
Serotonin-Related Functional Genetic Variants Affect the Occurrence of Psychiatric and Motor Adverse Events of Dopaminergic Treatment in Parkinson’s Disease: A Retrospective Cohort Study


Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants and Clinical Data
2.2. Single Nucleotide Polymorphism (SNP) Selection
2.3. DNA Isolation and Genotyping
2.4. Statistical Analysis
3. Results
3.1. Patients’ Characteristics
3.2. SNP Genotyping Analysis
3.3. Influence of Genetic Variability on the Risk for Psychiatric and Motor Adverse Events
3.4. Influence of Haplotypes on the Risk for Psychiatric and Motor Adverse Events
3.5. Influence of Gene–Gene Interactions on the Risk for Psychiatric and Motor Adverse Events
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bamalan, O.A.; Al Khalili, Y. Physiology, Serotonin. Available online: https://www.ncbi.nlm.nih.gov/books/NBK545168/ (accessed on 13 December 2021).
- Vegas-Suarez, S.; Paredes-Rodriguez, E.; Aristieta, A.; Lafuente, J.V.; Miguelez, C.; Ugedo, L. Dysfunction of serotonergic neurons in Parkinson’s disease and dyskinesia. Int. Rev. Neurobiol. 2019, 146, 259–279. [Google Scholar] [PubMed]
- Berger, M.; Gray, J.A.; Roth, B.L. The expanded biology of serotonin. Annu. Rev. Med. 2009, 60, 355–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huot, P.; Sgambato-Faure, V.; Fox, S.H.; McCreary, A.C. Serotonergic Approaches in Parkinson’s Disease: Translational Perspectives, an Update. ACS Chem. Neurosci. 2017, 8, 973–986. [Google Scholar] [CrossRef] [PubMed]
- Grosch, J.; Winkler, J.; Kohl, Z. Early Degeneration of Both Dopaminergic and Serotonergic Axons—A Common Mechanism in Parkinson’s Disease. Front. Cell Neurosci. 2016, 10, 293. [Google Scholar] [CrossRef] [Green Version]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Politis, M.; Niccolini, F. Serotonin in Parkinson’s disease. Behav. Brain Res. 2015, 277, 136–145. [Google Scholar] [CrossRef]
- Muñoz, A.; Lopez-Lopez, A.; Labandeira, C.M.; Labandeira-Garcia, J.L. Interactions Between the Serotonergic and Other Neurotransmitter Systems in the Basal Ganglia: Role in Parkinson’s Disease and Adverse Effects of L-DOPA. Front. Neuroanat. 2020, 14, 26. [Google Scholar] [CrossRef]
- Beucke, J.C.; Plotkin, M.; Winter, C.; Endrass, T.; Amthauer, H.; Juckel, G.; Kupsch, A. Midbrain serotonin transporters in de novo and L-DOPA-treated patients with early Parkinson’s disease—A [123 I]-ADAM SPECT study. Eur. J. Neurol. 2011, 18, 750–755. [Google Scholar] [CrossRef]
