
Multi-Omic Approaches in Colorectal Cancer beyond Genomic Data






Abstract
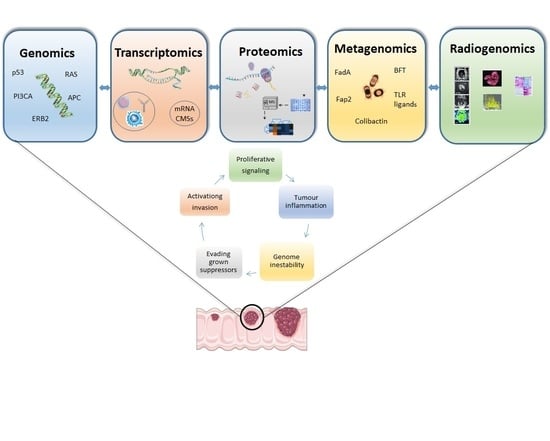
:
1. Introduction
2. Genomics
3. Transcriptomics
4. Proteomics
5. Metagenomics
6. Radiomics
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Chan, A.T.; Giovannucci, E.L. Primary Prevention of Colorectal Cancer. Gastroenterology 2010, 138, 2029–2043. [Google Scholar] [CrossRef] [Green Version]
- Fearon, E.R.; Vogelstein, B. A genetic modelfor colorectaltumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Fearon, E.R. Molecular Genetics of Colorectal Cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serebriiskii, I.G.; Connelly, C.; Frampton, G.; Newberg, J.; Cooke, M.; Miller, V.; Ali, S.; Ross, J.S.; Handorf, E.; Arora, S.; et al. Comprehensive characterization of RAS mutations in colon and rectal cancers in old and young patients. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Grasselli, J.; Elez, E.; Caratù, G.; Matito, J.; Santos, C.; Macarulla, T.; Vidal, J.; Garcia, M.; Viéitez, J.; Paéz, D.; et al. Concordance of blood- and tumor-based detection of RAS mutations to guide anti-EGFR therapy in metastatic colorectal cancer. Ann. Oncol. 2017, 28, 1294–1301. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantwell-Dorris, E.R.; O’Leary, J.; Sheils, O. BRAFV600E: Implications for Carcinogenesis and Molecular Therapy. Mol. Cancer Ther. 2011, 10, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Tie, J.; Gibbs, P.; Lipton, L.; Christie, M.; Jorissen, R.; Burgess, A.W.; Croxford, M.; Jones, I.; Langland, R.; Kosmider, S.; et al. Optimizing targeted therapeutic development: Analysis of a colorectal cancer patient population with the BRAFV600E mutation. Int. J. Cancer 2010, 128, 2075–2084. [Google Scholar] [CrossRef]
- Clarke, C.N.; Kopetz, E.S. BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: Clinical characteristics, clinical behavior, and response to targeted therapies. J. Gastrointest. Oncol. 2015, 6, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Valtorta, E.; Martino, C.; Sartore-Bianchi, A.; Penaullt-Llorca, F.; Viale, G.; Risio, M.; Rugge, M.; Grigioni, W.; Bencardino, K.; Lonardi, S.; et al. Assessment of a HER2 scoring system for colorectal cancer: Results from a validation study. Mod. Pathol. 2015, 28, 1481–1491. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.S.; Fakih, M.; Ali, S.M.; Elvin, J.A.; Schrock, A.B.; Suh, J.; Vergilio, J.-A.; Ramkissoon, S.; Severson, E.; Daniel, S.; et al. Targeting HER2 in colorectal cancer: The landscape of amplification and short variant mutations inERBB2andERBB. Cancer 2018, 124, 1358–1373. [Google Scholar] [CrossRef] [Green Version]
