
Cholesterol Management in Neurology: Time for Revised Strategies?

 ,
,  and
and
Abstract
:1. Introduction
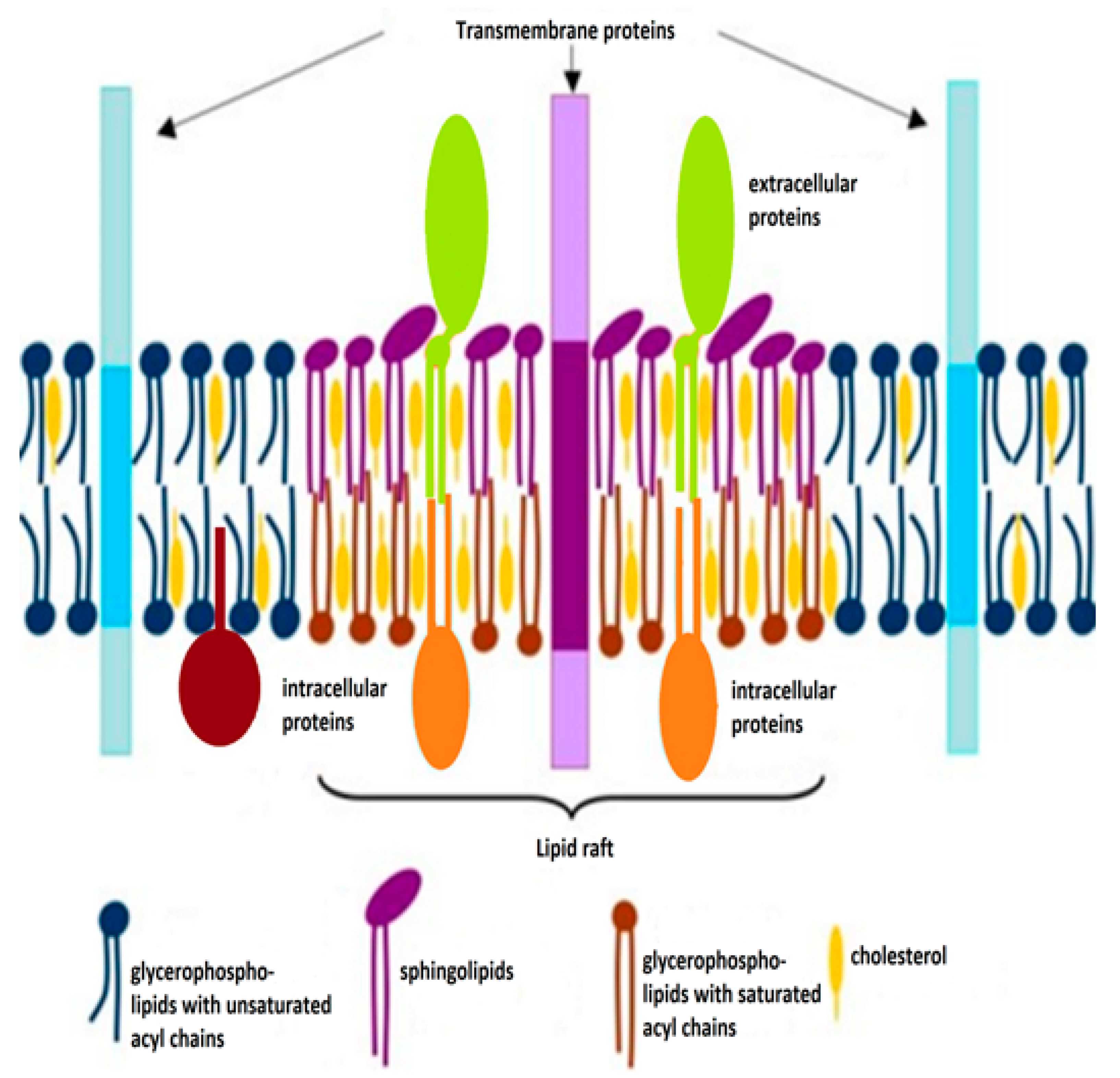
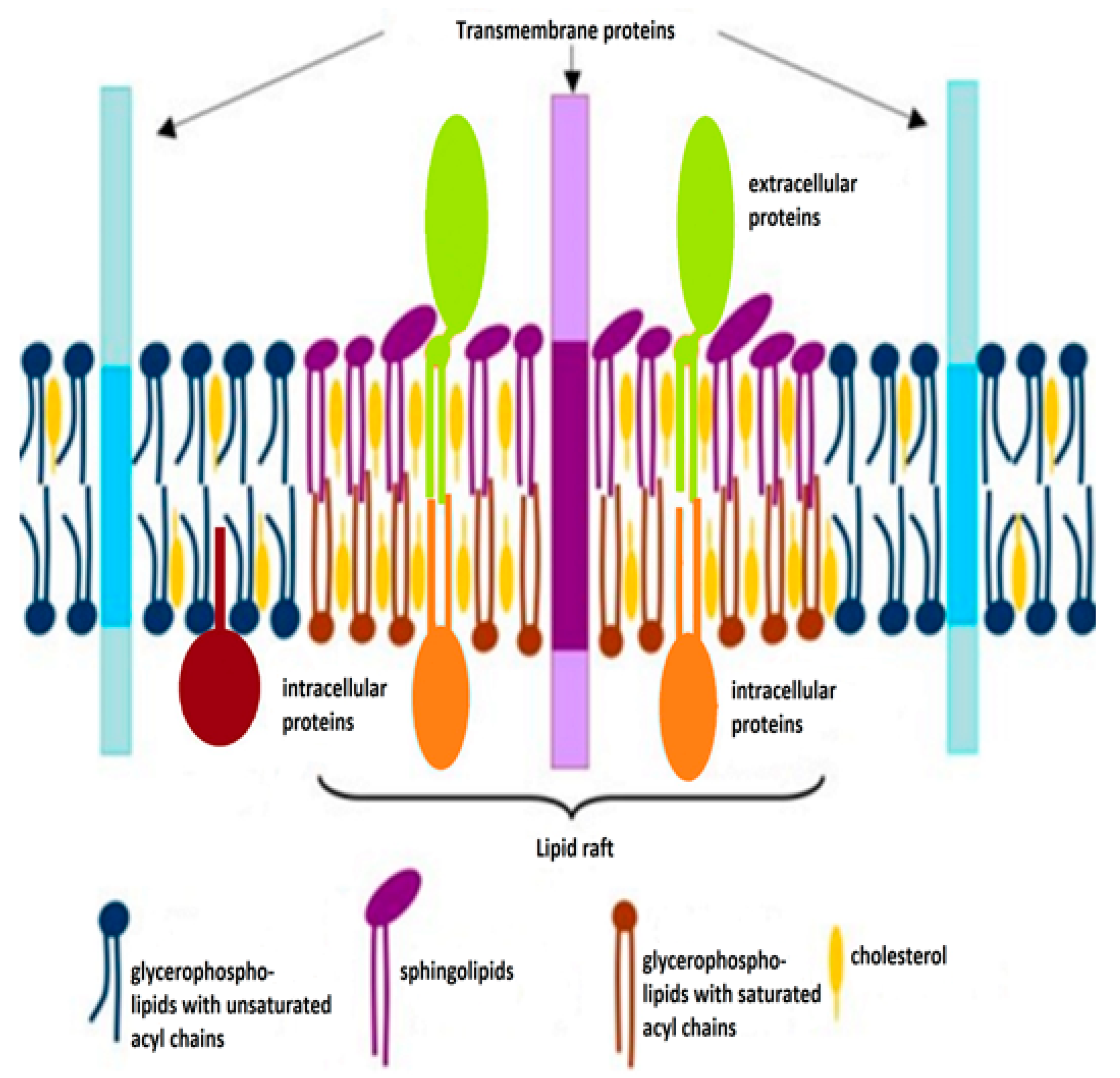
2. Cholesterol and Its Functions
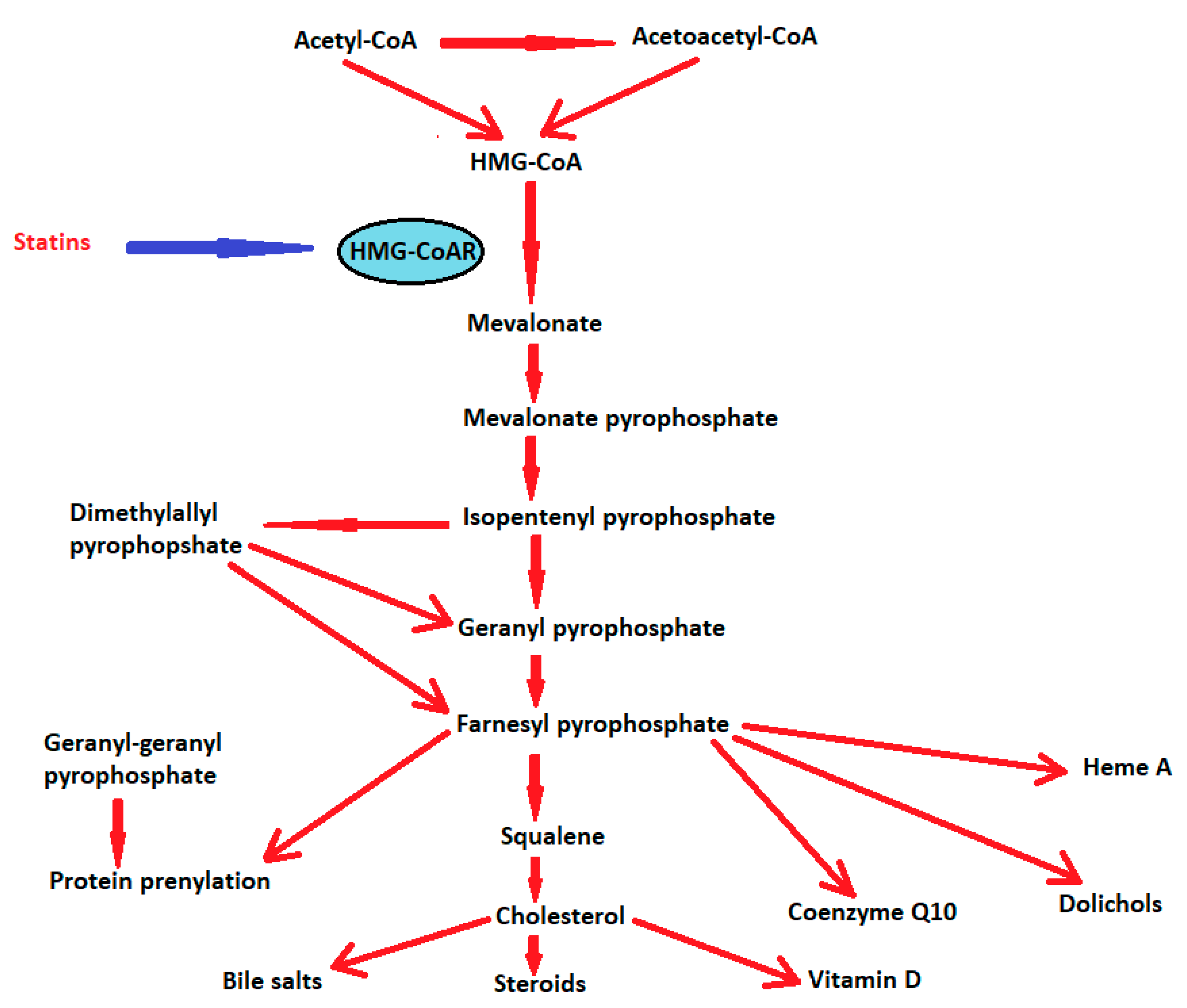
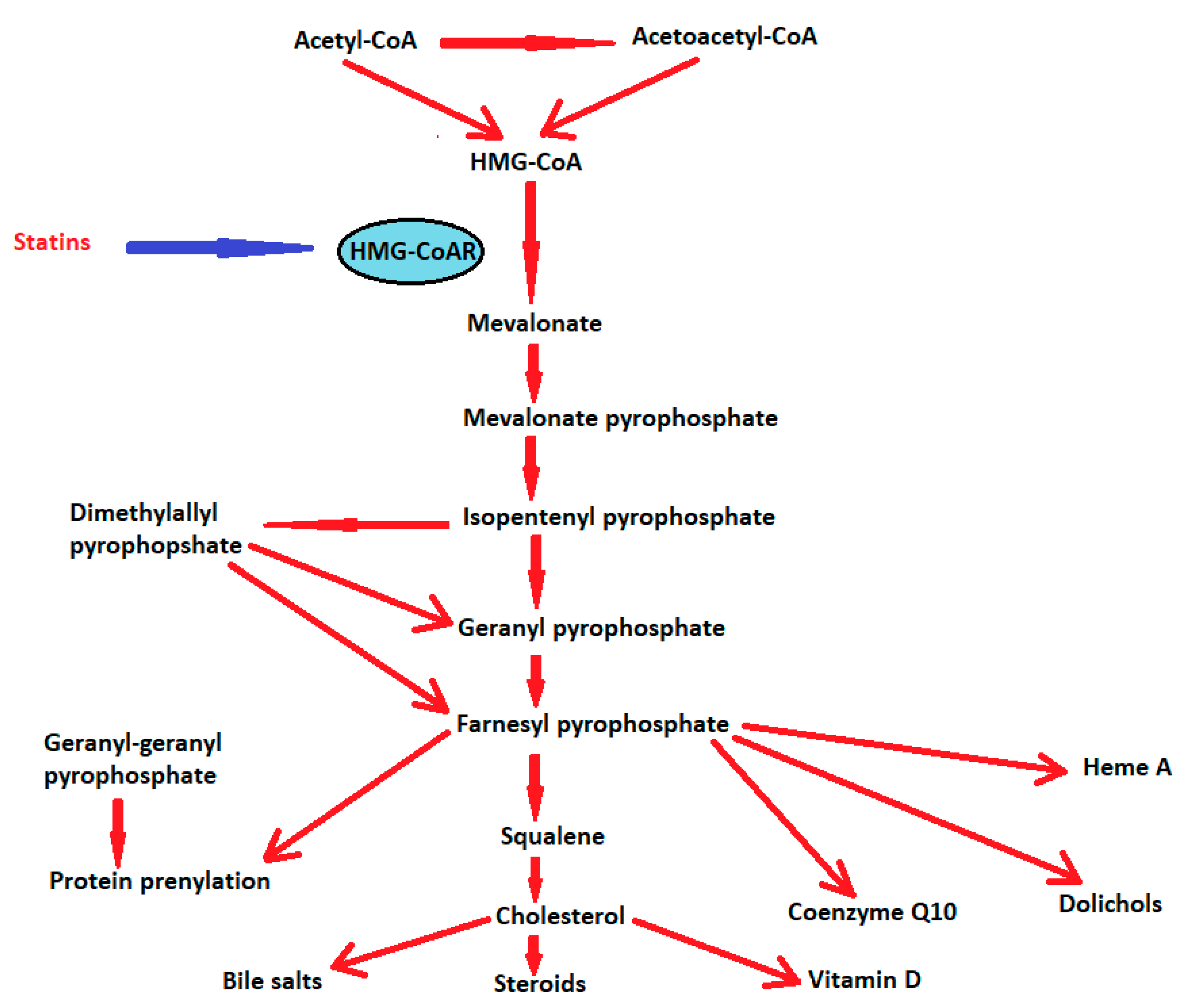
3. 3-Hydroxy-3-Methylglutaryl-Coenzyme A (HMG-CoA) Reductases or Statins
3.1. Myotoxic Effects of Statins
3.2. Cognitive Side-Effects of Statins
4. Statins in Clinical Practice in Specific Subgroups of Patients
4.1. Statins in Myasthenia Gravis
- -
- The statin’s myotoxic effects may exacerbate muscle weakness in patients with prior MG
- -
- via depletion of coenzyme Q10, statins could induce mitochondrial dysfunction with impairment of energy production at the presynaptic membrane [109]
- -
4.2. Statins in Muscular Dystrophies
- -
- Duchenne muscular dystrophy
- -
- Becker MD
- -
- congenital MD
- -
- distal MD
- -
- facioscapulohumeral MD
- -
- Emery-Dreifuss MD
- -
- limb-girdle MD
- -
- oculopharyngeal MD
- -
- myotonic MD
4.3. Statins and Cognitive Impairment
5. Conclusions
- -
- using non-statin lipid-lowering therapies in patients with muscular diseases, such as ezetimibe or PCSK9 inhibitors
- -
- extensive evaluation of patients with vascular events (ischemic strokes), with measurement of the LDL-cholesterol levels, neuropsychological evaluation, identification of the stroke subtype according to the TOAST criteria [158], and thorough risk stratification using, whenever possible, the coronary artery calcium (CAC) score [159]
- -
- when statins are necessary, the preference for a hydrophilic statin over a lipophilic one could avoid statin-induced cognitive impairment
- -
- the LDL-cholesterol levels should be regularly checked and the doses adjusted accordingly [148]
- -
- However, further clinical trials may help to develop efficient therapeutic strategies and set guidelines for particular subsets of patients.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Endo, A.; Kuroda, M.; Tsujita, Y. ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogenesis produced by penicillium citrinium. J. Antiobiot. 1976, 29, 1346–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet 1994, 344, 1383–1389. [Google Scholar]
- Byington, R.P.; Jukema, J.W.; Salonen, J.T.; Pitt, B.; Bruschke, A.V.; Hoen, H.; Furberg, C.D.; Mancini, G.B. Reduction in cardiovascular events during pravastatin therapy. Pooled analysis of clinical events of the Pravastatin Atherosclerosis Intervention Program. Circulation 1995, 92, 2419–2425. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the management of blood cholesterol: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar]
- Amarenco, P.; Kim, J.S.; Labreuche, J.; Charles, H.; Giroud, M.; Lee, B.C.; Mahagne, M.H.; Nighoghossian, N.; Gabriel Steg, P.; Vicaut, É.; et al. Treat Stroke to Target Investigators. Benefit of targeting a LDL (low-density lipoprotein) cholesterol <70 mg/dL during 5 years after ischemic stroke. Stroke 2020, 51, 1231–1239. [Google Scholar] [PubMed] [Green Version]
- Turner, R.M.; Pirmohamed, M. Statin-related myotoxicity: A comprehensive review of pharmacokinetic, pharmacogenomic and muscle components. J. Clin. Med. 2020, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Giugliano, R.P.; Cannon, C.P.; Blazing, M.A.; Nicolau, J.C.; Corbalán, R.; Špinar, J.; Park, J.-G.; White, J.A.; Bohula, E.A.; Braunwald, E.; et al. Benefit of adding ezetimibe to statin therapy on cardiovascular outcomes and and safety in patients with versus without diabetes mellitus. Circulation 2017, 137, 1571–1582. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wassermann, S.M.; et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, E. Mechanisms of cellular cholesterol compartmentalization: Recent insights. Curr. Opin. Cell Biol. 2018, 53, 77–83. [Google Scholar] [CrossRef]