- Pagano, G.; Yousaf, T.; Politis, M. PET Molecular Imaging Research of Levodopa-Induced Dyskinesias in Parkinson’s Disease. Curr. Neurol. Neurosci. Rep. 2017, 17, 11. [Google Scholar] [CrossRef] [Green Version]
- Politis, M.; Loane, C.; Wu, K.; Brooks, D.J.; Piccini, P. Serotonergic mediated body mass index changes in Parkinson’s disease. Neurobiol. Dis. 2011, 43, 609–615. [Google Scholar] [CrossRef]
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of Parkinson disease: A review. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef] [PubMed]
- Carta, M.; Carlsson, T.; Muñoz, A.; Kirik, D.; Björklund, A. Role of serotonin neurons in the induction of levodopa- and graft-induced dyskinesias in Parkinson’s disease. Mov. Disord. 2010, 25, 22792. [Google Scholar] [CrossRef] [PubMed]
- Carta, M.; Björklund, A. The serotonergic system in L-DOPA-induced dyskinesia: Pre-clinical evidence and clinical perspective. J. Neural. Transm. 2018, 125, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Ghiglieri, V.; Mineo, D.; Vannelli, A.; Cacace, F.; Mancini, M.; Pendolino, V.; Napolitano, F.; di Maio, A.; Mellone, M.; Stanic, J.; et al. Modulation of serotonergic transmission by eltoprazine in L-DOPA-induced dyskinesia: Behavioral, molecular, and synaptic mechanisms. Neurobiol. Dis. 2016, 86, 140–153. [Google Scholar] [CrossRef]
- Marin, C.; Aguilar, E.; Rodríguez-Oroz, M.C.; Bartoszyk, G.D.; Obeso, J.A. Local administration of sarizotan into the subthalamic nucleus attenuates levodopa-induced dyskinesias in 6-OHDA-lesioned rats. Psychopharmacology 2009, 204, 241–250. [Google Scholar] [CrossRef]
- Tan, S.K.; Hartung, H.; Sharp, T.; Temel, Y. Serotonin-dependent depression in Parkinson’s disease: A role for the subthalamic nucleus? Neuropharmacology 2011, 61, 387–399. [Google Scholar] [CrossRef]
- Gatto, E.M.; Aldinio, V. Impulse Control Disorders in Parkinson’s Disease. A Brief and Comprehensive Review. Front. Neurol. 2019, 10, 351. [Google Scholar] [CrossRef] [Green Version]
- Caligiore, D.; Montedori, F.; Buscaglione, S.; Capirchio, A. Increasing Serotonin to Reduce Parkinsonian Tremor. Front. Syst. Neurosci. 2021, 15, 682990. [Google Scholar] [CrossRef]
- Carta, M.; Carlsson, T.; Kirik, D.; Björklund, A. Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain 2007, 130, 1819–1833. [Google Scholar] [CrossRef] [Green Version]
