
Pancreatic Cancer: Beyond Brca Mutations

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Inclusion and Exclusion Criteria
2.2. Data Extraction
3. Results
3.1. GEne Alteration and Molecular Pathology of Pancreatic Cancer
3.2. Angiogenesis
3.3. EGFR
3.4. IGF1R
3.5. RAS
3.6. PI3K/AKT/mTOR
3.7. SRC
3.8. JAK/STAT
3.9. Notch
3.10. Hedgehog
3.11. WNT
3.12. TGFβ
3.13. Stromal Environment
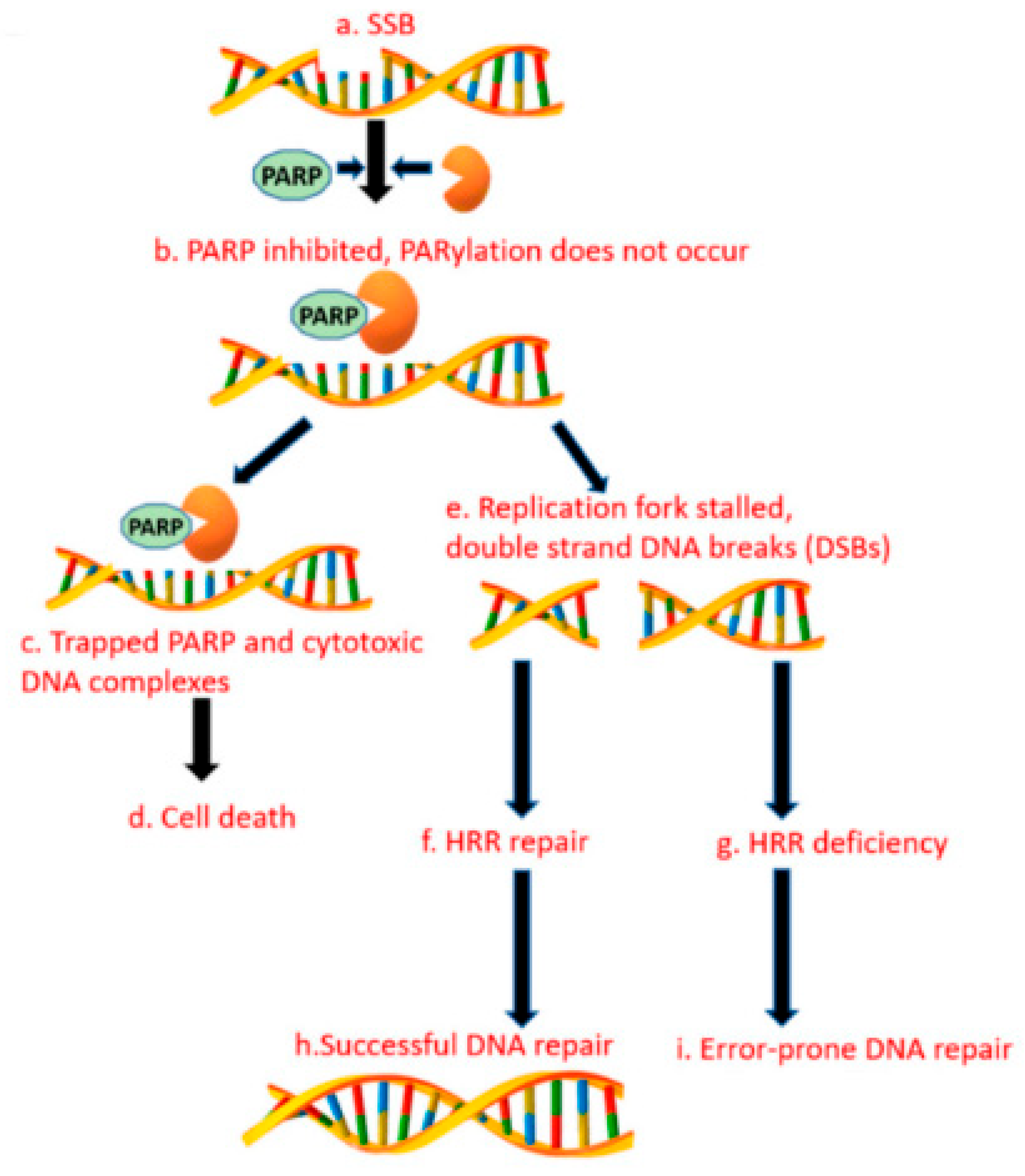
3.14. Alternative Poly (Adp-RiboSE) Polymerase Pathway
4. Discussion
5. Conclusions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Petersen, G.M. Familial pancreatic cancer. Semin. Oncol. 2016, 43, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–10494. [Google Scholar] [CrossRef] [PubMed]
- Thota, R.; Pauff, J.M.; Berlin, J.D. Treatment of metastatic pancreatic adenocarcinoma: A review. Oncology 2014, 28, 70–74. [Google Scholar] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Thompson, D.; Easton, D.F. Breast Cancer Linkage Consortium. Cancer Incidence in BRCA1 mutation carriers. J. Natl. Cancer Inst. 2002, 94, 1358–1365. [Google Scholar] [CrossRef]
- Takeuchi, S.; Doi, M.; Ikari, N.; Yamamoto, M.; Furukawa, T. Mutations in BRCA1, BRCA2, and PALB2, and a panel of 50 cancerassociated genes in pancreatic ductal adenocarcinoma. Sci. Rep. 2018, 8, 8105. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred reportingitems for systematic reviews and meta-analyses: The PRISMAstatement. Ann. Intern. Med. 2009, 151, 264–269. [Google Scholar] [CrossRef]
- Yashiro, M.; Carethers, J.M.; Laghi, L.; Saito, K.; Slezak, P.; Jaramillo, E.; Rubio, C.; Koizumi, K.; Hirakawa, K.; Boland, C.R. Genetic pathways in the evolution of morphologically distinct colorectal neoplasms. Cancer Res. 2001, 61, 2676–2683. [Google Scholar]
- Rozenblum, E.; Schutte, M.; Goggins, M.; Hahn, S.A.; Panzer, S.; Zahurak, M.; Goodman, S.N.; Sohn, T.A.; Hruban, R.H.; Yeo, C.J.; et al. Tumorsuppressive pathways in pancreatic carcinoma. Cancer Res. 1997, 57, 1731–1734. [Google Scholar] [PubMed]
- Moskaluk, C.A.; Hruban, R.H.; Kern, S.E. p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 1997, 57, 2140–2143. [Google Scholar] [PubMed]
- Hruban, R.H.; van Mansfeld, A.D.; Offerhaus, G.J.; van Weering, D.H.; Allison, D.C.; Goodman, S.N.; Kensler, T.W.; Bose, K.K.; Cameron, J.L.; Bos, J.L. K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am. J. Pathol. 1993, 143, 545–554. [Google Scholar] [PubMed]
- Schutte, M.; Hruban, R.H.; Geradts, J.; Maynard, R.; Hilgers, W.; Rabindran, S.K.; Moskaluk, C.A.; Hahn, S.A.; Schwarte-Waldhoff, I.; Schmiegel, W.; et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997, 57, 3126–3130. [Google Scholar] [PubMed]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Iacobuzio-Donahue, C.A.; Song, J.; Parmiagiani, G.; Yeo, C.J.; Hruban, R.H.; Kern, S.E. Missense mutations of MADH4: Characterization of the mutational hot spot and functional consequences in human tumors. Clin. Cancer Res. 2004, 10, 1597–1604. [Google Scholar] [CrossRef]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef]