- Sartore-Bianchi, A.; Lonardi, S.; Martino, C.; Fenocchio, E.; Tosi, F.; Ghezzi, S.; Leone, F.; Bergamo, F.; Zagonel, V.; Ciardiello, F.; et al. Pertuzumab and trastuzumab emtansine in patients with HER2-amplified metastatic colorectal cancer: The phase II HERACLES-B trial. ESMO Open 2020, 5, e000911. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Meric-Bernstam, F.; Swanton, C.; Hurwitz, H.; Spigel, D.R.; Sweeney, C.; Burris, H.A.; Bose, R.; Yoo, B.; Stein, A.; et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results From MyPathway, an Open-Label, Phase IIa Multiple Basket Study. J. Clin. Oncol. 2018, 36, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Pietrantonio, F.; Di Nicolantonio, F.; Schrock, A.B.; Lee, J.; Tejpar, S.; Sartore-Bianchi, A.; Hechtman, J.; Christiansen, J.; Novara, L.; Tebbutt, N.; et al. ALK, ROS1, and NTRK Rearrangements in Metastatic Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Tsuchiya, K.D.; Park, D., II; Fausel, R.; Welcsh, P.; Dzieciatkowski, S.; Wang, J.; Grady, W.M. RET is a potential tumor suppressor gene in colorectal cancer. Oncogene 2013, 32, 2037–2047. [Google Scholar] [CrossRef] [Green Version]
- Bian, J.; Dannappel, M.; Wan, C.; Firestein, R. Transcriptional Regulation of Wnt/β-Catenin Pathway in Colorectal Cancer. Cells 2020, 9, 2125. [Google Scholar] [CrossRef]
- Schneikert, J.; Behrens, J. The canonical Wnt signalling pathway and its APC partner in colon cancer development. Gut 2007, 56, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polakis, P. The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 2007, 17, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Ciriano, I.; Lee, S.; Park, W.-Y.; Kim, T.-M.; Park, P.J. A molecular portrait of microsatellite instability across multiple cancers. Nat. Commun. 2017, 8, 15180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic Review of Microsatellite Instability and Colorectal Cancer Prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Lindor, B.N.M.; Burgart, L.J.; Leontovich, O.; Goldberg, R.M.; Cunningham, J.M.; Sargent, D.J.; Walsh-Vockley, C.; Petersen, G.M.; Walsh, M.D.; Leggett, B.A.; et al. Immunohistochemistry versus microsatellite instability testing in phenotyping colorectal tumorsTesting in Phenotyping Colorectal Tumors. Society 2002, 20, 1043–1048. [Google Scholar]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Budinska, E.; Popovici, V.; Tejpar, S.; D’Ario, G.; Lapique, N.; Sikora, K.O.; Di Narzo, A.F.; Yan, P.; Hodgson, J.G.; Weinrich, S.; et al. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J. Pathol. 2013, 231, 63–76. [Google Scholar] [CrossRef]
- Dasari, A.; Morris, V.K.; Advani, S.; Menter, D.G.; Eng, C.; Luthra, R.; Loree, J.M.; Pereira, A.A.L.; Lam, M.; Willauer, A.N.; et al. Classifying colorectal cancer by tumor location rather than sidedness highlights a continuum in mutation profiles and consensus molecular subtypes. Clin. Cancer Res. 2018, 24, 1062–1072. [Google Scholar]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef]
- Becht, E.; De Reyniès, A.; Giraldo, N.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautes-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef] [Green Version]
