismal outcome of T-cell lymphoma patients failing first-line treatment: Results of a population-based study from the Modena Cancer Registry. Hematol. Oncol. 2015, 33, 147–151. [Google Scholar] [CrossRef]
- Bellei, M.; Foss, F.M.; Shustov, A.R.; Horwitz, S.M.; Marcheselli, L.; Kim, W.S.; Cabrera, M.E.; Dlouhy, I.; Nagler, A.; Advani, R.H.; et al. The outcome of peripheral T-cell lymphoma patients failing first-line therapy: A report from the prospective, international T-cell project. Haematologica 2018, 103, 1191–1197. [Google Scholar] [CrossRef]
- Vaklavas, C.; Forero-Torres, A. Safety and efficacy of brentuximab vedotin in patients with Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Ther. Adv. Hematol. 2012, 3, 209–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Sabattini, E.; Pizzi, M.; Tabanelli, V.; Baldin, P.; Sagramoso Sacchetti, C.; Agostinelli, C.; Luigi Zinzani, P.; Pileri, S.A. CD30 expression in peripheral T-cell lymphomas. Haematologica 2013, 98, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Onaindia, A.; Martínez, N.; Montes-Moreno, S.; Almaraz, C.; Rodríguez-Pinilla, S.M.; Cereceda, L.; Revert, J.B.; Ortega, C.; Tardio, A.; González, L.; et al. CD30 expression by B and T cells: A frequent finding in angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma-not otherwise specified. Am. J. Surg. Pathol. 2016, 40, 378–385. [Google Scholar] [CrossRef]
- Younes, A.; Bartlett, N.L.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.L.; Forero-Torres, A. Brentuximab Vedotin (SGN-35) for Relapsed CD30-Positive Lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821. [Google Scholar] [CrossRef] [Green Version]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase II study. J. Clin. Oncol. 2012, 30, 2190–2196. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, S.M.; Advani, R.H.; Bartlett, N.L.; Jacobsen, E.D.; Sharman, J.P.; O’Connor, O.A.; Siddiqi, T.; Kennedy, D.A.; Oki, Y. Objective responses in relapsed T-cell lymphomas with single-agent brentuximab vedotin. Blood 2014, 123, 3095–3100. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.H.; Tavallaee, M.; Sundram, U.; Salva, K.A.; Wood, G.S.; Li, S.; Rozati, S.; Nagpal, S.; Krathen, M.; Reddy, S.; et al. Phase II investigator-initiated study of brentuximab vedotin in mycosis fungoides and Sézary syndrome with variable CD30 expression level: A multi-institution collaborative project. J. Clin. Oncol. 2015, 33, 3750–3758. [Google Scholar] [CrossRef] [PubMed]
- Fanale, M.A.; Horwitz, S.M.; Forero-Torres, A.; Bartlett, N.L.; Advani, R.H.; Pro, B.; Chen, R.W.; Davies, A.; Illidge, T.; Uttarwar, M.; et al. Five-year outcomes for frontline brentuximab vedotin with CHP for CD30-expressing peripheral T-cell lymphomas. Blood 2018, 131, 2120–2124. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, S.; O’Connor, O.A.; Pro, B.; Trümper, L.; Iyer, S.; Advani, R.; Bartlett, N.L.; Christensen, J.H.; Morschhauser, F.; Domingo-Domenech, E.; et al. The ECHELON-2 Trial: 5-year results of a randomized, phase 3 study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann. Oncol. 2021. [Google Scholar] [CrossRef]