- Calhoun, E.S.; Jones, J.B.; Ashfaq, R.; Adsay, V.; Baker, S.J.; Valentine, V.; Hempen, P.M.; Hilgers, W.; Yeo, C.J.; Hruban, R.H.; et al. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: Potential therapeutic targets. Am. J. Pathol. 2003, 163, 1255–1260. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Hirakawa, T.; Yashiro, M.; Murata, A.; Hirata, K.; Kimura, K.; Amano, R.; Yamada, N.; Nakata, B.; Hirakawa, K. IGF-1 receptor and IGF binding protein-3 might predict prognosis of patients with resectable pancreatic cancer. BMC Cancer 2013, 13, 392. [Google Scholar] [CrossRef] [PubMed]
- Whipple, C.; Korc, M. Targeting angiogenesis in pancreatic cancer: Rationale and pitfalls. Langenbecks Arch. Surg. 2008, 393, 901–910. [Google Scholar] [CrossRef]
- Koch, S.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb. Perspect. Med. 2012, 2, a006502. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Baba, H.; Fukuda, T.; Takashima, M.; Sugimachi, K. High expression of vascular endothelial growth factor is associated with liver metastasis and a poor prognosis for patients with ductal pancreatic adenocarcinoma. Cancer 2000, 88, 2239–2245. [Google Scholar] [CrossRef]
- Kindler, H.L.; Niedzwiecki, D.; Hollis, D.; Sutherland, S.; Schrag, D.; Hurwitz, H.; Innocenti, F.; Mulcahy, M.F.; O’Reilly, E.; Wozniak, T.F.; et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: Phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J. Clin. Oncol. 2010, 28, 3617–3622. [Google Scholar] [CrossRef]
- Cardin, D.B.; Goff, L.; Li, C.I.; Shyr, Y.; Winkler, C.; DeVore, R.; Schlabach, L.; Holloway, M.; McClanahan, P.; Meyer, K.; et al. Phase II trial of sorafenib and erlotinib in advanced pancreatic cancer. Cancer Med. 2014, 3, 572–579. [Google Scholar] [CrossRef]
- Ioka, T.; Okusaka, T.; Ohkawa, S.; Boku, N.; Sawaki, A.; Fujii, Y.; Kamei, Y.; Takahashi, S.; Namazu, K.; Umeyama, Y.; et al. Efficacy and safety of axitinib in combination with gemcitabine in advanced pancreatic cancer: Subgroup analyses by region, including Japan, from the global randomized Phase III trial. Jpn. J. Clin. Oncol. 2015, 45, 439–448. [Google Scholar] [CrossRef]
- Kataoka, Y.; Mukohara, T.; Tomioka, H.; Funakoshi, Y.; Kiyota, N.; Fujiwara, Y.; Yashiro, M.; Hirakawa, K.; Hirai, M.; Minami, H. Foretinib (GSK1363089), a multi-kinase inhibitor of MET and VEGFRs, inhibits growth of gastric cancer cell lines by blocking inter-receptor tyrosine kinase networks. Investig. New Drugs 2012, 30, 1352–1360. [Google Scholar] [CrossRef]