- Politis, M.; Wu, K.; Loane, C.; Brooks, D.J.; Kiferle, L.; Turkheimer, F.E.; Bain, P.; Molloy, S.; Piccini, P. Serotonergic mechanisms responsible for levodopa-induced dyskinesias in Parkinson’s disease patients. J. Clin. Investig. 2014, 124, 1340–1349. [Google Scholar] [CrossRef]
- Pinna, A.; Costa, G.; Serra, M.; Contu, L.; Morelli, M. Neuroinflammation and L-dopa-induced abnormal involuntary movements in 6-hydroxydopamine-lesioned rat model of Parkinson’s disease are counteracted by combined administration of a 5-HT(1A/1B) receptor agonist and A(2A) receptor antagonist. Neuropharmacology 2021, 196, 108693. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.L. Adverse events from the treatment of Parkinson’s disease. Neurol. Clin. 2008, 26, S65–S83. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Jeon, B.S.; Kim, H.J.; Park, S.S. Genetic variant of HTR2A associates with risk of impulse control and repetitive behaviors in Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Kraemmer, J.; Smith, K.; Weintraub, D.; Guillemot, V.; Nalls, M.A.; Cormier-Dequaire, F.; Moszer, I.; Brice, A.; Singleton, A.B.; Corvol, J.C. Clinical-genetic model predicts incident impulse control disorders in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1106–1111. [Google Scholar] [CrossRef] [Green Version]
- Cilia, R.; Benfante, R.; Asselta, R.; Marabini, L.; Cereda, E.; Siri, C.; Pezzoli, G.; Goldwurm, S.; Fornasari, D. Tryptophan hydroxylase type 2 variants modulate severity and outcome of addictive behaviors in Parkinson’s disease. Parkinsonism Relat. Disord. 2016, 29, 96–103. [Google Scholar] [CrossRef]
- Zhang, X.; Cheng, X.; Hu, Y.B.; Lai, J.M.; You, H.; Hu, P.L.; Zou, M.; Zhu, J.H. Serotonin transporter polymorphic region 5-HTTLPR modulates risk for Parkinson’s disease. Neurobiol. Aging 2014, 35, 5. [Google Scholar] [CrossRef]
- Redenšek, S.; Flisar, D.; Kojović, M.; Gregorič Kramberger, M.; Georgiev, D.; Pirtošek, Z.; Trošt, M.; Dolžan, V. Dopaminergic Pathway Genes Influence Adverse Events Related to Dopaminergic Treatment in Parkinson’s Disease. Front. Pharmacol. 2019, 10, 8. [Google Scholar] [CrossRef]
- Alshogran, O.Y.; Al-Eitan, L.N.; Altawalbeh, S.M.; Aman, H.A. Association of DRD4 exon III and 5-HTTLPR VNTR genetic polymorphisms with psychiatric symptoms in hemodialysis patients. PLoS ONE 2021, 16, e0249284. [Google Scholar] [CrossRef]
- Terzić, T.; Kastelic, M.; Dolžan, V.; Plesničar, B.K. Influence of 5-HT1A and 5-HTTLPR genetic variants on the schizophrenia symptoms and occurrence of treatment-resistant schizophrenia. Neuropsychiatr. Dis. Treat. 2015, 11, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Taylor, J.A. SNPinfo: Integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res. 2009, 37, W600–W605. [Google Scholar] [CrossRef] [Green Version]