- Jagadeesh, D.; Sims, R.B.; Horowitz, S.M. Trial-in-progress: Frontline brentuximab vedotin and CHP (A+CHP) in patients with peripheral T-cell lymphoma with less than 10% CD30 expression. Blood 2020, 136, 30. [Google Scholar] [CrossRef]
- Schmitz, N.; Trümper, L.; Ziepert, M.; Nickelsen, M.; Ho, A.D.; Metzner, B.; Peter, N.; Loeffler, M.; Rosenwald, A.; Pfreundschuh, M. Treatment and prognosis of mature T-cell and NK-cell lymphoma: An analysis of patients with T-cell lymphoma treated in studies of the German High-Grade Non-Hodgkin Lymphoma Study Group. Blood 2010, 116, 3418–3425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, A.F.; Zain, J.; Savage, K.J.; Feldman, T.A.; Brammer, J.E.; Chen, L.; Popplewell, L.L.; Budde, L.E.; Peters, L.; Kurtzman, Y.; et al. Preliminary Results from a Phase 2 Trial of Brentuximab Vedotin Plus Cyclophosphamide, Doxorubicin, Etoposide, and Prednisone (CHEP-BV) Followed By BV Consolidation in Patients with CD30-Positive Peripheral T-Cell Lymphomas. Blood 2019, 134, 4023. [Google Scholar] [CrossRef]
- Chen, I.C.; Sethy, B.; Liou, J.P. Recent Update of HDAC Inhibitors in Lymphoma. Front. Cell Dev. Biol. 2020, 8, 1–14. [Google Scholar] [CrossRef]
- Palomero, T.; Couronné, L.; Khiabanian, H.; Kim, M.Y.; Ambesi-Impiombato, A.; Perez-Garcia, A.; Carpenter, Z.; Abate, F.; Allegretta, M.; Haydu, J.E.; et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat. Genet. 2014, 46, 166–170. [Google Scholar] [CrossRef]
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 171–175. [Google Scholar] [CrossRef]
- Duvic, M.; Vu, J. Vorinostat: A new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin. Investig. Drugs 2007, 16, 1111–1120. [Google Scholar] [CrossRef]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109, 31–39. [Google Scholar] [CrossRef]
- Olsen, E.A.; Kim, Y.H.; Kuzel, T.M.; Pacheco, T.R.; Foss, F.M.; Parker, S.; Frankel, S.R.; Chen, C.; Ricker, J.L.; Arduino, J.M.; et al. Phase IIB multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2007, 25, 3109–3115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oki, Y.; Younes, A.; Copeland, A.; Hagemeister, F.; Fayad, L.E.; Mclaughlin, P.; Shah, J.; Fowler, N.; Romaguera, J.; Kwak, L.W.; et al. Phase I study of vorinostat in combination with standard CHOP in patients with newly diagnosed peripheral T-cell lymphoma. Br. J. Haematol. 2013, 162, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Hopfinger, G.; Nösslinger, T.; Lang, A.; Linkesch, W.; Melchardt, T.; Weiss, L.; Egle, A.; Greil, R. Lenalidomide in combination with vorinostat and dexamethasone for the treatment of relapsed/refractory peripheral T cell lymphoma (PTCL): Report of a phase I/II trial. Ann. Hematol. 2014, 93, 459–462. [Google Scholar] [CrossRef]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Borchmann, P.; Morschhauser, F.; Wilhelm, M.; et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J. Clin. Oncol. 2012, 30, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Morschhauser, F.; Wilhelm, M.; Pinter-Brown, L.; et al. Romidepsin for the treatment of relapsed/refractory peripheral T-cell lymphoma: Pivotal study update demonstrates durable responses. J. Hematol. Oncol. 2014, 7, 1–9. [Google Scholar] [CrossRef]