- Kiehne, K.; Herzig, K.H.; Fölsch, U.R. c-met expression in pancreatic cancer and effects of hepatocyte growth factor on pancreatic cancer cell growth. Pancreas 1997, 15, 35–40. [Google Scholar] [CrossRef]
- Chen, H.M.; Tsai, C.H.; Hung, W.C. Foretinib inhibits angiogenesis, lymphangiogenesis and tumor growth of pancreatic cancer in vivo by decreasing VEGFR-2/3 and TIE-2 signaling. Oncotarget 2015, 6, 14940–14952. [Google Scholar] [CrossRef]
- Voldborg, B.R.; Damstrup, L.; Spang-Thomsen, M.; Poulsen, H.S. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol. 1997, 8, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, Y.; Friess, H.; Kobrin, M.S.; Buchler, M.; Beger, H.G.; Korc, M. Coexpression of epidermal growth factor receptor and ligands in human pancreatic cancer is associated with enhanced tumor aggressiveness. Anticancer Res. 1993, 13, 565–569. [Google Scholar] [PubMed]
- Philip, P.A.; Benedetti, J.; Corless, C.L.; Wong, R.; O’Reilly, E.M.; Flynn, P.J.; Rowland, K.M.; Atkins, J.N.; Mirtsching, B.C.; Rivkin, S.E.; et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J. Clin. Oncol. 2010, 28, 3605–3610. [Google Scholar] [CrossRef] [PubMed]
- Safran, H.; Iannitti, D.; Ramanathan, R.; Schwartz, J.D.; Steinhoff, M.; Nauman, C.; Hesketh, P.; Rathore, R.; Wolff, R.; Tantravahi, U.; et al. Herceptin and gemcitabine for metastatic pancreatic cancers that overexpress HER-2/neu. Cancer Investig. 2004, 22, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef]
- Da Cunha Santos, G.; Dhani, N.; Tu, D.; Chin, K.; Ludkovski, O.; Kamel-Reid, S.; Squire, J.; Parulekar, W.; Moore, M.J.; Tsao, M.S. Molecular predictors of outcome in a phase 3 study of gemcitabine and erlotinib therapy in patients with advanced pancreatic cancer: National Cancer Institute of Canada Clinical Trials Group Study PA.3. Cancer 2010, 116, 5599–5607. [Google Scholar] [CrossRef]
- Schultheis, B.; Reuter, D.; Ebert, M.P.; Siveke, J.; Kerkhoff, A.; Berdel, W.E.; Hofheinz, R.; Behringer, D.M.; Schmidt, W.E.; Goker, E.; et al. Gemcitabine combined with the monoclonal antibody nimotuzumab is an active first-line regimen in KRAS wildtype patients with locally advanced or metastatic pancreatic cancer: A multicenter, randomized phase IIb study. Ann. Oncol. 2017, 28, 2429–2435. [Google Scholar] [CrossRef]
- Philip, P.A.; Goldman, B.; Ramanathan, R.K.; Lenz, H.-J.; Lowy, A.M.; Whitehead, R.P.; Wakatsuki, T.; Iqbal, S.; Gaur, R.; Benedetti, J.K.; et al. Dual blockade of epidermal growth factor receptor and insulin-like growth factor receptor-1 signaling in metastatic pancreatic cancer: Phase Ib and randomized phase II trial of gemcitabine, erlotinib, and cixutumumab versus gemcitabine plus erlotinib (SWOG S0727). Cancer 2014, 120, 2980–2985. [Google Scholar] [CrossRef]
- Urtasun, N.; Vidal-Pla, A.; Pérez-Torras, S.; Mazo, A. Human pancreatic cancer stem cells are sensitive to dual inhibition of IGF-IR and ErbB receptors. BMC Cancer 2015, 15, 223. [Google Scholar] [CrossRef]