- Maxfield, F.R.; Tabas, I. Role of cholesterol and lipid organization in disease. Nature 2005, 438, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Gliozzi, M.; Musolino, V.; Bosco, F.; Scicchitano, M.; Scarano, F.; Nucera, S.; Zito, M.C.; Ruga, S.; Carresi, C.; Macrì, R.; et al. Cholesterol homeostasis: Researching a dialogue between the brain and peripheral tissues. Pharmacol. Res. 2021, 163, 105215. [Google Scholar] [PubMed]
- Subczynski, W.K.; Pasenkiewicz-Gierula, M.; Widomska, J.; Mainali, L.; Raguz, M. High cholesterol/low cholesterol: Effects in biological membranes: A review. Cell Biochem. Biophys. 2017, 75, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Morrill, G.A.; Kostellow, A.B. Molecular properties of globin channels and pores: Role of cholesterol in ligand binding and movement. Front. Physiol. 2016, 7, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.; Doktorova, M.; Molugu, T.R.; Heberle, F.A.; Scott, H.L.; Dzikovski, B.; Nagao, M.; Stingaciu, L.R.; Standaert, R.F.; Barrera, F.N.; et al. How cholesterol stiffens unsaturated lipid membranes. Proc. Natl. Acad. Sci. USA 2020, 117, 21896–21905. [Google Scholar] [CrossRef] [PubMed]
- Raghunathan, K.; Kenworthy, A.K. Dynamic pattern generation in cell membranes: Current insights into membrane organization. Biochim. Biophys. Acta Biomembr. 2018, 1860, 2018–2031. [Google Scholar] [CrossRef]
- Paukner, K.; Králová Lesná, I.; Poledne, R. Cholesterol in the cell membrane-an emerging player in atherogenesis. Int. J. Mol. Sci. 2022, 23, 533. [Google Scholar] [CrossRef]
- Ouweneel, A.B.; Thomas, M.J.; Sorci-Thomas, M.G. The ins and outs of lipid rafts: Functions in intracellular cholesterol homeostasis, microparticles, and cell membranes: Thematic Review Series: Biology of Lipid Rafts. J. Lipid Res. 2020, 61, 676–686. [Google Scholar] [CrossRef] [Green Version]
- Fabiani, C.; Georgiev, V.N.; Peñalva, D.A.; Sigaut, L.; Pietrasanta, L.; Corradi, J.; Dimova, R.; Antollini, S.S. Membrane lipid organization and nicotinic acetylcholine receptor function: A two-way physiological relationship. Arch. Biochem. Biophys. 2022, 730, 109413. [Google Scholar] [CrossRef]
- Berghoff, S.A.; Spieth, L.; Saher, G. Local cholesterol metabolism orchestrates remyelination. Trends Neurosci. 2022, 45, 272–283. [Google Scholar] [CrossRef]
- Poitelon, Y.; Kopec, A.M.; Belin, S. Myelin fat facts: An overview of lipids and fatty acid metabolism. Cells 2020, 9, 812. [Google Scholar] [CrossRef] [Green Version]
- Rhea, E.M.; Banks, W.A. Interactions of lipids, lipoproteins, and apolipoproteins with the blood-brain barrier. Pharm. Res. 2021, 38, 1469–1475. [Google Scholar] [CrossRef] [PubMed]
- Molina-Gonzalez, I.; Miron, V.E. Astrocytes in myelination and remyelination. Neurosci. Lett. 2019, 713, 134532. [Google Scholar] [CrossRef] [PubMed]
- Binotti, B.; Jahn, R.; Pérez-Lara, Á. An overview of the synaptic vesicle lipid composition. Arch. Biochem. Biophys. 2021, 709, 108966. [Google Scholar] [CrossRef]
- Geda, O.; Tábi, T.; Lakatos, P.P.; Szökő, É. Differential ganglioside and cholesterol depletion by various cyclodextrin derivatives and their effect on synaptosomal glutamate release. Int. J. Mol. Sci. 2022, 23, 9460. [Google Scholar] [CrossRef] [PubMed]
- Waseem, T.V.; Kolos, V.A.; Lapatsina, L.P.; Fedorovich, S.V. Influence of cholesterol depletion in plasma membrane of rat synaptosomes on calcium-dependent and calcium-independent exocytosis. Neurosci. Lett. 2006, 405, 106–110. [Google Scholar] [CrossRef]