- Dupuis, J.; Morschhauser, F.; Ghesquières, H.; Tilly, H.; Casasnovas, O.; Thieblemont, C.; Ribrag, V.; Bossard, C.; Le Bras, F.; Bachy, E.; et al. Combination of romidepsin with cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated patients with peripheral T-cell lymphoma: A non-randomised, phase 1b/2 study. Lancet Haematol. 2015, 2, e160–e165. [Google Scholar] [CrossRef]
- Bachy, E.; Camus, V.; Thieblemont, C.; Casasnovas, R.-O.; Ysebaert, L.; Damaj, G.L.; Guidez, S.; Pica, G.-M.; Kim, W.S.; Lim, S.T.; et al. Final Analysis of the Ro-CHOP Phase III Study (Conducted by LYSA): Romidepsin Plus CHOP in Patients with Peripheral T-Cell Lymphoma. Blood 2020, 136, 32–33. [Google Scholar] [CrossRef]
- Bachy, E.; Camus, V.; Thieblemont, C.; Sibon, D.; Casasnovas, R.-O.; Ysebaert, L.; Damaj, G.; Guidez, S.; Pica, G.M.; Kim, W.S.; et al. Romidepsin Plus CHOP Versus CHOP in Patients with Previously Untreated Peripheral T-Cell Lymphoma: Results of the Ro-CHOP Phase III Study (Conducted by LYSA). J. Clin. Oncol. 2021, 1–11. [Google Scholar] [CrossRef]
- Chiappella, A.; Carniti, C.; Re, A.; Castellino, C.; Evangelista, A.; Tabanelli, V.; Ciancia, R.; Orsucci, L.; Pinto, A.; Usai, S.V.; et al. Adding Romidepsin to CHOEP in First Line Treatment of Peripheral T-Cell Lymphomas Does Not Improve the Response Rate: Final Analysis of Phase II PTCL13 Study. Blood 2021, 138, 134. [Google Scholar] [CrossRef]
- Foss, F.; Advani, R.; Duvic, M.; Hymes, K.B.; Intragumtornchai, T.; Lekhakula, A.; Shpilberg, O.; Lerner, A.; Belt, R.J.; Jacobsen, E.D.; et al. A Phase II trial of Belinostat (PXD101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma. Br. J. Haematol. 2015, 168, 811–819. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Horwitz, S.; Masszi, T.; Van Hoof, A.; Brown, P.; Doorduijn, J.; Hess, G.; Jurczak, W.; Knoblauch, P.; Chawla, S.; et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the pivotal phase II BELIEF (CLN-19) study. J. Clin. Oncol. 2015, 33, 2492–2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, P.B.; Cashen, A.F.; Nikolinakos, P.G.; Beaven, A.W.; Barta, S.K.; Bhat, G.; Hasal, S.J.; De Vos, S.; Oki, Y.; Deng, C.; et al. Belinostat in combination with standard cyclophosphamide, doxorubicin, vincristine and prednisone as first-line treatment for patients with newly diagnosed peripheral T-cell lymphoma. Exp. Hematol. Oncol. 2021, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Howman, R.A.; Prince, H.M. New drug therapies in peripheral T-cell lymphoma. Expert Rev. Anticancer Ther. 2011, 11, 457–472. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Hamlin, P.A.; Portlock, C.; Moskowitz, C.H.; Noy, A.; Straus, D.J.; MacGregor-Cortelli, B.; Neylon, E.; Sarasohn, D.; Dumetrescu, O.; et al. Pralatrexate, a novel class of antifol with high affinity for the reduced folate carrier-type 1, produces marked complete and durable remissions in a diversity of chemotherapy refractory cases of T-cell lymphoma. Br. J. Haematol. 2007, 139, 425–428. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Pro, B.; Pinter-Brown, L.; Bartlett, N.; Popplewell, L.; Coiffier, B.; Lechowicz, M.J.; Savage, K.J.; Shustov, A.R.; Gisselbrecht, C.; et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: Results from the pivotal PROPEL study. J. Clin. Oncol. 2011, 29, 1182–1189. [Google Scholar] [CrossRef]
- Advani, R.H.; Ansell, S.M.; Lechowicz, M.J.; Beaven, A.W.; Loberiza, F.; Carson, K.R.; Evens, A.M.; Foss, F.; Horwitz, S.; Pro, B.; et al. A phase II study of cyclophosphamide, etoposide, vincristine and prednisone (CEOP) Alternating with Pralatrexate (P) as front line therapy for patients with peripheral T-cell lymphoma (PTCL): Final results from the T- cell consortium trial. Br. J. Haematol. 2016, 172, 535–544. [Google Scholar] [CrossRef] [Green Version]
- Moore, D.C.; Elmes, J.B.; Shibu, P.A.; Larck, C.; Park, S.I. Mogamulizumab: An Anti-CC Chemokine Receptor 4 Antibody for T-Cell Lymphomas. Ann. Pharmacother. 2020, 54, 371–379. [Google Scholar] [CrossRef]
- Ishida, T.; Inagaki, H.; Utsunomiya, A.; Takatsuka, Y.; Komatsu, H.; Iida, S.; Takeuchi, G.; Eimoto, T.; Nakamura, S.; Ueda, R. CXC chemokine receptor 3 and CC chemokine receptor 4 expression in T-cell and NK-cell lymphomas with special reference to clinicopathological significance for peripheral T-cell lymphoma, unspecified. Clin. Cancer Res. 2004, 10, 5494–5500. [Google Scholar] [CrossRef] [Green Version]
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M.; et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 2003, 278, 3466–3473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Utsunomiya, A.; Tobinai, K.; Tsukasaki, K.; Uike, N.; Uozumi, K.; Yamaguchi, K.; Yamada, Y.; Hanada, S.; Tamura, K.; et al. Phase I study of KW-0761, a defucosylated humanized anti-CCR4 antibody, in relapsed patients with adult T-cell leukemia-lymphoma and peripheral T-cell lymphoma. J. Clin. Oncol. 2010, 28, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Zinzani, P.L.; Karlin, L.; Radford, J.; Caballero, D.; Fields, P.; Chamuleau, M.E.D.; D’Amore, F.; Haioun, C.; Thieblemont, C.; González-Barca, E.; et al. European phase II study of mogamulizumab, an anti-CCR4 monoclonal antibody, in relapsed/refractory peripheral T-cell lymphoma. Haematologica 2016, 101, e407–e410. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Jo, T.; Takemoto, S.; Suzushima, H.; Uozumi, K.; Yamamoto, K.; Uike, N.; Saburi, Y.; Nosaka, K.; Utsunomiya, A.; et al. Dose-intensified chemotherapy alone or in combination with mogamulizumab in newly diagnosed aggressive adult T-cell leukaemia-lymphoma: A randomized phase II study. Br. J. Haematol. 2015, 169, 672–682. [Google Scholar] [CrossRef]