- Angelova, M.; Charoentong, P.; Hackl, H.; Fischer, M.L.; Snajder, R.; Krogsdam, A.M.; Waldner, M.J.; Bindea, G.; Mlecnik, B.; Galon, J.; et al. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015, 16, 1–17. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.H.; Pagès, F.; et al. Clinical Impact of Different Classes of Infiltrating T Cytotoxic and Helper Cells (Th1, Th2, Treg, Th17) in Patients with Colorectal Cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okita, A.; Takahashi, S.; Ouchi, K.; Inoue, M.; Watanabe, M.; Endo, M.; Honda, H.; Yamada, Y.; Ishioka, C. CMS classification of CRCas a predictive factor for chemotherapeutic efficacy against metastatic CRC. Oncotarget 2018, 9, 18698–18711. [Google Scholar] [CrossRef] [PubMed]
- Stintzing, S.; Wirapati, P.; Lenz, H.-J.; Neureiter, D.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Kaiser, F.; Al-Batran, S.; Heintges, T.; et al. Consensus molecular subgroups (CMS) of colorectal cancer (CRC) and first-line efficacy of FOLFIRI plus cetuximab or bevacizumab in the FIRE3 (AIO KRK-0306) trial. Ann. Oncol. 2019, 30, 1796–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-C.; Hsu, C.-W.; Chen, C.-D.; Yu, C.-J.; Chang, K.-P.; Tai, D.-I.; Liu, H.-P.; Su, W.-H.; Chang, Y.-S.; Yu, J.-S. Candidate Serological Biomarkers for Cancer Identified from the Secretomes of 23 Cancer Cell Lines and the Human Protein Atlas. Mol. Cell. Proteom. 2010, 9, 1100–1117. [Google Scholar] [CrossRef] [Green Version]
- Boisvert, F.-M.; Lam, Y.W.; Lamont, D.; Lamond, A.I. A Quantitative Proteomics Analysis of Subcellular Proteome Localization and Changes Induced by DNA Damage. Mol. Cell. Proteom. 2010, 9, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Fijneman, R.J.; de Wit, M.; Pourghiasian, M.; Piersma, S.R.; Pham, T.V.; Warmoes, M.; Lavaei, M.; Piso, C.; Smit, F.; Diemen, P.M.D.-V.; et al. Proximal Fluid Proteome Profiling of Mouse Colon Tumors Reveals Biomarkers for Early Diagnosis of Human Colorectal Cancer. Clin. Cancer Res. 2012, 18, 2613–2624. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.; Noonan, S.; Mullen, M.P.; Scaife, C.; Tosetto, M.; Nolan, B.; Wynne, K.; Hyland, J.; Sheahan, K.; Elia, G.; et al. Predicting response to vascular endothelial growth factor inhibitor and chemotherapy in metastatic colorectal cancer. BMC Cancer 2014, 14, 887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsila, T.; Juliachs, M.; Gregori, J.; Macarulla, T.; Villarreal, L.; Bardelli, A.; Torrance, C.; Elez, E.; Tabernero, J.; Villanueva, J. Circulating pEGFR Is a Candidate Response Biomarker of Cetuximab Therapy in Colorectal Cancer. Clin. Cancer Res. 2014, 20, 6346–6356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Zhu, C.; Yang, K.; Li, J.; Du, N.; Zong, M.; Zhou, J.; He, J. Poly(C)-binding protein 1 mediates drug resistance in colorectal cancer. Oncotarget 2017, 8, 13312–13319. [Google Scholar] [CrossRef] [Green Version]