- Dolzan, V.; Serretti, A.; Mandelli, L.; Koprivsek, J.; Kastelic, M.; Plesnicar, B.K. Acute antipyschotic efficacy and side effects in schizophrenia: Association with serotonin transporter promoter genotypes. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1562–1566. [Google Scholar] [CrossRef] [PubMed]
- Tregouet, D.A.; Garelle, V. A new JAVA interface implementation of THESIAS: Testing haplotype effects in association studies. Bioinformatics 2007, 23, 1038–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le François, B.; Czesak, M.; Steubl, D.; Albert, P.R. Transcriptional regulation at a HTR1A polymorphism associated with mental illness. Neuropharmacology 2008, 55, 977–985. [Google Scholar] [CrossRef]
- Albert, P.R. Transcriptional regulation of the 5-HT1A receptor: Implications for mental illness. Philos Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 2402–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savitz, J.; Lucki, I.; Drevets, W.C. 5-HT(1A) receptor function in major depressive disorder. Prog. Neurobiol. 2009, 88, 17–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, L.A.; Sun, E.W.; Martin, A.M.; Keating, D.J. The ever-changing roles of serotonin. Int. J. Biochem. Cell Biol. 2020, 125, 29. [Google Scholar] [CrossRef] [PubMed]
- Hrovatin, K.; Kunej, T.; Dolžan, V. Genetic variability of serotonin pathway associated with schizophrenia onset, progression, and treatment. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2020, 183, 113–127. [Google Scholar] [CrossRef]
- Stępnicki, P.; Kondej, M.; Kaczor, A.A. Current Concepts and Treatments of Schizophrenia. Molecules 2018, 23, 2087. [Google Scholar] [CrossRef] [Green Version]
- Burstein, E.S. Relevance of 5-HT(2A) Receptor Modulation of Pyramidal Cell Excitability for Dementia-Related Psychosis: Implications for Pharmacotherapy. CNS Drugs 2021, 35, 727–741. [Google Scholar] [CrossRef]
- Rothenberg, K.G.; Rajaram, R. Advances in Management of Psychosis in Neurodegenerative Diseases. Curr. Treat. Options Neurol. 2019, 21, 3. [Google Scholar] [CrossRef]