- Kim, Y.H.; Bagot, M.; Pinter-Brown, L.; Rook, A.H.; Porcu, P.; Horwitz, S.M.; Whittaker, S.; Tokura, Y.; Vermeer, M.; Zinzani, P.L.; et al. Mogamulizumab versus vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): An international, open-label, randomised, controlled phase 3 trial. Lancet Oncol. 2018, 19, 1192–1204. [Google Scholar] [CrossRef]
- Went, P.; Agostinelli, C.; Gallamini, A.; Piccaluga, P.P.; Ascani, S.; Sabattini, E.; Bacci, F.; Falini, B.; Motta, T.; Paulli, M.; et al. Marker expression in peripheral T-cell lymphoma: A proposed clinical-pathologic prognostic score. J. Clin. Oncol. 2006, 24, 2472–2479. [Google Scholar] [CrossRef]
- Wulf, G.G.; Altmann, B.; Ziepert, M.; D’Amore, F.; Held, G.; Greil, R.; Tournilhac, O.; Relander, T.; Viardot, A.; Wilhelm, M.; et al. Alemtuzumab plus CHOP versus CHOP in elderly patients with peripheral T-cell lymphoma: The DSHNHL2006-1B/ACT-2 trial. Leukemia 2021, 35, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Yuan, C.M.; Hubacheck, J.; Janik, J.E.; Wilson, W.; Morris, J.C.; Jasper, G.A.; Stetler-Stevenson, M. Variable CD52 expression in mature T cell and NK cell malignancies: Implications for alemtuzumab therapy. Br. J. Haematol. 2009, 145, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Gallamini, A.; Zaja, F.; Patti, C.; Billio, A.; Specchia, M.R.; Tucci, A.; Levis, A.; Manna, A.; Secondo, V.; Rigacci, L.; et al. Alemtuzumab (Campath-1H) and CHOP chemotherapy as first-line treatment of peripheral T-cell lymphoma: Results of a GITIL (Gruppo Italiano Terapie Innovative nei Linfomi) prospective multicenter trial. Blood 2007, 110, 2316–2323. [Google Scholar] [CrossRef] [Green Version]
- Binder, C.; Ziepert, M.; Pfreundschuh, M.; Dührsen, U.; Eimermacher, H.; Aldaoud, A.; Rosenwald, A.; Loeffler, M.; Schmitz, N.; Truemper, L. CHO(E)P-14 followed by alemtuzumab consolidation in untreated peripheral T cell lymphomas: Final analysis of a prospective phase II trial. Ann. Hematol. 2013, 92, 1521–1528. [Google Scholar] [CrossRef] [Green Version]
- Kluin-Nelemans, H.C.; van Marwijk Kooy, M.; Lugtenburg, P.J.; van Putten, W.L.J.; Luten, M.; Oudejans, J.; van Imhoff, G.W. Intensified alemtuzumab-CHOP therapy for peripheral T-cell lymphoma. Ann. Oncol. 2011, 22, 1595–1600. [Google Scholar] [CrossRef] [PubMed]
- Toumishey, E.; Prasad, A.; Dueck, G.; Chua, N.; Finch, D.; Johnston, J.; Van Der Jagt, R.; Stewart, D.; White, D.; Belch, A.; et al. Final report of a phase 2 clinical trial of lenalidomide monotherapy for patients with T-Cell lymphoma. Cancer 2015, 121, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Lunning, M.; Horwitz, S.; Advani, R.; Vose, J.; Lee, H.; Mehta-Shah, N.; Zain, J.; Haverkos, B.; Lechowicz, B.; Moskowitz, A.; et al. Phase I/II study of CHOEP plus lenalidomide as initial therapy for patients with stage II-IV peripheral T-cell lymphoma: Phase II results. Hematol. Oncol. 2019, 227, 280–281. [Google Scholar] [CrossRef] [Green Version]
- Lemonnier, F.; Safar, V.; Beldi-Ferchiou, A.; Cottereau, A.S.; Bachy, E.; Cartron, G.; Fataccioli, V.; Pelletier, L.; Robe, C.; Letourneau, A.; et al. Integrative analysis of a phase 2 trial combining lenalidomide with CHOP in angioimmunoblastic T-cell lymphoma. Blood Adv. 2021, 5, 539–548. [Google Scholar] [CrossRef]