- Chauvin, A.; Boisvert, F.-M. Clinical Proteomics in Colorectal Cancer, a Promising Tool for Improving Personalised Medicine. Proteomes 2018, 6, 49. [Google Scholar] [CrossRef] [Green Version]
- Croner, R.S.; Sevim, M.; Metodiev, M.V.; Jo, P.; Ghadimi, M.; Schellerer, V.; Brunner, M.; Geppert, C.; Rau, T.; Stürzl, M.; et al. Identification of Predictive Markers for Response to Neoadjuvant Chemoradiation in Rectal Carcinomas by Proteomic Isotope Coded Protein Label (ICPL) Analysis. Int. J. Mol. Sci. 2016, 17, 209. [Google Scholar] [CrossRef] [Green Version]
- Fraher, M.H.; O’Toole, P.W.; Quigley, E.M.M. Techniques used to characterize the gut microbiota: A guide for the clinician. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 312–322. [Google Scholar] [CrossRef]
- Peterson, J.; Garges, S.; Giovanni, M.; McInnes, P.; Wang, L.; Schloss, J.A.; Bonazzi, V.; McEwen, J.E.; Wetterstrand, K.A.; Deal, C.; et al. The NIH Human Microbiome Project. Genome Res. 2009, 19, 2317–2323. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aries, V.; Crowther, J.S.; Drasar, B.S.; Hill, M.J.; E Williams, R. Bacteria and the aetiology of cancer of the large bowel. Gut 1969, 10, 334–335. [Google Scholar] [CrossRef] [Green Version]
- Fulbright, L.E.; Ellermann, M.; Arthur, J.C. The microbiome and the hallmarks of cancer. PLoS Pathog. 2017, 13, e1006480. [Google Scholar] [CrossRef] [PubMed]
- Garrett, W.S. The gut microbiota and colon cancer. Science 2019, 364, 1133–1135. [Google Scholar] [CrossRef]
- Scott, A.J.; Alexander, J.L.; Merrifield, C.A.; Cunningham, D.; Jobin, C.; Brown, R.; Alverdy, J.; O’Keefe, S.J.; Gaskins, H.R.; Teare, J.; et al. International Cancer Microbiome Consortium consensus statement on the role of the human microbiome in carcinogenesis. Gut 2019, 68, 1624–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019, 363, eaar7785. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.; Orberg, E.T.; Geis, A.L.; Chan, J.L.; Fu, K.; Shields, C.E.D.; Dejea, C.M.; Fathi, P.; Chen, J.; Finard, B.B.; et al. Bacteroides fragilis Toxin Coordinates a Pro-carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe 2018, 23, 203–214.e5. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.A.; Garrett, W.S. Fusobacterium nucleatum—Symbiont, opportunist and oncobacterium. Nat. Rev. Microbiol. 2019, 17, 156–166. [Google Scholar] [CrossRef]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Routy, B.; le Chatelier, E.; DeRosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; Kang, J.; Baik, S.H.; Lee, K.Y.; Lim, B.J.; Jeon, T.J.; Ryu, T.H.; Sohn, S.-K. Relationship between 18F-fluorodeoxyglucose uptake and v-ki-Ras2 kirsten rat sarcoma viral oncogene homolog mutation in colorectal cancer patients variability depending on c-reactive protein level. Medicine 2016, 95, 1–7. [Google Scholar] [CrossRef]