- De Luca, A.; Maiello, M.R.; D’Alessio, A.; Pergameno, M.; Normanno, N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: Role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets 2012, 16 (Suppl. 2), S17–S27. [Google Scholar] [CrossRef]
- Infante, J.R.; Somer, B.G.; Park, J.O.; Li, C.-P.; Scheulen, M.E.; Kasubhai, S.M.; Oh, D.-Y.; Liu, Y.; Redhu, S.; Steplewski, K.; et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 2014, 50, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.H.; Bekaii-Saab, T.; Van Ziffle, J.; Mirzoeva, O.M.; Joseph, N.M.; Talasaz, A.; Kuhn, P.; Tempero, M.A.; Collisson, E.A.; Kelley, R.K.; et al. A Multicenter, Open-Label Phase II Clinical Trial of Combined MEK plus EGFR Inhibition for Chemotherapy-Refractory Advanced Pancreatic Adenocarcinoma. Clin. Cancer Res. 2016, 22, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Willems, L.; Tamburini, J.; Chapuis, N.; Lacombe, C.; Mayeux, P.; Bouscary, D. PI3K and mTOR signaling pathways in cancer: New data on targeted therapies. Curr. Oncol. Rep. 2012, 14, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Schlieman, M.G.; Fahy, B.N.; Ramsamooj, R.; Beckett, L.; Bold, R.J. Incidence, mechanism and prognostic value of activated AKT in pancreas cancer. Br. J. Cancer 2003, 89, 2110–2115. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Yao, Y.; Zhu, J.; Li, D.; Abbruzzese, J.L.; Reddy, S.A. The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-kappaB and c-Myc in pancreatic cancer cells. Oncogene 2004, 23, 8571–8580. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Yashiro, M.; Kaizaki, R.; Yasuda, K.; Doi, Y.; Sawada, T.; Ohira, M.; Hirakawa, K. Synergistic antiproliferative effect of mTOR inhibitors in combination with 5-fluorouracil in scirrhous gastric cancer. Cancer Sci. 2009, 100, 2402–2410. [Google Scholar] [CrossRef]
- Kordes, S.; Klümpen, H.J.; Weterman, M.J.; Schellens, J.H.; Richel, D.J.; Wilmink, J.W. Phase II study of capecitabine and the oral mTOR inhibitor everolimus in patients with advanced pancreatic cancer. Cancer Chemother. Pharmacol. 2015, 75, 1135–1141. [Google Scholar] [CrossRef]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by Src family kinases. Annu Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef]
- Chee, C.E.; Krishnamurthi, S.; Nock, C.J.; Meropol, N.J.; Gibbons, J.; Fu, P.; Bokar, J.; Teston, L.; O’Brien, T.; Gudena, V.; et al. Phase II Study of Dasatinib (BMS-354825) in Patients with Metastatic Adenocarcinoma of the Pancreas. Oncolpgist 2013, 18, 1091–1092. [Google Scholar] [CrossRef]
- George, T.J.; Ali, A.; Wang, Y.; Lee, J.-H.; Ivey, A.M.; DeRemer, D.; Daily, K.C.; Allegra, C.J.; Hughes, S.J.; Fan, Z.H.; et al. Phase II Study of 5-Fluorouracil, Oxaliplatin plus Dasatinib (FOLFOX-D) in First-Line Metastatic Pancreatic Adenocarcinoma. Oncolpgist 2021, 26, 825-e1674. [Google Scholar] [CrossRef]