- Sahli, Z.T.; Tarazi, F.I. Pimavanserin: Novel pharmacotherapy for Parkinson’s disease psychosis. Expert Opin. Drug Discov. 2018, 13, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Chendo, I.; Ferreira, J.J. Pimavanserin for the treatment of Parkinson’s disease psychosis. Expert Opin. Pharm. 2016, 17, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Latsko, M.S.; Gilman, T.L.; Matt, L.M.; Nylocks, K.M.; Coifman, K.G.; Jasnow, A.M. A Novel Interaction between Tryptophan Hydroxylase 2 (TPH2) Gene Polymorphism (rs4570625) and BDNF Val66Met Predicts a High-Risk Emotional Phenotype in Healthy Subjects. PLoS ONE 2016, 11, e0162585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossu, G.; Rinaldi, R.; Colosimo, C. The rise and fall of impulse control behavior disorders. Parkinsonism Relat. Disord. 2018, 46 (Suppl. 1), S24–S29. [Google Scholar] [CrossRef]
- Wigner, P.; Czarny, P.; Synowiec, E.; Bijak, M.; Białek, K.; Talarowska, M.; Galecki, P.; Szemraj, J.; Sliwinski, T. Association between single nucleotide polymorphisms of TPH1 and TPH2 genes, and depressive disorders. J. Cell Mol. Med. 2018, 22, 1778–1791. [Google Scholar] [CrossRef] [Green Version]
- Tao, S.; Chattun, M.R.; Yan, R.; Geng, J.; Zhu, R.; Shao, J.; Lu, Q.; Yao, Z. TPH-2 Gene Polymorphism in Major Depressive Disorder Patients with Early-Wakening Symptom. Front. Neurosci. 2018, 12, 827. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.A.; Li, J.T.; Dai, W.J.; Liao, X.M.; Dong, L.C.; Lu, T.L.; Bousman, C.; Si, T.M. Genetic variation in the tryptophan hydroxylase 2 gene moderates depressive symptom trajectories and remission over 8 weeks of escitalopram treatment. Int. Clin. Psychopharmacol. 2016, 31, 127–133. [Google Scholar] [CrossRef]
- Plemenitaš, A.; Kores Plesničar, B.; Kastelic, M.; Porcelli, S.; Serretti, A.; Dolžan, V. Genetic variability in tryptophan hydroxylase 2 gene in alcohol dependence and alcohol-related psychopathological symptoms. Neurosci. Lett. 2015, 604, 86–90. [Google Scholar] [CrossRef]
- Powell, A.; Ireland, C.; Lewis, S.J.G. Visual Hallucinations and the Role of Medications in Parkinson’s Disease: Triggers, Pathophysiology, and Management. J. Neuropsychiatry Clin. Neurosci. 2020, 32, 334–343. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- AlShimemeri, S.; Fox, S.H.; Visanji, N.P. Emerging drugs for the treatment of L-DOPA-induced dyskinesia: An update. Expert Opin. Emerg. Drugs 2020, 25, 131–144. [Google Scholar] [CrossRef]
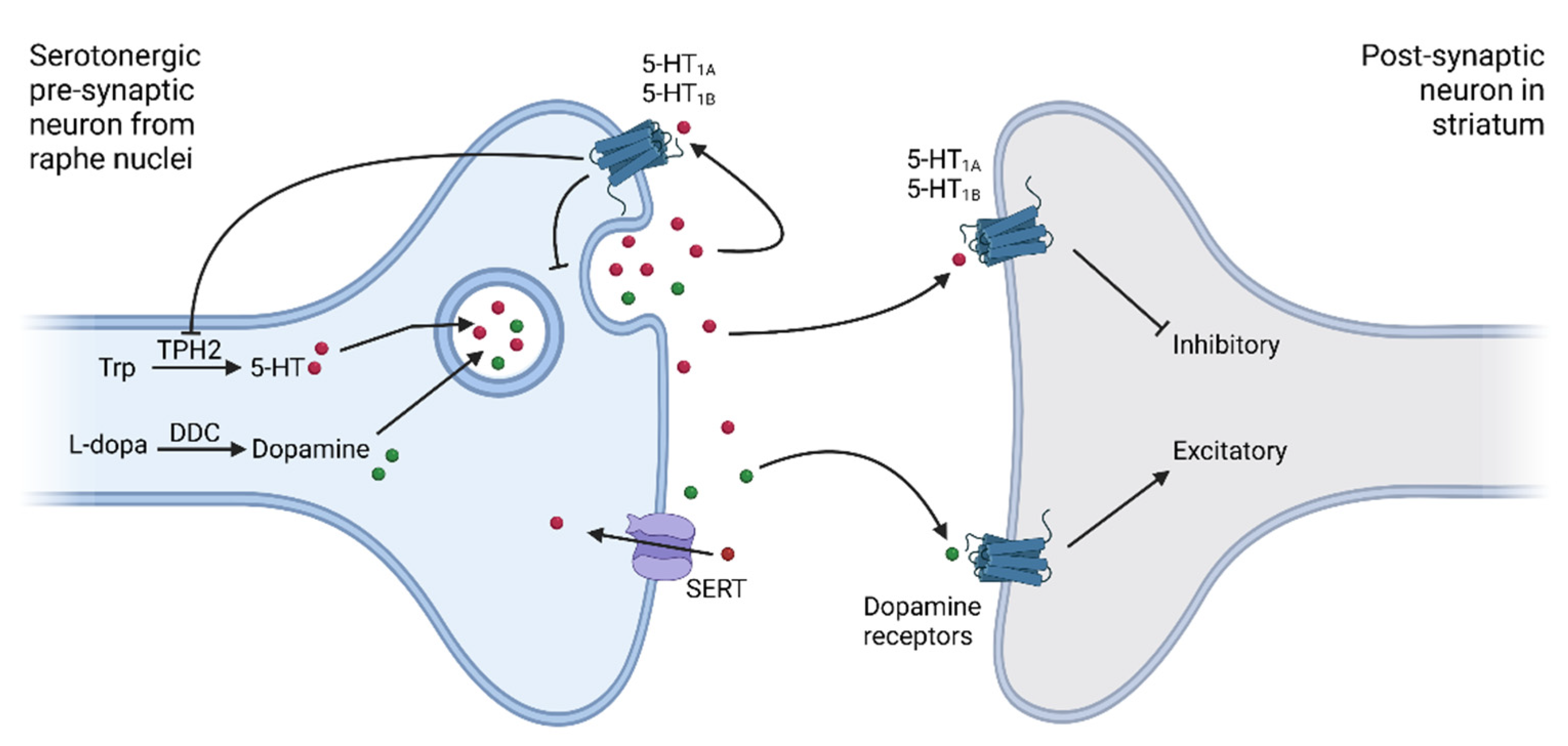

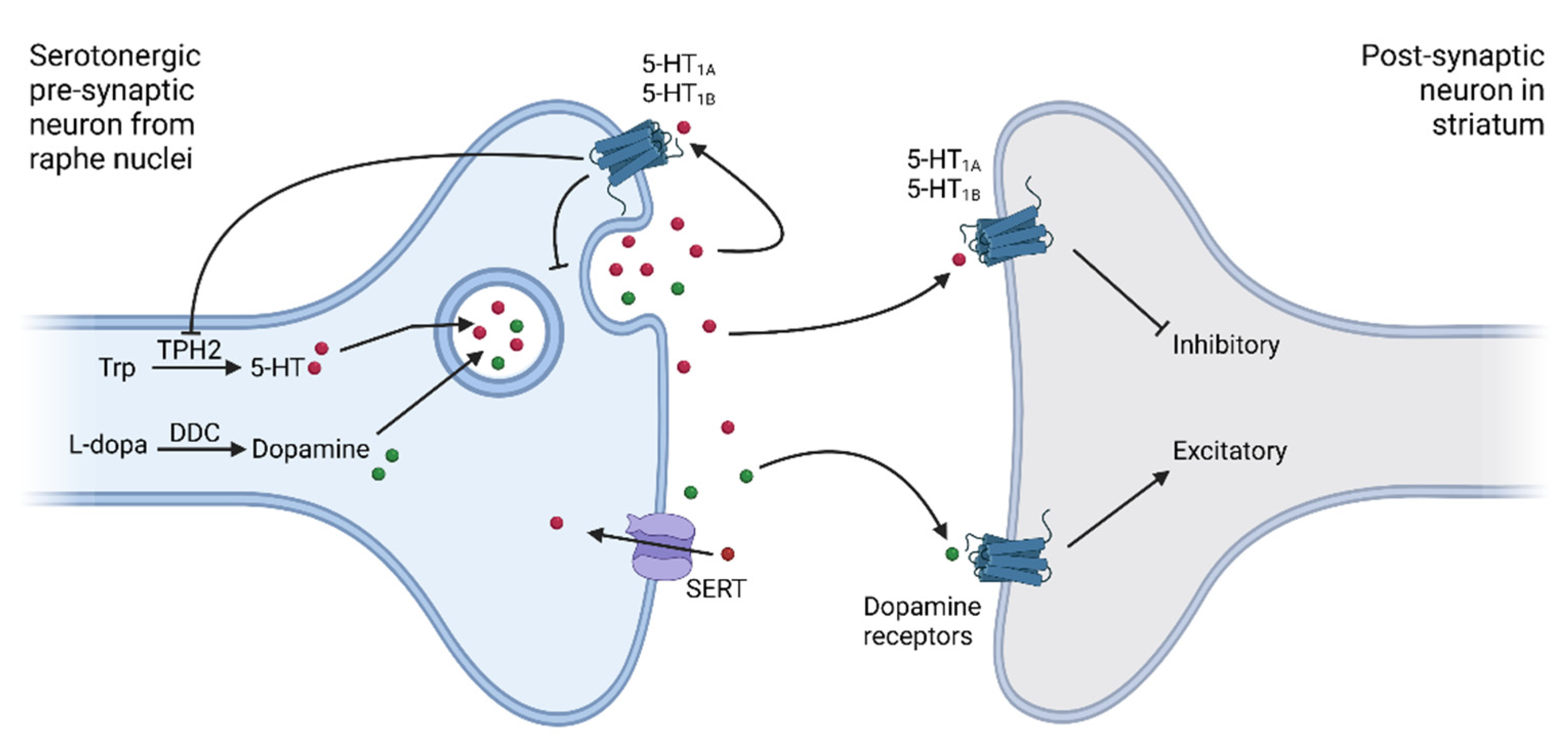{kind=link}
| Gene | Polymorphism | Location in Gene | MAF | Function Prediction * | Genotype | N (%) | HWE Equilibrium p Value |
|---|---|---|---|---|---|---|---|