- Arslan, E.; Aksoy, T.; Gürsu, R.U.; Dursun, N.; Çakar, E.; Çermik, T.F. The Prognostic Value of 18F-FDG PET/CT and KRAS Mutation in Colorectal Cancers. Mol. Imaging Radionucl. Ther. 2020, 29, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-W.; Shen, W.-C.; Chen, W.T.-L.; Hsieh, T.-C.; Yen, K.-Y.; Chang, J.-G.; Kao, C.-H. Metabolic Imaging Phenotype Using Radiomics of [18F]FDG PET/CT Associated with Genetic Alterations of Colorectal Cancer. Mol. Imaging Biol. 2018, 21, 183–190. [Google Scholar] [CrossRef]
- Oh, J.E.; Kim, M.J.; Lee, J.; Hur, B.Y.; Kim, B.; Kim, D.Y.; Baek, J.Y.; Chang, H.J.; Park, S.C.; Oh, J.H.; et al. Magnetic Resonance-Based Texture Analysis Differentiating KRAS Mutation Status in Rectal Cancer. Cancer Res. Treat. 2020, 52, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Xu, Q.; Ma, Y.; Duan, J.; Zhang, H.; Liu, T.; Li, L.; Sun, H.; Shi, K.; Xie, S.; et al. Characterizing MRI features of rectal cancers with different KRAS status. BMC Cancer 2019, 19, 1111. [Google Scholar] [CrossRef]
- González-Castro, V.; Cernadas, E.; Huelga, E.; Fernández-Delgado, M.; Porto, J.; Antunez, J.R.; Souto-Bayarri, M. CT Radiomics in Colorectal Cancer: Detection of KRAS Mutation Using Texture Analysis and Machine Learning. Appl. Sci. 2020, 10, 6214. [Google Scholar] [CrossRef]
- Taguchi, N.; Oda, S.; Yokota, Y.; Yamamura, S.; Imuta, M.; Tsuchigame, T.; Nagayama, Y.; Kidoh, M.; Nakaura, T.; Shiraishi, S.; et al. CT texture analysis for the prediction of KRAS mutation status in colorectal cancer via a machine learning approach. Eur. J. Radiol. 2019, 118, 38–43. [Google Scholar] [CrossRef]
- Krikelis, D.; Skoura, E.; Kotoula, V.; Rondogianni, P.; Pianou, N.; Samartzis, A.; Xanthakis, I.; Fountzilas, G.; Datseris, I.E. Lack of association between KRAS mutations and 18F-FDG PET/CT in Caucasian metastatic colorectal cancer patients. Anticancer Res. 2014, 34, 2571–2579. [Google Scholar]
- Hong, H.-S.; Kim, S.H.; Park, H.-J.; Park, M.-S.; Kim, K.W.; Kim, W.H.; Kim, N.K.; Lee, J.M.; Cho, H.J. Correlations of Dynamic Contrast-Enhanced Magnetic Resonance Imaging with Morphologic, Angiogenic, and Molecular Prognostic Factors in Rectal Cancer. Yonsei Med. J. 2013, 54, 123–130. [Google Scholar] [CrossRef]
- Kawada, K.; Nakamoto, Y.; Kawada, M.; Hida, K.; Matsumoto, T.; Murakami, T.; Hasegawa, S.; Togashi, K.; Sakai, Y. Relationship between 18F-Fluorodeoxyglucose Accumulation and KRAS/BRAF Mutations in Colorectal Cancer. Clin. Cancer Res. 2012, 18, 1696–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Dong, D.; Fang, M.; Zhu, Y.; Zang, Y.; Liu, Z.; Zhang, H.; Ying, J.; Zhao, X.; Tian, J. Can CT-based radiomics signature predict KRAS/NRAS/BRAF mutations in colorectal cancer? Eur. Radiol. 2018, 28, 2058–2067. [Google Scholar] [CrossRef] [PubMed]