- Jatiani, S.S.; Baker, S.J.; Silverman, L.R.; Reddy, E.P. Jak/STAT pathways in cytokine signaling and myeloproliferative disorders: Approaches for targeted therapies. Genes Cancer 2010, 1, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Seidel, H.M.; Lamb, P.; Rosen, J. Pharmaceutical intervention in the JAK/STAT signaling pathway. Oncogene 2000, 19, 2645–2656. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Raulefs, S.; Bruns, P.; Afonso-Grunz, F.; Plötner, A.; Thermann, R.; Jäger, C.; Schlitter, A.M.; Kong, B.; Regel, I.; et al. Next-generation sequencing reveals novel differentially regulated mRNAs, lncRNAs, miRNAs, sdRNAs and a piRNA in pancreatic cancer. Mol. Cancer 2015, 14, 94. [Google Scholar] [CrossRef]
- Lili, L.N.; Matyunina, L.V.; Walker, L.D.; Daneker, G.W.; McDonald, J.F. Evidence for the importance of personalized molecular profiling in pancreatic cancer. Pancreas 2014, 43, 198–211. [Google Scholar] [CrossRef]
- Hurwitz, H.I.; Uppal, N.; Wagner, S.A.; Bendell, J.C.; Beck, J.T.; Wade, S.M., 3rd; Nemunaitis, J.J.; Stella, P.J.; Pipas, J.M.; Wainberg, Z.A.; et al. Randomized, Double-Blind, Phase II Study of Ruxolitinib or Placebo in Combination with Capecitabine in Patients with Metastatic Pancreatic Cancer for Whom Therapy with Gemcitabine Has Failed. J. Clin. Oncol. 2015, 33, 4039–4047. [Google Scholar] [PubMed]
- Leach, S.D. Epithelial differentiation in pancreatic development and neoplasia: New niches for nestin and Notch. J. Clin. Gastroenterol. 2005, 39, S78–S82. [Google Scholar] [CrossRef]
- Avila, J.L.; Kissil, J.L. Notch signaling in pancreatic cancer: Oncogene or tumor suppressor? Trends Mol. Med. 2013, 19, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Ristorcelli, E.; Lombardo, D. Targeting Notch signaling in pancreatic cancer. Expert Opin. Ther. Targets 2010, 14, 541–552. [Google Scholar] [CrossRef]
- Hu, Z.I.; Bendell, J.C.; Bullock, A.; LoConte, N.K.; Hatoum, H.; Ritch, P.; Hool, H.; Leach, J.W.; Sanchez, J.; Sohal, D.P.S.; et al. A randomized phase II trial of nab-paclitaxel and gemcitabine with tarextumab or placebo in patients with untreated metastatic pancreatic cancer. Cancer Med. 2019, 8, 5148–5157. [Google Scholar] [CrossRef]
- Cook, N.; Basu, B.; Smith, D.M.; Gopinathan, A.; Evans, J.; Steward, W.P.; Palmer, D.; Propper, D.; Venugopal, B.; Hategan, M.; et al. A phase I trial of the γ-secretase inhibitor MK-0752 in combination with gemcitabine in patients with pancreatic ductal adenocarcinoma. Br. J. Cancer 2018, 118, 793–801. [Google Scholar] [CrossRef]
- Chen, J.K.; Taipale, J.; Young, K.E.; Maiti, T.; Beachy, P.A. Small molecule modulation of Smoothened activity. Proc. Natl. Acad. Sci. USA 2002, 99, 14071–14076. [Google Scholar] [CrossRef] [PubMed]