- Funfschilling, U.; Jockusch, W.J.; Sivakumar, N.; Mobius, W.; Corthals, K.; Li, S.; Quintes, S.; Kim, Y.; Schaap, I.A.; Rhee, J.S.; et al. Critical time window of neuronal cholesterol synthesis during neurite outgrowth. J. Neurosci. 2012, 32, 7632–7645. [Google Scholar] [CrossRef] [Green Version]
- Pfrieger, F.W.; Ungerer, N. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 2011, 50, 357–371. [Google Scholar] [CrossRef]
- Mauch, D.H.; Nagler, K.; Schumacher, S.; Goritz, C.; Muller, E.C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar] [CrossRef]
- Valdez, C.M.; Smith, M.A.; Perry, G.; Phelix, C.F.; Santamaria, F. Cholesterol homeostasis markers are localized to mouse hippocampal pyramidal and granule layers. Hippocampus 2010, 20, 902–905. [Google Scholar] [CrossRef] [Green Version]
- Moutinho, M.; Nunes, M.J.; Rodrigues, E. The mevalonate pathway in neurons: It’s not just about cholesterol. Exp. Cell Res. 2017, 360, 55–60. [Google Scholar] [CrossRef]
- Seabra, M.C. Membrane association and targeting of prenylated Ras-like GTPases. Cell. Signal. 1998, 10, 167–172. [Google Scholar] [CrossRef]
- Govek, E.E.; Newey, S.E.; Van Aelst, L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005, 19, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Gonsiorek, E.A.; Barnhart, C.; Davare, M.A.; Engebose, A.J.; Lauridsen, H.; Bruun, D.; Lesiak, A.; Wayman, G.; Bucelli, R.; et al. Statins decrease dendritic arborization in rat sympathetic neurons by blocking RhoA activation. J. Neurochem. 2009, 108, 1057–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, D.W.; Halford, R.W.; Ramirez, D.M.; Shah, R.; Kotti, T. Cholesterol 24- hydroxylase: An enzyme of cholesterol turnover in the brain. Annu. Rev. Biochem. 2009, 78, 1017–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega, G.L.; Weiner, M.F.; Lipton, A.M.; Von Bergmann, K.; Lutjohann, D.; Moore, C.; Svetlik, D. Reduction in levels of 24S-hydroxycholesterol by statin treatment in patients with Alzheimer disease. Arch. Neurol. 2003, 60, 510–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukui, K.; Ferris, H.; Kahn, C.R. Effect of cholesterol reduction on receptor signaling in neurons. J. Biol. Chem. 2015, 290, 26383–26392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosqueira, A.; Camino, P.A.; Barrantes, F.J. Cholesterol modulates acetylcholine receptor diffusion by tuning confinement sojourns and nanocluster stability. Sci. Rep. 2018, 8, 11974. [Google Scholar] [CrossRef] [PubMed]
- Woods, K.; Sharp, L.; Brannigan, G. Untangling direct and domain-mediated interactions between nicotinic acetylcholine receptors in DHA-rich membranes. J. Membr. Biol. 2019, 252, 385–396. [Google Scholar] [CrossRef] [Green Version]
- Barrantes, F.J. Fluorescence studies of nicotinic acetylcholine receptor and its associated lipid milieu: The influence of Erwin London’s methodological approaches. J. Membr. Biol. 2022, 255, 563–574. [Google Scholar] [CrossRef]
- Krivoi, I.I.; Petrov, A.M. Cholesterol and the safety factor for neuromuscular transmission. Int. J. Mol. Sci. 2019, 20, 1046. [Google Scholar] [CrossRef] [Green Version]
- Borroni, V.; Barrantes, F.J. Cholesterol modulates the rate and mechanism of acetylcholine receptor internalization. J. Biol. Chem. 2011, 286, 17122–17132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paciullo, F.; Gresele, P. Effect of statins on measures of coagulation: Potential role of low-density lipoprotein receptors. Eur. Heart J. 2019, 40, 392. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Qiao, L.; Jiang, Z.; Dong, X.; Feng, H.; Gui, Q.; Lu, Y.; Liang, Y. Significant reduction in the LDL cholesterol increases the risk of intracerebral hemorrhage: A systematic review and meta-analysis of 33 randomized controlled trials. Am. J. Transl. Res. 2020, 12, 463–477. [Google Scholar] [PubMed]