| HTR1A | rs6295 c.-1019G>C | 5′UTR | 0.46 | Influences transcription factor binding | GG | 60 (26.0) | 0.632 |
| GC | 119 (51.5) | ||||||
| CC | 52 (22.5) | ||||||
| HTR1B | rs13212041 c.* 824G>A | 3′UTR | 0.19 | Influences miRNA binding | TT | 154 (66.7) | 0.778 |
| CT | 70 (30.3) | ||||||
| CC | 7 (3.0) | ||||||
| TPH2 | rs1843809 c.608+5263G>T | Intron | 0.15 | Possibly in LD with a causative variant | TT | 164 (71.0) | 0.908 |
| GT | 61 (26.4) | ||||||
| GG | 6 (2.6) | ||||||
| rs7305115 g.45237A>G | Coding region | 0.42 | Influences splicing | GG | 80 (34.6) | 0.888 | |
| AG | 111 (48.1) | ||||||
| AA | 40 (17.3) | ||||||
| rs4290270 p.Ala375= | Coding region | 0.38 | Influences splicing | TT | 83 (35.9) | 0.172 | |
| AT | 119 (51.5) | ||||||
| AA | 29 (12.6) | ||||||
| rs4570625 c.-844G>T | 5′UTR | 0.21 | Influences transcription factor binding | GG | 133 (57.6) | 0.335 | |
| GT | 88 (38.1) | ||||||
| TT | 10 (4.3) | ||||||
| SLC6A4 | 5-HTTLPR | 5′UTR | 0.43 | Affects transcriptional efficiency [30] | LL | 84 (36.4) | 0.454 |
| LS | 106 (45.9) | ||||||
| SS | 41 (17.7) | ||||||
| 5-HTTLPR rs25531 c.-1936A>G | 5′UTR | 0.45 | Affects transcriptional activity [29] and transcription factor binding | LALA | 75 (31.2) | 0.970 | |
| LAS, LALG | 113 (48.9) | ||||||
| SS, SLG, LGLG | 43 (18.6) |
| Association | Genotype | OR Adj. ** | 95% CI Adj. | p Value Adj. | ||
|---|---|---|---|---|---|---|
| Adverse Event | Adjusted for * | SNP | ||||
| Visual hallucinations | Age at diagnosis | HTR1A rs6295 | GG | Ref. | ||
| GC | 2.58 | 1.15–5.78 | 0.021 | |||
| CC | 1.23 | 0.46–3.31 | 0.676 | |||
| TPH2 rs4290270 | TT | Ref. | ||||
| AT | 1.14 | 0.58–2.27 | 0.702 | |||
| AA | 2.78 | 1.08–7.03 | 0.034 | |||
| TPH2 rs4570625 | GG | Ref. | ||||
| GT+TT | 1.86 | 1.00–3.44 | 0.047 | |||
| SLC6A4 5HTTLPR | LL | Ref. | ||||
| LS+SS | 0.52 | 0.28–0.96 | 0.037 | |||
| SLC6A4 5HTTLPR rs25531 | LALA | Ref. | ||||
| LAS, LALG | 0.54 | 0.28–1.05 | 0.069 | |||
| SS, SLG, LGLG | 0.37 | 0.14–0.97 | 0.044 | |||
| Impulse control disorders | Age at diagnosis Ever being treated with DA | TPH2 rs4570625 | GG | Ref. | ||
| GT | 3.00 | 1.27–7.07 | 0.012 | |||
| TT | 4.37 | 0.74–25.97 | 0.105 | |||
| GT+TT | 3.10 | 1.34–7.18 | 0.008 | |||
| TPH2 Haplotype * | Frequency | Visual Hallucinations | Impulse Control Disorders | Motor Fluctuations | Dyskinesia | |
|---|---|---|---|---|---|---|
| TGTG | 51.6 | Reference | ||||
| TAAT | 15.6 | OR | 1.94 | 1.77 | 0.80 | 1.30 |
| 95%CI | 1.06–3.55 | 0.74–4.21 | 0.46–1.39 | 0.75–2.25 | ||