- Thayer, S.P.; Pasca di Magliano, M.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernández-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Pasca di Magliano, M.; Sekine, S.; Ermilov, A.; Ferris, J.; Dlugosz, A.A.; Hebrok, M. Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev. 2006, 20, 3161–3173. [Google Scholar] [CrossRef]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- De Jesus-Acosta, A.; Sugar, E.A.; O’Dwyer, P.J.; Ramanathan, R.K.; Von Hoff, D.D.; Rasheed, Z.; Zheng, L.; Begum, A.; Anders, R.; Maitra, A.; et al. Phase 2 study of vismodegib, a hedgehog inhibitor, combined with gemcitabine and nab-paclitaxel in patients with untreated metastatic pancreatic adenocarcinoma. Br. J. Cancer 2020, 122, 498–505. [Google Scholar] [CrossRef]
- Baarsma, H.A.; Königshoff, M.; Gosens, R. The WNT signaling pathway from ligand secretion to gene transcription: Molecular mechanisms and pharmacological targets. Pharmacol. Ther. 2013, 138, 66–83. [Google Scholar] [CrossRef]
- Espada, J.; Calvo, M.B.; Díaz-Prado, S.; Medina, V. Wnt signalling and cancer stem cells. Clin. Transl. Oncol. 2009, 11, 411–427. [Google Scholar] [CrossRef]
- Cui, J.; Jiang, W.; Wang, S.; Wang, L.; Xie, K. Role of Wnt/β-catenin signaling in drug resistance of pancreatic cancer. Curr. Pharm. Des. 2012, 18, 2464–2471. [Google Scholar] [CrossRef]
- Davis, S.L.; Cardin, D.B.; Shahda, S.; Lenz, H.J.; Dotan, E.; O’Neil, B.H.; Kapoun, A.M.; Stagg, R.J.; Berlin, J.; Messersmith, W.A.; et al. A phase 1b dose escalation study of Wnt pathway inhibitor vantictumab in combination with nab-paclitaxel and gemcitabine in patients with previously untreated metastatic pancreatic cancer. Investig. New Drugs 2020, 38, 821–830. [Google Scholar] [CrossRef]
- Fuxe, J.; Karlsson, M.C. TGF-β-induced epithelial-mesenchymal transition: A link between cancer and inflammation. Semin. Cancer Biol. 2012, 22, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small-molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Devel. Ther. 2015, 9, 4479–4499. [Google Scholar] [PubMed]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Rosen, M.N.; Goodwin, R.A.; Vickers, M.M. BRCA mutated pancreatic cancer: A change is coming. World J. Gastroenterol. 2021, 27, 1943–1958. [Google Scholar] [CrossRef]
- Holter, S.; Borgida, A.; Dodd, A.; Grant, R.; Semotiuk, K.; Hedley, D.; Dhani, N.; Narod, S.; Akbari, M.; Moore, M.; et al. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients with Pancreatic Adenocarcinoma. J. Clin. Oncol. 2015, 33, 3124–3129. [Google Scholar] [CrossRef]
- Lowery, M.A.; Wong, W.; Jordan, E.J.; Lee, J.W.; Kemel, Y.; Vijai, J.; Mandelker, D.; Zehir, A.; Capanu, M.; Salo-Mullen, E.; et al. Prospective Evaluation of Germline Alterations in Patients with Exocrine Pancreatic Neoplasms. J. Natl. Cancer Inst. 2018, 110, 1067–1074. [Google Scholar] [CrossRef]