- März, W.; Kleber, M.E.; Scharnagl, H.; Speer, T.; Zewinger, S.; Ritsch, A.; Parhofer, K.G.; von Eckardstein, A.; Landmesser, U.; Laufs, U. HDL cholesterol: Reappraisal of its clinical relevance. Clin. Res. Cardiol. 2017, 106, 663–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorabi, A.M.; Kiaie, N.; Hajighasemi, S.; Banach, M.; Penson, P.E.; Jamialahmadi, T.; Sahebkar, A. Statin-induced nitric oxide signaling: Mechanisms and therapeutic implications. J. Clin. Med. 2019, 8, 2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic effects of statins on the cardiovascular system. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [Green Version]
- DeGorter, M.K.; Tirona, R.G.; Schwarz, U.I.; Choi, Y.-H.; Dresser, G.K.; Suskin, N.; Myers, K.; Zou, G.; Iwuchukwu, O.; Wei, W.-Q.; et al. Clinical and pharmacogenetic predictors of circulating atorvastatin and rosuvastatin concentration in routine clinical care. Circ. Cardiovasc. Genet. 2013, 6, 400–408.48. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, D.; Banach, M.; Chianetta, R.; Luzzu, L.M.; Pantea Stoian, A.; Diaconu, C.C.; Citarrella, R.; Montalto, G.; Rizzo, M. An overview of statin-induced myopathy and perspectives for the future. Expert Opin. Drug Saf. 2020, 19, 601–615.49. [Google Scholar] [CrossRef]
- Tournadre, A. Statins, myalgia, and rhabdomyolysis. Jt. Bone Spine 2020, 87, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Chrysant, S.G. New onset diabetes mellitus induced by statins: Current evidence. Postgrad. Med. 2017, 129, 430–435. [Google Scholar] [CrossRef]
- Mollazadeh, H.; Tavana, E.; Fanni, G.; Bo, S.; Banach, M.; Pirro, M.; von Haehling, S.; Jamialahmadi, T.; Sahebkar, A. Effects of statins on mitochondrial pathways. J. Cachexia Sarcopenia Muscle 2021, 12, 237–251. [Google Scholar] [CrossRef]
- Samaras, K.; Makkar, S.R.; Crawford, J.D.; Kochan, N.A.; Slavin, M.J.; Wen, W.; Trollor, J.N.; Brodaty, H.; Sachdev, P.S. Effects of statins on memory, cognition, and brain volume in the elderly. J. Am. Coll. Cardiol. 2019, 74, 2554–2568. [Google Scholar] [CrossRef] [PubMed]
- Adhyaru, B.B.; Jacobson, T.A. Safety and efficacy of statin therapy. Nat. Rev. Cardiol. 2018, 15, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Brinton, E.A.; Ito, M.K.; Jacobson, T.A. Understanding Statin use in America and Gaps in Patient Education (USAGE): An internet-based survey of 10,138 current and former statin users. J. Clin. Lipidol. 2012, 6, 208–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Cai, R.; Yuan, Y.; Varghese, Z.; Moorhead, J.; Ruan, X.Z. Association between reductions in low-density lipoprotein cholesterol with statin therapy and the risk of new-onset diabetes: A meta-analysis. Sci. Rep. 2017, 7, 39982. [Google Scholar] [CrossRef] [Green Version]
- Galicia-Garcia, U.; Jebari, S.; Larrea-Sebal, A.; Uribe, K.B.; Siddiqi, H.; Ostolaza, H.; Benito-Vicente, A.; Martín, C. Statin treatment-induced development of type 2 diabetes: From clinical evidence to mechanistic insights. Int. J. Mol. Sci. 2020, 21, 4725. [Google Scholar] [CrossRef]
- Schultz, B.G.; Patten, D.K.; Berlau, D.J. The role of statins in both cognitive impairment and protection against dementia: A tale of two mechanisms. Transl. Neurodegener. 2018, 7, 5. [Google Scholar] [CrossRef]
- Jurcau, A.; Simion, A. Oxidative stress in the pathogenesis of Alzheimer’s disease and cerebrovascular disease with therapeutic implications. CNS Neurol. Disord. Drug Targets 2020, 19, 94–108. [Google Scholar]
- Jacobson, T.A.; Cheeley, M.K.; Jones, P.H.; La Forge, R.; Maki, K.C.; López, A.G.; Xiang, P.; Bushnell, D.M.; Martin, M.L.; Cohen, J.D. The Statin Adverse Treatment Experience Survey: Experience of patients reporting side effects of statin therapy. J. Clin. Lipidol. 2019, 13, 415–424. [Google Scholar] [CrossRef]