- Ruan, J.; Zain, J.M.; Palmer, B.; Jovanovic, B.; Mi, X.; Swaroop, A.; Winter, J.; Gordon, L.I.; Karmali, R.; Pro, B. Multicenter phase II study of romidepsin plus lenalidomide for patients with previously untreated peripheral T-cell lymphoma (PTCL). J. Clin. Oncol. 2021, 39, 7514. [Google Scholar] [CrossRef]
- Derissen, E.J.; Beijnen, J.H.; Schellens, J.H. Concise drug review: Azacitidine and decitabine. Oncologist 2013, 18, 619–624. [Google Scholar] [CrossRef] [Green Version]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Lemonnier, F.; Dupuis, J.; Sujobert, P.; Tournillhac, O.; Cheminant, M.; Sarkozy, C.; Pelletier, L.; Marçais, A.; Robe, C.; Fataccioli, V.; et al. Treatment with 5-azacytidine induces a sustained response in patients with angioimmunoblastic T-cell lymphoma. Blood 2018, 132, 2305–2309. [Google Scholar] [CrossRef] [Green Version]
- Ruan, J.; Moskowitz, A.J.; Mehta-Shah, N.; Sokol, L.; Chen, Z.; Rahim, R.; Song, W.; Van Besien, K.; Horwitz, S.M.; Rutherford, S.C.; et al. Multi-Center Phase II Study of Oral Azacitidine (CC-486) Plus CHOP As Initial Treatment for Peripheral T-Cell Lymphoma (PTCL). Blood 2020, 136, 33–34. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, S.M.; Koch, R.; Porcu, P.; Oki, Y.; Moskowitz, A.; Perez, M.; Myskowski, P.; Officer, A.; Jaffe, J.D.; Morrow, S.N.; et al. Activity of the PI3K-δ,g inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma. Blood 2018, 131, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, S.M.; Moskowitz, A.J.; Mehta-Shah, N.; Jacobsen, E.D.; Khodadoust, M.S.; Ganesan, N.; Drill, E.; Hancock, H.; Davey, T.; Myskowski, P.; et al. The combination of duvelisib and romidepsin (DR) is highly active against relapsed/refractory peripheral T-cell lymphoma with low rates of transaminitis: Final results and biomarker analysis. In Proceedings of the 2021 ASH Annual Meeting and Exposition, Atlanta, GA, USA, 11–14 December 2021; p. 619. [Google Scholar]
- Brammer, J.E.; Zinzani, P.L.; Zain, J.; Mead, M.; Casulo, C.; Jacobsen, E.D.; Gritti, G.; Litwak, D.; Cohan, D.; Katz, D.J.; et al. Duvelisib in Patients with Relapsed/Refractory Peripheral T-Cell Lymphoma from the Phase 2 Primo Trial: Results of an Interim Analysis. Blood 2021, 138, 2456. [Google Scholar] [CrossRef]
- Stiff, P.J.; Unger, J.M.; Cook, J.R.; Constine, L.S.; Couban, S.; Stewart, D.A.; Shea, T.C.; Porcu, P.; Winter, J.N.; Kahl, B.S.; et al. Autologous Transplantation as Consolidation for Aggressive Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2013, 369, 1681–1690. [Google Scholar] [CrossRef] [Green Version]
- Al-Mansour, Z.; Li, H.; Cook, J.R.; Constine, L.S.; Couban, S.; Stewart, D.A.; Shea, T.C.; Porcu, P.; Winter, J.N.; Kahl, B.S.; et al. Autologous transplantation as consolidation for high risk aggressive T-cell non-Hodgkin lymphoma: A SWOG 9704 intergroup trial subgroup analysis. Leuk. Lymphoma 2019, 60, 1934–1941. [Google Scholar] [CrossRef]