- Negreros-Osuna, A.A.; Parakh, A.; Corcoran, R.B.; Pourvaziri, A.; Kambadakone, A.; Ryan, D.P.; Sahani, D.V. Radiomics Texture Features in Advanced Colorectal Cancer: Correlation with BRAF Mutation and 5-year Overall Survival. Radiol. Imaging Cancer 2020, 2, e190084. [Google Scholar] [CrossRef]
- Pernicka, J.S.G.; Gagniere, J.; Chakraborty, J.; Yamashita, R.; Nardo, L.; Creasy, J.M.; Petkovska, I.; Do, R.R.K.; Bates, D.D.B.; Paroder, V.; et al. Radiomics-based prediction of microsatellite instability in colorectal cancer at initial computed tomography evaluation. Abdom. Radiol. 2019, 44, 3755–3763. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zhang, G.; Zhang, J.; Yang, Y.; Ren, J.; Yan, X.; Wang, Z.; Zhao, Z.; Huang, X.; Bao, H.; et al. Predicting Microsatellite Instability Status in Colorectal Cancer Based on Triphasic Enhanced Computed Tomography Radiomics Signatures: A Multicenter Study. Front. Oncol. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Li, Z.; Zhong, Q.; Zhang, L.; Wang, M.; Xiao, W.; Cui, F.; Yu, F.; Huang, C.; Feng, Z. Computed Tomography-Based Radiomics Model to Preoperatively Predict Microsatellite Instability Status in Colorectal Cancer: A Multicenter Study. Front. Oncol. 2021, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bedrikovetski, S.; Dudi-Venkata, N.N.; Kroon, H.M.; Seow, W.; Vather, R.; Carneiro, G.; Moore, J.W.; Sammour, T. Artificial intelligence for pre-operative lymph node staging in colorectal cancer: A systematic review and meta-analysis. BMC Cancer 2021, 21, 1058. [Google Scholar] [CrossRef] [PubMed]
- Großerüschkamp, F.; Schörner, S.; Kraeft, A.-L.; Schuhmacher, D.; Sternemann, C.; Jütte, H.; Feder, I.; Wisser, S.; Lugnier, C.; Overheu, O.; et al. 385O Automated detection of microsatellite status in early colon cancer (CC) using artificial intelligence (AI) integrated infrared (IR) imaging on unstained samples from the AIO ColoPredictPlus 2.0 (CPP) registry study. Ann. Oncol. 2021, 32, S531–S532. [Google Scholar] [CrossRef]


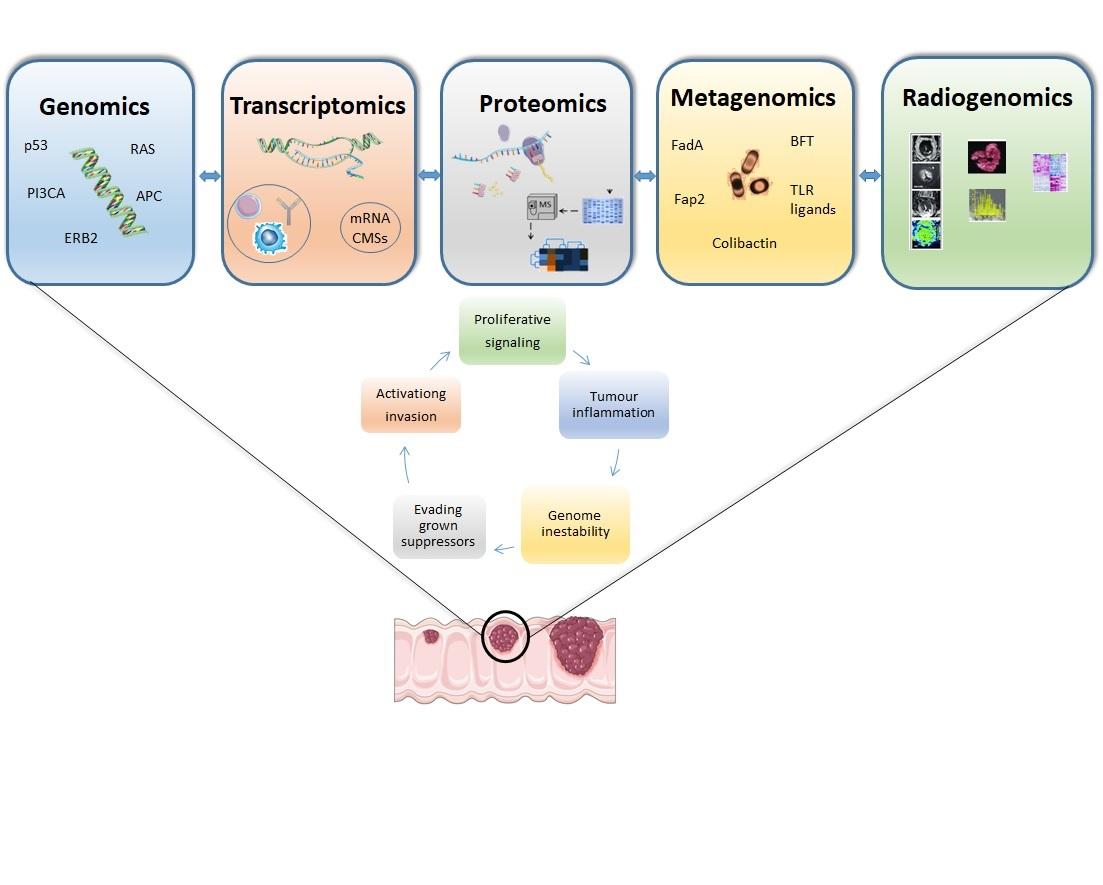{kind=link}
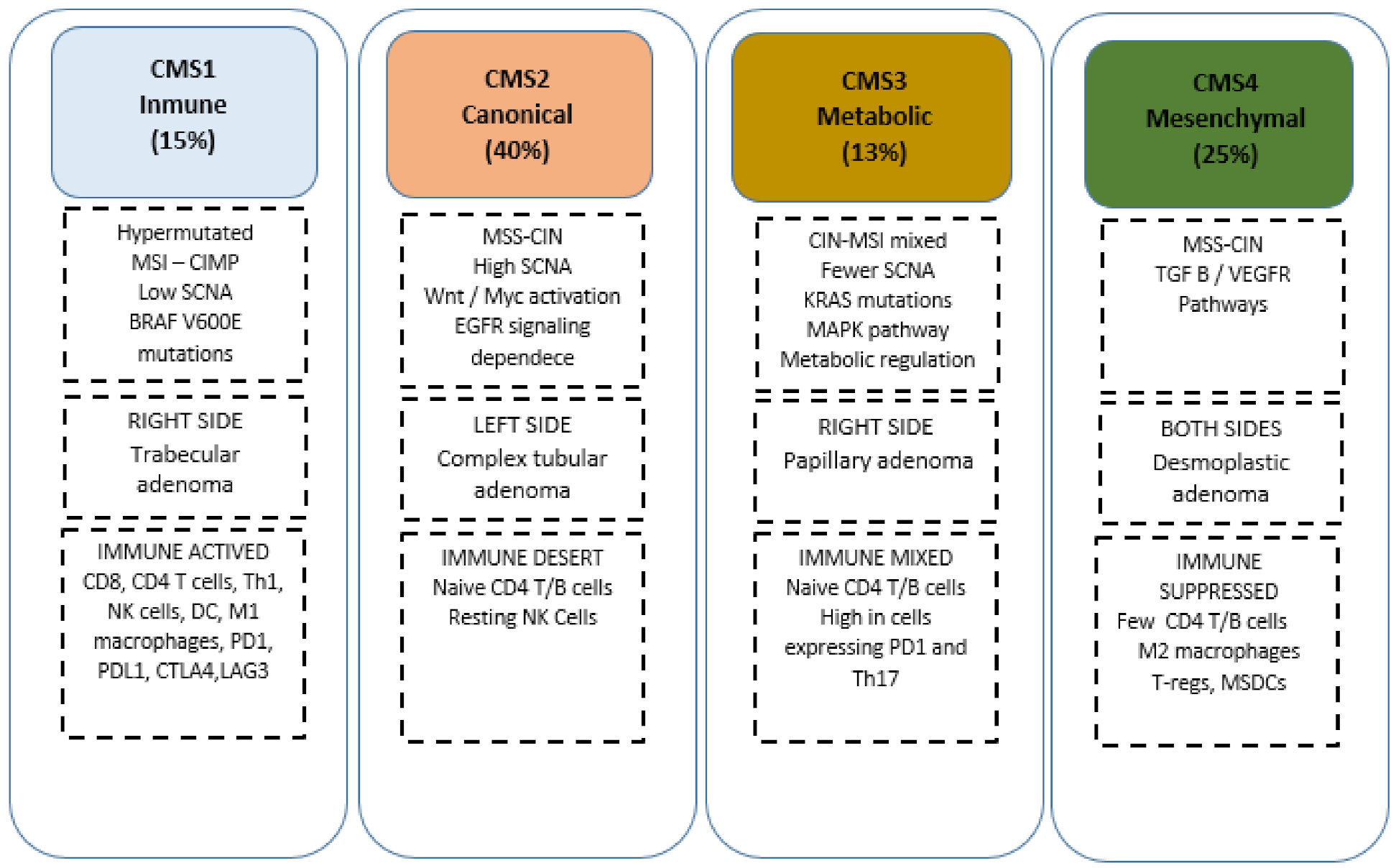{kind=link}
| C1 Wound Healing | Elevated expression of angiogenic genes High proliferation rate Low Th1/Th2 ratio related to the adaptive immune infiltrate. |
| C2 IFN-y dominant | High proliferation rate Highest intratumoral heterogeneity Macrophages M1/M2 polarisation CD8 T cell population TCR diversity. |
| C3 Inflammatory | Elevated Th17 and Th1 genesLow to moderate proliferation Lower levels of aneuploidy Higher somatic copy-number alterations |
| C4 Lymphocyte Depleted | Moderate cell proliferation and intratumoral heterogeneity Prominent macrophage signature with Th1 suppressed and a high M2 response |
| C5 Immunologically quiet | Lowest lymphocyte and highest macrophage, dominated by M2 Low rates of proliferation and heterogeneity. |
| C6 TGF- β | Mixed tumours with the highest TGF-b signature High lymphocytic infiltrate with a balanced Th1:Th2 ratio. |