- Alfirevic, A.; Neely, D.; Armitage, J.; Chinoy, H.; Cooper, R.G.; Laaksonen, R.; Carr, D.F.; Bloch, K.M.; Fahy, J.; Hanson, A.; et al. Phenotype standardization for statin-induced myotoxicity. Clin. Pharmacol. Ther. 2014, 96, 470–476. [Google Scholar] [CrossRef]
- Furberg, C.D.; Pitt, B. Withdrawal of cerivastatin from the world market. Curr. Control Trials Cardiovasc. Med. 2001, 2, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Birmingham, B.K.; Bujac, S.R.; Elsby, R.; Azumaya, C.T.; Wei, C.; Chen, Y.; Mosqueda-Garcia, R.; Ambrose, H.J. Impact of ABCG2 and SLCO1B1 polymorphisms on pharmacokinetics of rosuvastatin, atorvastatin and simvastatin acid in Caucasian and Asian subjects: A class effect? Eur. J. Clin. Pharmacol. 2015, 71, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Kozyra, M.; Ingelman-Sundberg, M.; Lauschke, V.M. Rare genetic variants in cellular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Genet. Med. 2017, 19, 20–29. [Google Scholar] [CrossRef]
- Bellosta, S.; Corsini, A. Statin drug interactions and related adverse reactions: An update. Expert Opin. Drug Saf. 2018, 17, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef] [PubMed]
- Schirris, T.J.; Renkema, G.H.; Ritschel, T.; Voermans, N.C.; Bilos, A.; van Engelen, B.G.; Brandt, U.; Koopman, W.J.; Beyrath, J.D.; Rodenburg, R.J.; et al. Statin-induced myopathy is associated with mitochondrial complex III inhibition. Cell Metab. 2015, 22, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Kwak, H.B.; Thalacker-Mercer, A.; Anderson, E.J.; Lin, C.T.; Kane, D.A.; Lee, N.S.; Cortright, R.N.; Bamman, M.M.; Neufer, P.D. Simvastatin impairs ADP-stimulated respiration and increases mitochondrial oxidative stress in primary human skeletal myotubes. Free Radic. Biol. Med. 2012, 52, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Saito, S.; Nakanishi, T.; Shirasaki, Y.; Nakajima, M.; Tamai, I. Association of miR-145 with statin-induced skeletal muscle toxicity in human rhabdomyosarcoma RD Cells. J. Pharm. Sci. 2017, 106, 2873–2880. [Google Scholar] [CrossRef] [Green Version]
- Lotteau, S.; Ivarsson, N.; Yang, Z.; Restagno, D.; Colyer, J.; Hopkins, P.; Weightman, A.; Himori, K.; Yamada, T.; Bruton, J.; et al. A mechanism for statin-induced susceptibility to myopathy. JACC Basic Transl. Sci. 2019, 4, 509–523. [Google Scholar] [CrossRef]
- Jurcau, A. Insights into the pathogenesis of neurodegenerative diseases: Focus on mitochondrial dysfunction and oxidative stress. Int. J. Mol. Sci. 2021, 22, 11847. [Google Scholar] [CrossRef]
- Draeger, A.; Monastyrskaya, K.; Mohaupt, M.; Hoppeler, H.; Savolainen, H.; Allemann, C.; Babiychuk, E.B. Statin therapy induces ultrastructural damage in skeletal muscle in patients without myalgia. J. Pathol. 2006, 210, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, P.M.; Thompson, P.D.; Cannon, C.P.; Guyton, J.R.; Bergeron, J.; Zieve, F.J.; Bruckert, E.; Jacobson, T.A.; Kopecky, S.L.; Baccara-Dinet, M.T.; et al. ODYSSEY ALTERNATIVE Investigators. Efficacy and safety of alirocumab vs ezetimibe in statin-intolerant patients, with a statin rechallenge arm: The ODYSSEY ALTERNATIVE randomized trial. J. Clin. Lipidol. 2015, 9, 758–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selva-O’Callaghan, A.; Alvarado-Cardenas, M.; Pinal-Fernández, I.; Trallero-Araguás, E.; Milisenda, J.C.; Martínez, M.Á.; Marín, A.; Labrador-Horrillo, M.; Juárez, C.; Grau-Junyent, J.M. Statin-induced myalgia and myositis: An update on pathogenesis and clinical recommendations. Expert Rev. Clin. Immunol. 2018, 14, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Mammen, A.L. Statin-associated autoimmune myopathy. N. Engl. J. Med. 2016, 374, 664–669. [Google Scholar] [CrossRef]