- Sibon, D.; Fournier, M.; Brière, J.; Lamant, L.; Haioun, C.; Coiffier, B.; Bologna, S.; Morel, P.; Gabarre, J.; Hermine, O.; et al. Long-term outcome of adults with systemic anaplastic large-cell lymphoma treated within the Groupe d’Étude des Lymphomes de l’Adulte Trials. J. Clin. Oncol. 2012, 30, 3939–3946. [Google Scholar] [CrossRef]
- Parrilla Castellar, E.R.; Jaffe, E.S.; Said, J.W.; Swerdlow, S.H.; Ketterling, R.P.; Knudson, R.A.; Sidhu, J.S.; Hsi, E.D.; Karikehalli, S.; Jiang, L.; et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood 2014, 124, 1473–1480. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, M.B.; Hamilton-Dutoit, S.J.; Bendix, K.; Ketterling, R.P.; Bedroske, P.P.; Luoma, I.M.; Sattler, C.A.; Boddicker, R.L.; Bennani, N.N.; Nørgaard, P.; et al. DUSP22 and TP63 rearrangements predict outcome of ALK-negative anaplastic large cell lymphoma: A Danish cohort study. Blood 2017, 130, 554–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, B.J.; Mok, T.; Kim, D.-W.; Wu, Y.-L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-Line Crizotinib versus Chemotherapy in ALK -Positive Lung Cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossé, Y.P.; Lim, M.S.; Voss, S.D.; Wilner, K.; Ruffner, K.; Laliberte, J.; Rolland, D.; Balis, F.M.; Maris, J.M.; Weigel, B.J.; et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: A Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013, 14, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Passerini, C.G.; Farina, F.; Stasia, A.; Redaelli, S.; Ceccon, M.; Mologni, L.; Messa, C.; Guerra, L.; Giudici, G.; Sala, E.; et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J. Natl. Cancer Inst. 2014, 106, 2–5. [Google Scholar] [CrossRef]
- Fukano, R.; Mori, T.; Sekimizu, M.; Choi, I.; Kada, A.; Saito, A.M.; Asada, R.; Takeuchi, K.; Terauchi, T.; Tateishi, U.; et al. Alectinib for relapsed or refractory anaplastic lymphoma kinase-positive anaplastic large cell lymphoma: An open-label phase II trial. Cancer Sci. 2020, 111, 4540–4547. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
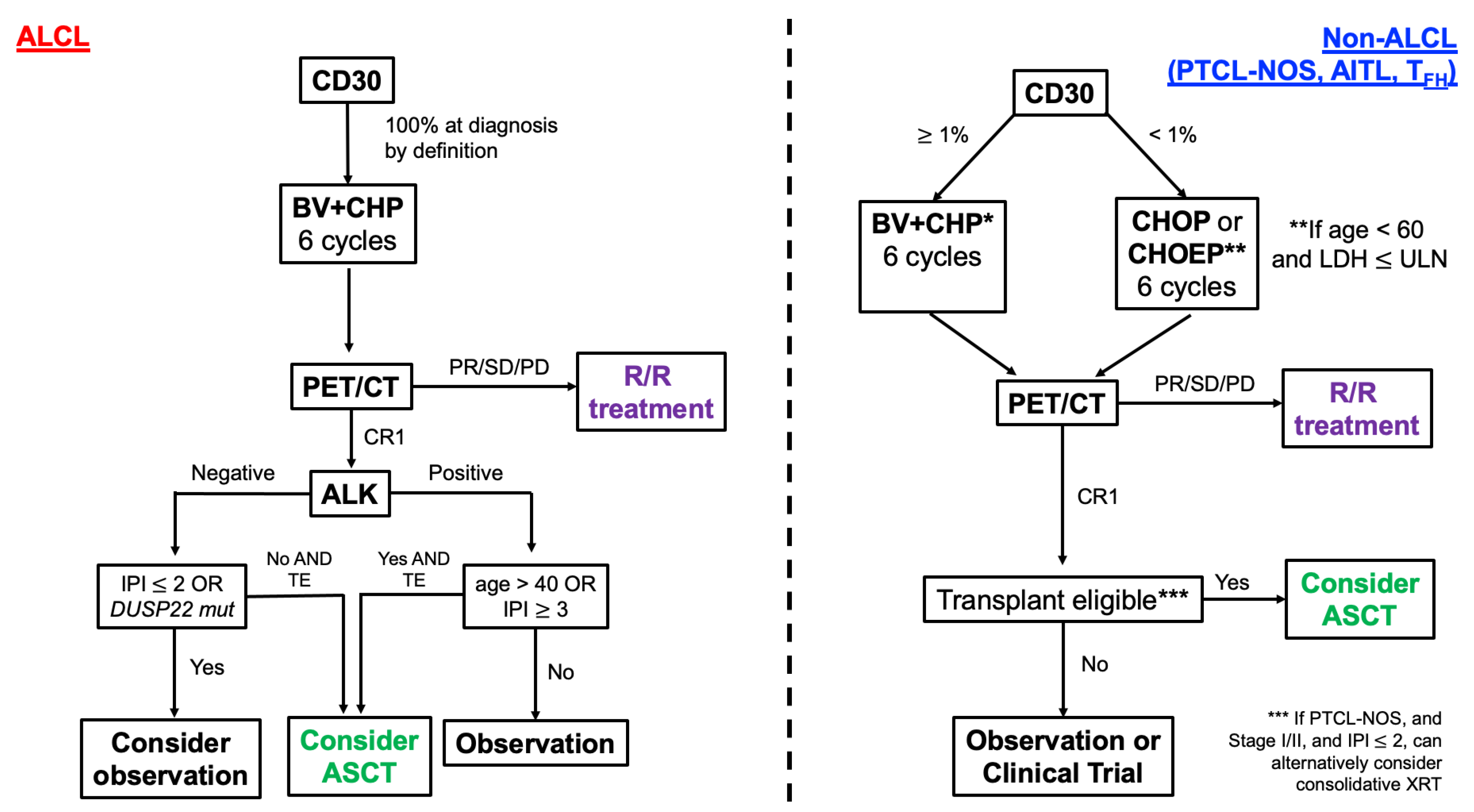

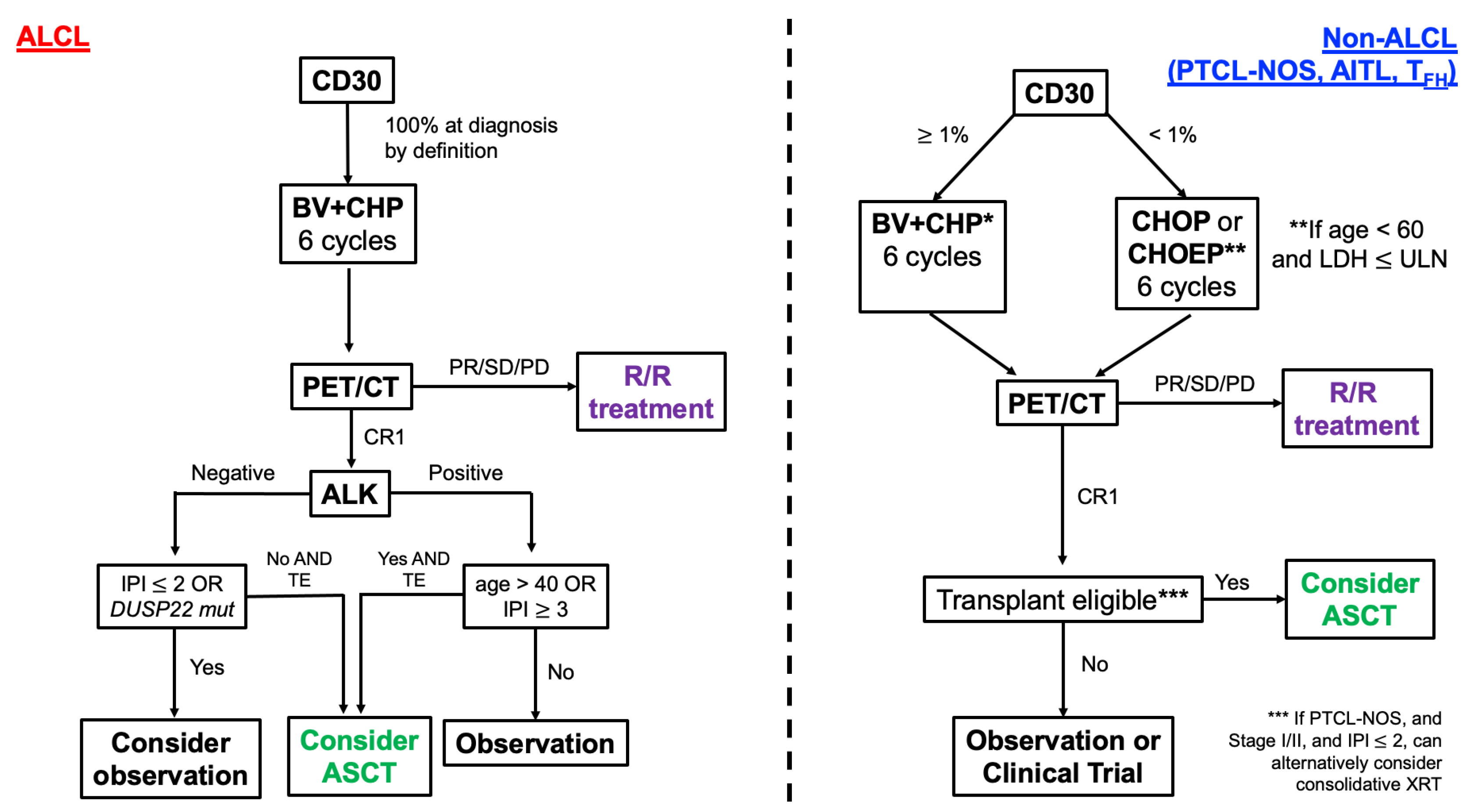{kind=link}
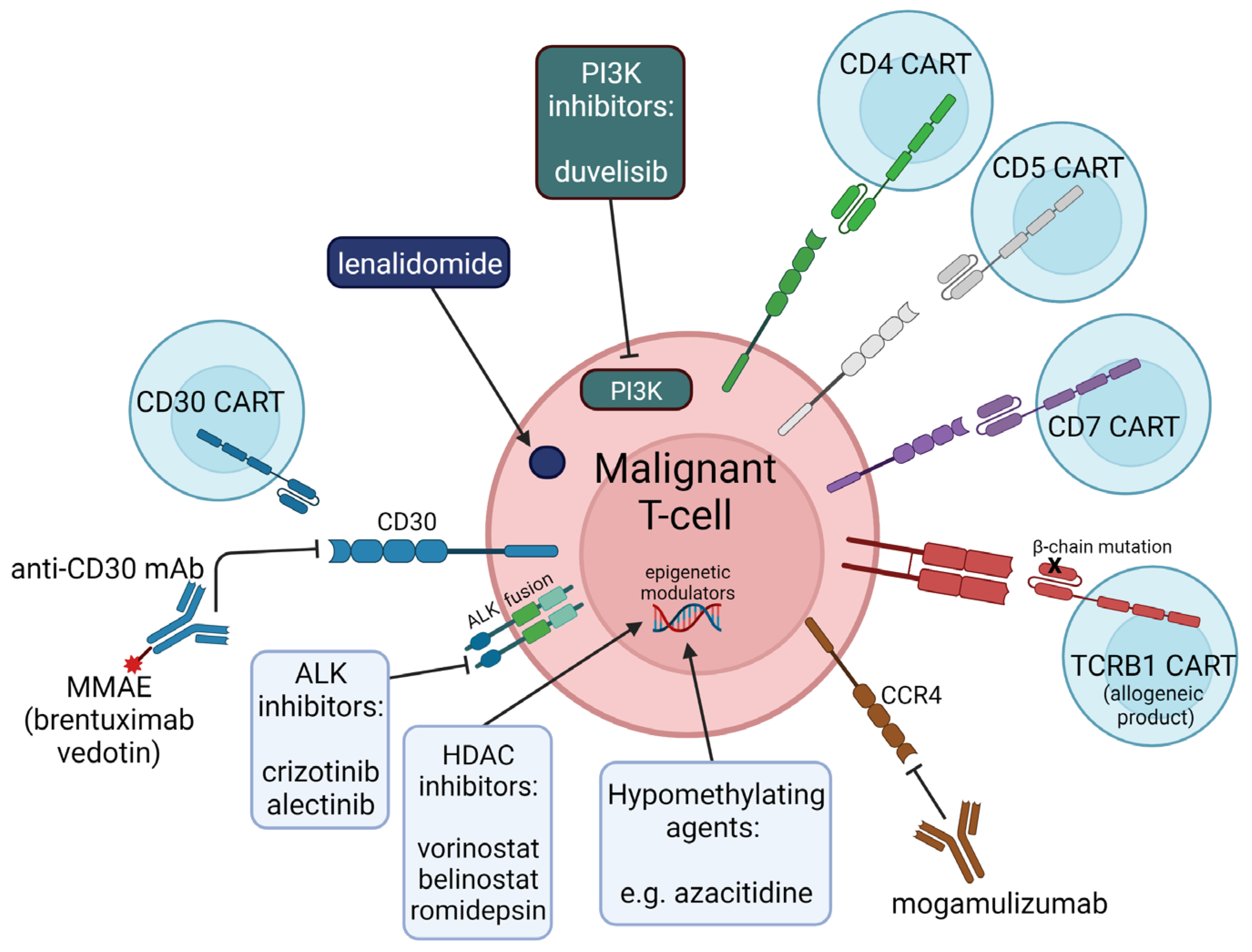
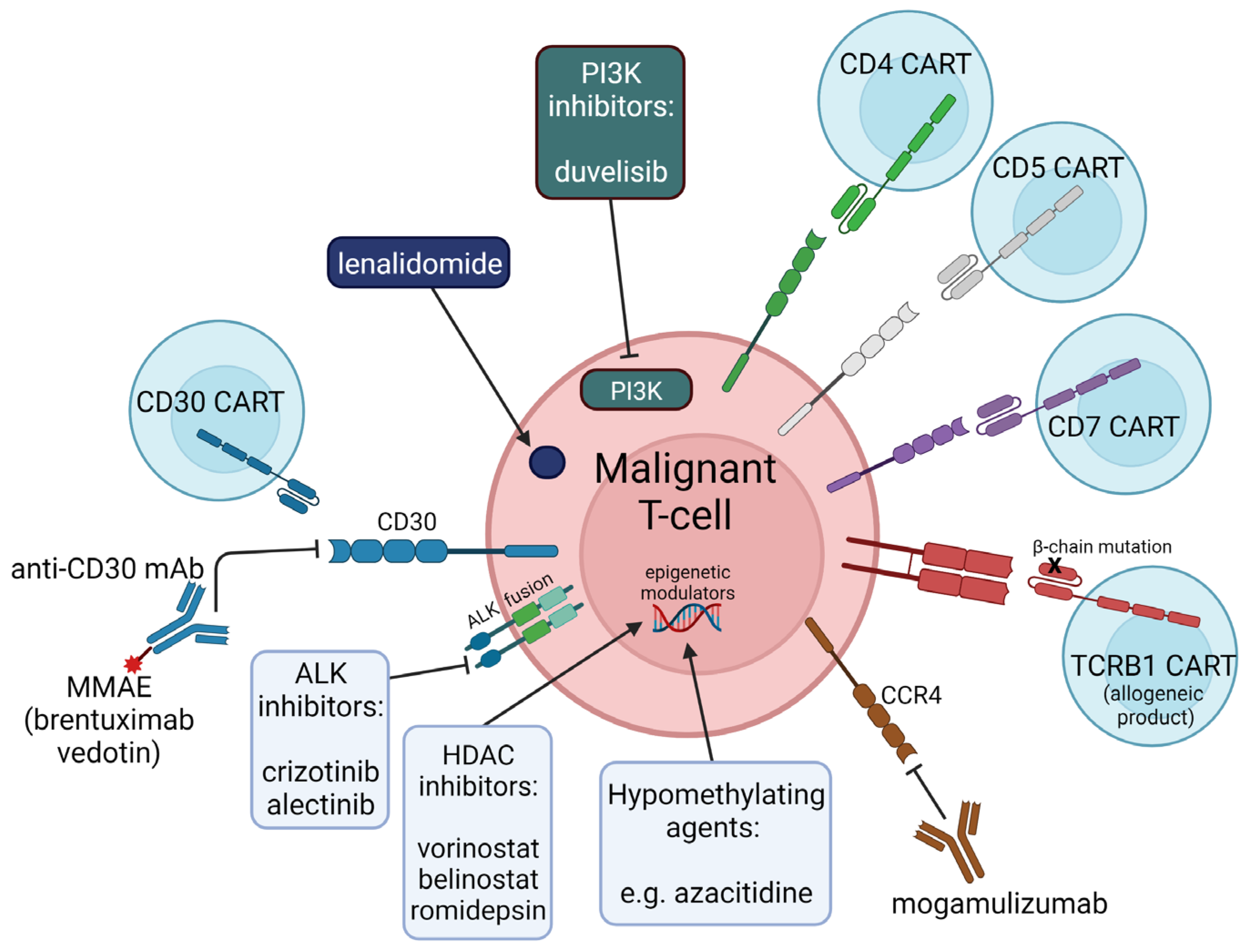{kind=link}
| Trial Number | Sponsor | Experimental Treatment | Phase | Status | PTCL Status |
|---|---|---|---|---|---|
| NCT04334174 * | Univ. of Kansas | BV after ASCT | II | Recruiting | BV + CHP induction in CD30+ PTCL |
| NCT03719105 * | New York Medical College | Pralatrexate + BV + chemotherapy | I | Recruiting | PTCL (non-ALCL or non-NK leukemia/lymphoma) |
| NCT01716806 * | Seagen, Inc. | BV | II | Recruiting | CD30+ PTCL |
| NCT04569032 * | Seagen, Inc. | BV + CHP | II | Recruiting | CD30+ PTCL < 10% |
| NCT04803201 * | Alliance for Clinical Trials in Oncology | Duvelisib or azacitidine (CC-486) + CHO(E)P | II | Recruiting | CD30+ PTCL < 10% |
| NCT03728972 * | MSKCC | Pembrolizumab | II | Recruiting | NK/T-cell lymphoma |