| Biomarkers | Relevance | References |
|---|---|---|
| Apolipoprotein E 180 (APOE) Angiotensinogen (AGT) Vitamin D binding protein (DBP) | Survival outcomes in Bevacizumab-treated patients | Martin et al. (2014) [45] |
| Phosphorylated EGFR (pEGFR) | Response to Cetuximab | Katsila et al. (2014) [46] |
| Poly (C) binding protein 1 (PCBP1) | Oxaliplatin resistance | Guo et al. (2017) [47] |
| FAST Kinase Domains 2 (FASTKD2) Caldesmon 1 (CALD1) Carboxypeptidase A3 (CPA3) Receptor interacting serine/threonine-protein kinase 1 (RIPK1) Mast cell carboxypeptidase 4 (CPA3) Beta-1,3-galactosyltransferase 5 (B3GALT5) CD177 antigen (CD177 Dihydropyrimidine dehydrogenase (DPYD) | Response to neoadjuvant treatment (5-Fu/Capecitabine ± oxaliplatin) for rectal cancer | Chauvin et al. (2018) [48] |
| Plectin-1 (PLEC 1) Transketolase (TKT) Trifunctional enzyme subunit mitochondrial precursor (HADHA) Transgelin (TAGLN) | Response to 5-FU ± oxaliplatin | Croner et al. (2016) [47] |
| Fibrinogen B chain (FGB) Serpin B5–B9 Peroxiredoxin-4 (PRDX4) Cathepsin D (CTSD) | Response to 5-FU ± oxaliplatin | Repetto et al. (2017) |
| Year | Author | Complementary Imaging Method | Study | N | Study Population | Aim | Conclusion |
|---|---|---|---|---|---|---|---|
| 2021 | Cao et al. [75] | CT scan | R | 502 | Stage II–III | Prediction of MSI status | 32 radiomics features show correlation with MSI status, the combined model (Clinical risk factors + radiomic features) is better to predict MSI status |
| 2021 | Li et al. [76] | CT scan | R | 368 | Prediction of MSI status | The combined model (tumour location + 8 radiomic features) can predict MSI status. | |
| 2020 | Arslan et al. [63] | FDG-PET/CT | R | 83 | All stages | Prediction of KRAS status | SUVmax was higher in KRASmt |
| 2020 | Oh et al. [65] | MRI | R | 60 | Rectal tumours All stages | Prediction of KRAS status | MRI imaging features (Skewness, médium texture) could predict KRASmt |
| 2020 | Gonzalez-Castro et al. [67] | CT scan | R | 47 | All stages | Prediction of KRAS status | Radiomics features (texture in the tumour region + standard intensity) can predict the presence of the KRASmt |
| 2020 | Negreros-Osuna [73] | CT scan | R | 145 | Stage IV | Prediction of BRAF status | Standard deviation (SD) and mean value of positive pixels (MPP) were lower in the BRAFmt group. |
| 2020 | Cui et al. [74] | MRI | R | 304 | Rectal tumours All stages | Prediction of KRAS status | Seven radiomics features were moderated predicting KRAS status |
| 2019 | Chen et al. [64] | FDG-PET/CT | R | 74 | All stages | Prediction of KRAS status | KRASmt tumours had an increased value of SUVmax |
| 2019 | Xu et al. [66] | MRI | R | 158 | Rectal Tumours stages II–III | Prediction of KRAS status | Six radiomic features were higher in the KRASmt group |
| 2019 | Taguchi et al. [68] | CT scan | R | 40 | Stage II–IV | Prediction of KRAS status | CT textures can predict the KRASmt |