- Pasnoor, M.; Barohn, R.J.; Dimachkie, M.M. Toxic myopathies. Curr. Opin. Neurol. 2018, 31, 575–582. [Google Scholar] [CrossRef]
- Kuswanto, W.; Burzyn, D.; Panduro, M.; Wang, K.K.; Jang, Y.C.; Wagers, A.J.; Benoist, C.; Mathis, D. Poor repair of skeletal muscle in aging mice reflects a defect in local interleukin-33-dependent accumulation of regulatory T cells. Immunity 2016, 44, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Liu, R.T.; Du, T.; Yang, C.L.; Liu, Y.D.; Ge, M.R.; Zhang, M.; Li, X.L.; Li, H.; Dou, Y.C.; et al. Exosomes derived from statin-modified bone marrow dendritic cells increase thymus-derived natural regulatory T cells in experimental autoimmune myasthenia gravis. J. Neuroinflammation 2019, 16, 202. [Google Scholar] [CrossRef]
- Golomb, B.A.; Criqui, M.H.; White, H.; Dimsdale, J.E. Conceptual foundations of the UCSD statin study. Arch. Intern. Med. 2004, 164, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Hussain, G.; Wang, J.; Rasul, A.; Anwar, H.; Imran, A.; Qasim, M.; Zafar, S.; Kamran, S.K.S.; Razzaq, A.; Aziz, N.; et al. Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis. 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Berghoff, S.A.; Gerndt, N.; Winchenbach, J.; Stumpf, S.K.; Hosang, L.; Odoardi, F.; Ruhwedel, T.; Böhler, C.; Barrette, B.; Stassart, R.; et al. Dietary cholesterol promotes repair of demyelinated lesions in the adult brain. Nat. Commun. 2017, 8, 14241. [Google Scholar] [CrossRef]
- Klopfleisch, S.; Merkler, D.; Schmitz, M.; Klöppner, S.; Schedensack, M.; Jeserich, G.; Althaus, H.H.; Brück, W. Negative impact of statins on oligodendrocytes and myelin formation in vitro and in vivo. J. Neurosci. 2008, 28, 13609–13614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miron, V.E.; Zehntner, S.P.; Kuhlmann, T.; Ludwin, S.K.; Owens, T.; Kennedy, T.E.; Bedell, B.J.; Antel, J.P. Statin therapy inhibits remyelination in the central nervous system. Am. J. Pathol. 2009, 174, 1880–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, L.; Zhang, T.; Fan, K.; Cai, W.; Liu, H. Astrocyte-neuron signaling in synaptogenesis. Front. Cell Dev. Biol. 2021, 9, 680301. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.L.; Song, W.; Sansom, M.S.P. Lipid-dependent regulation of ion channels and G protein-coupled receptors: Insights from structures and simulations. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 31–50. [Google Scholar] [CrossRef]
- Gimpl, G. Interaction of G protein coupled receptors and cholesterol. Chem. Phys. Lipids 2016, 199, 61–73. [Google Scholar] [CrossRef]
- Herron, C.E.; Metais, C. Effects of chronic and acute simvastatin on neuronal excitability and LTP in APPswe/PS1dE9 mice. Alzheimers Dement. 2010, 6, S561. [Google Scholar] [CrossRef]
- Bukiya, A.; Blank, N.P.S.; Rosenhouse-Dantsker, A. Cholesterol intake and statin use regulate neuronal G protein-gated inwardly rectifying potassium channels. J. Lipid Res. 2019, 60, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Langsjoen, P.H.; Langsjoen, A.M. The clinical use of HMG CoA-reductase inhibitors and the associated depletion of coenzyme Q10. A review of animal and human publications. Biofactors 2003, 18, 101–111. [Google Scholar] [CrossRef]
- Derosa, G.; D’Angelo, A.; Maffioli, P. Coenzyme q10 liquid supplementation in dyslipidemic subjects with statin-related clinical symptoms: A double-blind, randomized, placebo-controlled study. Drug Des. Devel. Ther. 2019, 13, 3647–3655. [Google Scholar] [CrossRef] [Green Version]
- Bouitbir, J.; Charles, A.L.; Echaniz-Laguna, A.; Kindo, M.; Daussin, F.; Auwerx, J.; Piquard, F.; Geny, B.; Zoll, J. Opposite effects of statins on mitochondria of cardiac and skeletal muscles: A ‘mitohormesis’ mechanism involving reactive oxygen species and PGC-1. Eur. Heart J. 2012, 33, 1397–1407. [Google Scholar] [CrossRef]