- Zhen, D.B.; Rabe, K.G.; Gallinger, S.; Syngal, S.; Schwartz, A.G.; Goggins, M.G.; Hruban, R.H.; Cote, M.L.; McWilliams, R.R.; Roberts, N.J.; et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: A PACGENE study. Genet. Med. 2015, 17, 569–577. [Google Scholar] [CrossRef]
- Helleday, T.; Bryant, H.E.; Schultz, N. Poly(ADP-ribose) polymerase (PARP-1) in homologous recombination and as a target for cancer therapy. Cell Cycle 2005, 4, 1176–1178. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Lee, J.W.; Zalupski, M.; Capanu, M.; Park, J.; Golan, T.; Tahover, E.; Lowery, M.A.; Chou, J.F.; Sahai, V.; et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin with or without Veliparib in Patients with Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. J. Clin. Oncol. 2020, 38, 1378–1388. [Google Scholar] [CrossRef]
- Bendell, J.; O’Reilly, E.M.; Middleton, M.R.; Chau, I.; Hochster, H.; Fielding, A.; Burke, W.; Burris, H. Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann. Oncol. 2015, 26, 804–811. [Google Scholar] [CrossRef]
- Oberstein, P.E.; Olive, K.P. Pancreatic cancer: Why is it so hard to treat? Therap. Adv. Gastroenterol. 2013, 6, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Rucki, A.A.; Zheng, L. Pancreatic cancer stroma: Understanding biology leads to new therapeutic strategies. World J. Gastroenterol. 2014, 20, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. HALO 109-301 Investigators. Randomized Phase III Trial of Pegvorhyaluronidase Alfa with Nab-Paclitaxel Plus Gemcitabine for Patients with Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients with Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062–1069. [Google Scholar] [CrossRef]
- Zhou, H.; Roy, S.; Cochran, E.; Zouaoui, R.; Chu, C.L.; Duffner, J.; Zhao, G.; Smith, S.; Galcheva-Gargova, Z.; Karlgren, J.; et al. M402, a novel heparan sulfate mimetic, targets multiple pathways implicated in tumor progression and metastasis. PLoS ONE 2011, 6, e21106. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Overall survival from the phase 3 POLO trial: Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. J. Clin. Oncol. 2021, 39 (Suppl. 3), 378. [Google Scholar] [CrossRef]
- Shroff, R.T.; Hendifar, A.; McWilliams, R.R.; Geva, R.; Epelbaum, R.; Rolfe, L.; Goble, S.; Lin, K.K.; Biankin, A.V.; Giordano, H.; et al. Rucaparib Monotherapy in Patients with Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis. Oncol. 2018, 2, 1–15. [Google Scholar] [CrossRef]
- Reiss, K.A.; Mick, R.; O’Hara, M.H.; Teitelbaum, U.; Karasic, T.B.; Schneider, C.; Cowden, S.; Southwell, T.; Romeo, J.; Izgur, N.; et al. Phase II Study of Maintenance Rucaparib in Patients with Platinum-Sensitive Advanced Pancreatic Cancer and a Pathogenic Germline or Somatic Variant in BRCA1, BRCA2, or PALB2. J. Clin. Oncol. 2021, 39, 2497–2505. [Google Scholar] [CrossRef]