| 2019 | Pernicka et al. [74] | CT scan | R | 198 | Stage II–III | Prediction of MSI status | The combined model (Clinical + radiomic features) is better at predicting MSI |
| 2018 | Yang et al. [72] | CT scan | R | 117 | All Stages | Prediction of KRAS/NRAS/BRAF status | Three radiomics features could be useful for predicting KRASmt/NRASmt/BRAFmt |
| 2017 | Coner et al. [47] | FDG-PET/CT | R | 55 | Prediction of KRAS status | No significant association between KRAS gene mutation and SUVmax, MTV, TLG and haematological parameters. | |
| 2016 | Lee et al. [62] | FDG-PET/CT | P | 179 | All stages | Prediction of the KRAS status depending on CRP level | Higher SUVmax in KRASmt patients with normal CRP |
| 2016 | Lovinfosse et al. | FDG-PET/CT | R | 151 | All stages | Prediction of KRAS, NRAS, BRAF | No significant association between quantitative parameters and KRAS, NRAS, BRAF status |
| 2015 | Kawada et al. [69] | FDG-PET/CT | R | 55 | Stage IV | Prediction of KRAS status | SUVmax remained significantly associated with KRASmt in tumours larger than 10mm |
| 2014 | Krikelis et al. [67] | FDG-PET/CT | R | 44 | Stage IV | Prediction of KRAS status | No significant correlation between SUVmax values and KRASmt |
| 2013 | Hong et al. [70] | MRI | R | 29 | Rectal Tumours Stages II–III | Prediction of KRAS status | No significant correlations between MRI parameters and KRASmt |
| 2012 | Kawada et al. [69] | FDG-PET/CT | R | 51 | All stages | Prediction of KRAS-BRAF status | Higher FDG accumulation in patients with KRASmt and BRAFmt and can be used to predict mutations |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sardo, E.; Napolitano, S.; Della Corte, C.M.; Ciardiello, D.; Raucci, A.; Arrichiello, G.; Troiani, T.; Ciardiello, F.; Martinelli, E.; Martini, G. Multi-Omic Approaches in Colorectal Cancer beyond Genomic Data. J. Pers. Med. 2022, 12, 128. https://doi.org/10.3390/jpm12020128
Sardo E, Napolitano S, Della Corte CM, Ciardiello D, Raucci A, Arrichiello G, Troiani T, Ciardiello F, Martinelli E, Martini G. Multi-Omic Approaches in Colorectal Cancer beyond Genomic Data. Journal of Personalized Medicine. 2022; 12(2):128. https://doi.org/10.3390/jpm12020128
Chicago/Turabian StyleSardo, Emilia, Stefania Napolitano, Carminia Maria Della Corte, Davide Ciardiello, Antonio Raucci, Gianluca Arrichiello, Teresa Troiani, Fortunato Ciardiello, Erika Martinelli, and Giulia Martini. 2022. "Multi-Omic Approaches in Colorectal Cancer beyond Genomic Data" Journal of Personalized Medicine 12, no. 2: 128. https://doi.org/10.3390/jpm12020128
APA StyleSardo, E., Napolitano, S., Della Corte, C. M., Ciardiello, D., Raucci, A., Arrichiello, G., Troiani, T., Ciardiello, F., Martinelli, E., & Martini, G. (2022). Multi-Omic Approaches in Colorectal Cancer beyond Genomic Data. Journal of Personalized Medicine, 12(2), 128. https://doi.org/10.3390/jpm12020128