- Mikus, C.R.; Boyle, L.J.; Borengasser, S.J.; Oberlin, D.J.; Naples, S.P.; Fletcher, J.; Meers, G.M.; Ruebel, M.; Laughlin, M.H.; Dellsperger, K.C.; et al. Simvastatin impairs exercise training adaptations. J. Am. Coll. Cardiol. 2013, 62, 709–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouitbir, J.; Sanvee, G.M.; Panajatovic, M.V.; Singh, F.; Krähenbühl, S. Mechanisms of statin-associated skeletal muscle-associated symptoms. Pharmacol. Res. 2020, 154, 104201. [Google Scholar] [CrossRef] [PubMed]
- Stringer, H.A.; Sohi, G.K.; Maguire, J.A.; Côté, H.C. Decreased skeletal muscle mitochondrial DNA in patients with statin-induced myopathy. J. Neurol. Sci. 2013, 325, 142–147. [Google Scholar] [CrossRef]
- Jurcau, A.; Ardelean, I.A. Molecular pathophysiological mechanisms of ischemia/reperfusion injuries after recanalization therapy for acute ischemic stroke. J. Integr. Neurosci. 2021, 20, 727–744. [Google Scholar] [PubMed]
- Roy, A.; Jana, M.; Kundu, M.; Corbett, G.T.; Rangaswamy, S.B.; Mishra, R.K.; Luan, C.-H.; Gonzalez, F.J.; Pahan, K. HMG-CoA reductase inhibitors bind to PPARalpha to upregulate neurotrophin expression in the brain and improve memory in mice. Cell Metab. 2015, 22, 253–265. [Google Scholar] [CrossRef] [Green Version]
- Okudan, N.; Belviranli, M. High dose simvastatin and rosuvastatin impair cognitive abilities of healthy rats via decreasing hippocampal neurotrophins and irisin. Brain Res. Bull. 2020, 165, 81–89. [Google Scholar] [CrossRef]
- Tan, B.; Rosenfeldt, F.; Ou, R.; Stough, C. Evidence and mechanisms for statin-induced cognitive decline. Expert Rev. Clin. Pharmacol. 2019, 12, 397–406. [Google Scholar] [CrossRef]
- Deveau, C.M.; Rodriguez, E.; Schroering, A.; Yamamoto, B.K. Serotonin transporter regulation by cholesterol-independent lipid signaling. Biochem. Pharmacol. 2021, 183, 114349. [Google Scholar] [CrossRef]
- Virgo, J.; Wong, S.; Rantell, K.; Plant, G. Statins can cause myasthenia gravis: Fact or fiction? J. Neurol. Neurosurg. Psychiatry 2013, 84, e2. [Google Scholar] [CrossRef]
- Deenen, J.C.; Horlings, C.G.; Verschuuren, J.J.; Verbeek, A.L.; van Engelen, B.G. The epidemiology of neuromuscular disorders: A comprehensive overview of the literature. J. Neuromuscul. Dis. 2015, 2, 3–85. [Google Scholar] [CrossRef] [Green Version]
- Westerberg, E.; Punga, A.R. Mortality rates and causes of death in Swedish myasthenia gravis patients. Neuromuscul. Disord. 2020, 30, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Basta, I.; Pekmezović, T.; Peric, S.; Nikolić, A.; Rakoćević-Stojanović, V.; Stević, Z.; Lavrnić, D. Survival and mortality of adult-onset myasthenia gravis in the population of Belgrade, Serbia. Muscle Nerve 2018, 58, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, S.; Alvi, U.; Soliven, B.; Rezania, K. Drugs that induce or cause deterioration of myasthenia gravis: An update. J. Clin. Med. 2021, 10, 1537. [Google Scholar] [CrossRef]
- Gale, J.; Danesh-Meyer, H.V. Statins can induce myasthenia gravis. J. Clin. Neurosci. 2014, 21, 195–197. [Google Scholar] [CrossRef]
- Crisan, E.; Patil, V.K. Neuromuscular complications of statin therapy. Curr. Neurol. Neurosci Rep. 2020, 20, 47. [Google Scholar] [CrossRef] [PubMed]
- Purvin, V.; Kawasaki, A.; Smith, K.H.; Kesler, A. Statin-associated myasthenia gravis: Report of 4 cases and review of the literature. Medicine 2006, 85, 82–85. [Google Scholar] [CrossRef]
- Gras-Champel, V.; Masmoudi, I.; Batteux, B.; Merle, P.E.; Liabeuf, S.; Masmoudi, K. Statin-associated myasthenia: A case report and literature review. Therapies 2020, 75, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Gras-Champel, V.; Batteux, B.; Masmoudi, K.; Liabeuf, S. Statin-induced myasthenia: A disproportionality analysis of the WHO’s VigiBase pharmacovigilance database. Muscle Nerve 2019, 60, 382–386. [Google Scholar] [CrossRef] [Green Version]