- Peyraud, F.; Italiano, A. Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers 2020, 12, 1502. [Google Scholar] [CrossRef]
- Samstein, R.M.; Krishna, C.; Ma, X.; Pei, X.; Lee, K.W.; Makarov, V.; Kuo, F.; Chung, J.; Srivastava, R.M.; Purohit, T.A.; et al. Mutations in BRCA1 and BRCA2 differentially affect the tumor microenvironment and response to checkpoint blockade immunotherapy. Nat. Cancer 2020, 1, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.J.; Reiss, K.A. PARP Inhibitors in Pancreatic Cancer. Cancer J. 2021, 27, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Dooley, J.; Tian, L.; Schonefeldt, S.; Delghingaro-Augusto, V.; Garcia-Perez, J.E.; Pasciuto, E.; Di Marino, D.; Carr, E.J.; Oskolkov, N.; Lyssenko, V.; et al. Genetic predisposition for beta cell fragility underlies type 1 and type 2 diabetes. Nat. Genet. 2016, 48, 519–527. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Pathway | Target | Incidence | Ref. |
|---|---|---|---|
| Angiogenesis | VEGFR, RON, c-Met, c-KIT, FLT-3, PDGF | 90% | [22,23,24,25,26,27,28,29,30] |
| EGFR | EGFR | 30–95% | [31,32,33,34,35,36,37] |
| IGF1R | IGF1R | 60% | [38,39] |
| RAS | RAS/RAF/MEK/ERK | 90–95% | [40,41,42] |
| PI3K/AKT/mTOR | PI3K/AKT/mTOR | 55–60% | [43,44,45,46,47] |
| SRC | C-SRC | 70% | [48,49,50] |
| JAK/STAT | JAK1, JAK2, JAK3, Tyk2, Stat3 | - | [51,52,53,54,55] |
| NOTCH | NOTCH2/3R | - | [56,57,58,59,60] |
| HEDGEHOG | HH ligands | - | [61,62,63,64,65,66] |
| WNT | FZD1,2,5,7,8 R | - | [67,68,69,70] |
| TGFβ | TGFβ | - | [71,72,73] |
| ADP-RIBOSE | BRCA1/2 | 5–7% | [74,75,76,77,78,79,80] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricci, V.; Fabozzi, T.; Bareschino, M.A.; Barletta, E.; Germano, D.; Paciolla, I.; Tinessa, V.; Grimaldi, A.M. Pancreatic Cancer: Beyond Brca Mutations. J. Pers. Med. 2022, 12, 2076. https://doi.org/10.3390/jpm12122076
Ricci V, Fabozzi T, Bareschino MA, Barletta E, Germano D, Paciolla I, Tinessa V, Grimaldi AM. Pancreatic Cancer: Beyond Brca Mutations. Journal of Personalized Medicine. 2022; 12(12):2076. https://doi.org/10.3390/jpm12122076
Chicago/Turabian StyleRicci, Vincenzo, Teresa Fabozzi, Maria Anna Bareschino, Emiddio Barletta, Domenico Germano, Immacolata Paciolla, Vincenza Tinessa, and Antonio Maria Grimaldi. 2022. "Pancreatic Cancer: Beyond Brca Mutations" Journal of Personalized Medicine 12, no. 12: 2076. https://doi.org/10.3390/jpm12122076
APA StyleRicci, V., Fabozzi, T., Bareschino, M. A., Barletta, E., Germano, D., Paciolla, I., Tinessa, V., & Grimaldi, A. M. (2022). Pancreatic Cancer: Beyond Brca Mutations. Journal of Personalized Medicine, 12(12), 2076. https://doi.org/10.3390/jpm12122076