| p value | 0.032 | 0.197 | 0.433 | 0.343 | ||
| GAAG | 14.0 | OR | 0.80 | 1.09 | 0.93 | 0.88 |
| 95%CI | 0.40–1.58 | 0.40–2.96 | 0.51–1.66 | 0.49–1.60 | ||
| p value | 0.516 | 0.871 | 0.795 | 0.679 | ||
| TATT | 6.5 | OR | 1.20 | 5.20 | 1.79 | 1.99 |
| 95%CI | 0.45–3.21 | 1.86–14.50 | 0.82–3.90 | 0.92–4.32 | ||
| p value | 0.720 | 0.002 | 0.143 | 0.080 | ||
| TGAG | 5.2 | OR | 1.27 | 2.24 | 1.35 | 1.51 |
| 95%CI | 0.44–3.67 | 0.61–8.26 | 0.53–3.43 | 0.60–3.80 | ||
| p value | 0.652 | 0.227 | 0.533 | 0.382 |
| Interaction | Visual Hallucinations | Impulse Control Disorders | Motor Fluctuations | Dyskinesia | |
|---|---|---|---|---|---|
| TPH2 rs4290270 and 5-HTTLPR | OR | 0.98 | 0.78 | 0.63 | 0.29 |
| 95%CI | 0.27–3.58 | 0.17–3.57 | 0.20–1.93 | 0.09–0.91 | |
| p value | 0.972 | 0.743 | 0.416 | 0.034 | |
| TPH2 rs7305115 and HTR1A rs6295 | OR | / | 0.15 | 0.20 | 0.16 |
| 95%CI | / | 0.02–1.59 | 0.06–0.71 | 0.04–0.60 | |
| p value | / | 0.116 | 0.013 | 0.006 | |
| TPH2 rs4570625 and HTR1A rs6295 | OR | 0.26 | 0.13 | 0.28 | 0.37 |
| 95%CI | 0.04–1.54 | 0.01–1.30 | 0.08–0.95 | 0.11–1.28 | |
| p value | 0.137 | 0.082 | 0.041 | 0.116 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Redenšek, S.; Blagus, T.; Trošt, M.; Dolžan, V. Serotonin-Related Functional Genetic Variants Affect the Occurrence of Psychiatric and Motor Adverse Events of Dopaminergic Treatment in Parkinson’s Disease: A Retrospective Cohort Study. J. Pers. Med. 2022, 12, 266. https://doi.org/10.3390/jpm12020266
Redenšek S, Blagus T, Trošt M, Dolžan V. Serotonin-Related Functional Genetic Variants Affect the Occurrence of Psychiatric and Motor Adverse Events of Dopaminergic Treatment in Parkinson’s Disease: A Retrospective Cohort Study. Journal of Personalized Medicine. 2022; 12(2):266. https://doi.org/10.3390/jpm12020266
Chicago/Turabian StyleRedenšek, Sara, Tanja Blagus, Maja Trošt, and Vita Dolžan. 2022. "Serotonin-Related Functional Genetic Variants Affect the Occurrence of Psychiatric and Motor Adverse Events of Dopaminergic Treatment in Parkinson’s Disease: A Retrospective Cohort Study" Journal of Personalized Medicine 12, no. 2: 266. https://doi.org/10.3390/jpm12020266
APA StyleRedenšek, S., Blagus, T., Trošt, M., & Dolžan, V. (2022). Serotonin-Related Functional Genetic Variants Affect the Occurrence of Psychiatric and Motor Adverse Events of Dopaminergic Treatment in Parkinson’s Disease: A Retrospective Cohort Study. Journal of Personalized Medicine, 12(2), 266. https://doi.org/10.3390/jpm12020266