| NCT04639843 * | National Cancer Institute (NCI) | Doxorubicin + azacitidine + romidepsin + duvelisib | I | Not yet recruiting | New and R/R PTCL |
| NCT02737046 * | University of Miami | Belinostat + zidovudine | II | Recruiting | ATLL |
| NCT03264131 * | UNC | BV-CHEP | II | Recruiting | ATLL |
| NCT04301076 * | National Cancer Institute | Lenalidomide + EPOCH | I | Recruiting | ATLL |
| NCT04795869 | Northwestern University | BV + pembrolizumab | II | Not yet recruiting | R/R PTCL |
| NCT04747236 | Univ. of Virginia | Azacitidine + romidepsin | II | Recruiting | R/R PTCL |
| NCT03240211 | Univ. of Virginia | Pembrolizumab + Decitabine and/or Pralatrexate | I | Recruiting | R/R PTCL |
| NCT03598998 | City of Hope | Pralatrexate + pembrolizumab | I/II | Recruiting | R/R PTCL |
| NCT03534180 | City of Hope | Romidepsin + venetoclax | II | Recruiting | R/R PTCL |
| NCT03278782 | M.D. Anderson | Romidepsin + pembrolizumab | I/II | Recruiting | R/R PTCL |
| NCT03011814 | City of Hope | Durvalumab +/− lenalidomide | I/II | Recruiting | R/R PTCL |
| NCT04447027 | National Cancer Institute | Romidepsin + azacitidine + dexamethasone + lenalidomide | I | Recruiting | R/R PTCL |
| NCT04703192 | Daiichi Sankyo, Inc. | Valemetostat | II | Recruiting | R/R PTCL |
| Cellular Therapy for R/R PTCL | |||||
| NCT04712864 | Legend Biotech USA, Inc. | CD4 CAR-T | I | Recruiting | R/R CD4+ PTCL |
| NCT03690011 | Baylor College of Medicine | CD7 CAR-T | I | Recruiting | R/R CD7+ PTCL |
| NCT04984356 | Wugen, Inc. | CD7 CAR-T | I | Recruiting | R/R CD7+ PTCL |
| NCT04004637 | PersonGen BioTherapeutics | CD7 CAR-T | I | Recruiting | NK/T-cell lymphoma |
| NCT04083495 | UNC | CD30 CAR-T | II | Recruiting | R/R CD30+ PTCL |
| NCT04526834 | Tessa Therapeutics | CD30 CAR-T | I | Recruiting | R/R CD30+ PTCL |
| NCT02917083 | Baylor College of Medicine | CD30 CAR-T | I | Recruiting | R/R CD30+ PTCL |
| NCT04136275 | Massachusetts General Hospital | CD37 CAR-T | I | Recruiting | R/R CD37+ PTCL |
| NCT03590574 | Autolus Limited | TRBC1 CAR-T | I/II | Recruiting | R/R TRBC1+ PTCL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angelos, M.G.; Ballard, H.J.; Barta, S.K. Advances and Personalized Approaches in the Frontline Treatment of T-Cell Lymphomas. J. Pers. Med. 2022, 12, 267. https://doi.org/10.3390/jpm12020267
Angelos MG, Ballard HJ, Barta SK. Advances and Personalized Approaches in the Frontline Treatment of T-Cell Lymphomas. Journal of Personalized Medicine. 2022; 12(2):267. https://doi.org/10.3390/jpm12020267
Chicago/Turabian StyleAngelos, Mathew G., Hatcher J. Ballard, and Stefan K. Barta. 2022. "Advances and Personalized Approaches in the Frontline Treatment of T-Cell Lymphomas" Journal of Personalized Medicine 12, no. 2: 267. https://doi.org/10.3390/jpm12020267
APA StyleAngelos, M. G., Ballard, H. J., & Barta, S. K. (2022). Advances and Personalized Approaches in the Frontline Treatment of T-Cell Lymphomas. Journal of Personalized Medicine, 12(2), 267. https://doi.org/10.3390/jpm12020267