- Attardo, S.; Musumeci, O.; Velardo, D.; Toscano, A. Statins neuromuscular adverse effects. Int. J. Mol. Sci. 2022, 23, 8364. [Google Scholar] [CrossRef]
- Milani, M.; Ostlie, N.; Wang, W.; Conti-Fine, B.M. T cells and cytokines in the pathogenesis of acquired myasthenia gravis. Ann. N. Y. Acad. Sci. 2003, 998, 284–307. [Google Scholar] [CrossRef]
- Youssef, S.; Stüve, O.; Patarroyo, J.C.; Ruiz, P.J.; Radosevich, J.L.; Hur, E.M.; Bravo, M.; Mitchell, D.J.; Sobel, R.A.; Steinman, L.; et al. The HMG-CoA reductase inhibitor atorvastatin promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature 2002, 420, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Ragbourne, S.C.; Crook, M.A. Use of lipid-lowering medications in myasthenia gravis: A case report and literature review. J. Clin. Lipidol. 2015, 9, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.N.; Kesari, A.; Yokota, T.; Pandey, G.S. Muscular dystrophy: Disease mechanisms and therapies. Biomed. Res. Int. 2015, 2015, 456348. [Google Scholar] [CrossRef] [PubMed]
- Colvin, M.K.; Poysky, J.; Kinnett, K.; Damiani, M.; Gibbons, M.; Hoskin, J.; Moreland, S.; Trout, C.J.; Weidner, N. Psychosocial management of the patient with Duchenne muscular dystrophy. Pediatrics 2018, 142, S99–S109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Trifirò, G. Global epidemiology of Duchenne muscular dystrophy: An updated systematic review and meta-analysis. Orphanet J. Rare Dis. 2020, 15, 141. [Google Scholar] [CrossRef]
- Ryder, S.; Leadley, R.M.; Armstrong, N.; Westwood, M.; de Kock, S.; Butt, T.; Jain, M.; Kleijnen, J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet J. Rare Dis. 2017, 12, 79. [Google Scholar] [CrossRef] [Green Version]
- Machuca-Tzili, L.; Brook, D.; Hilton-Jones, D. Clinical and molecular aspects of the myotonic dystrophies: A review. Muscle Nerve 2005, 32, 1–18. [Google Scholar] [CrossRef]
- Wahbi, K.; Furling, D. Cardiovascular manifestations of myotonic dystrophy. Trends Cardiovasc. Med. 2020, 30, 232–238. [Google Scholar] [CrossRef]
- Shakir, M.K.M.; Shin, T.; Hoang, T.D.; Mai, V.Q. Successful treatment of a patient with statin-induced myopathy and myotonic dystrophy type II with proprotein convertase subtilisin/kexin type 9 inhibitor, alirocumab (Praluent). J. Clin. Lipidol. 2017, 11, 1485–1487. [Google Scholar] [CrossRef]
- Triplett, J.D.; Shelly, S.; Livne, G.; Milone, M.; Kassardjian, C.D.; Liewluck, T.; Kelly, C.; Naddaf, E.; Laughlin, R.S.; Lamb, C.J.; et al. Diagnostic modelling and therapeutic monitoring of immune-mediated necrotizing myopathy: Role of electrical myotonia. Brain Commun. 2020, 2, fcaa191. [Google Scholar] [CrossRef]
- Meriggioli, M.N.; Barboi, A.C.; Rowin, J.; Cochran, E.J. HMG-CoA reductase inhibitor myopathy: Clinical, electrophysiological, and pathologic data in five patients. J. Clin. Neuromuscul. Dis. 2001, 2, 129–134. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, D.F.; Lissa, T.V.; Melo, A.C., Jr. Myotonic potentials in statin-induced rhabdomyolysis. Arq. Neuropsiquiatr. 2008, 66, 891–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camerino, G.M.; Musumeci, O.; Conte, E.; Musaraj, K.; Fonzino, A.; Barca, E.; Marino, M.; Rodolico, C.; Tricarico, D.; Camerino, C.; et al. Risk of myopathy in patients in therapy with statins: Identification of biological markers in a pilot study. Front. Pharmacol. 2017, 8, 500. [Google Scholar] [CrossRef]
- Screen, M.; Jonson, P.H.; Raheem, O.; Palmio, J.; Laaksonen, R.; Lehtimäki, T.; Sirito, M.; Krahe, R.; Hackman, P.; Udd, B. Abnormal splicing of NEDD4 in myotonic dystrophy type 2: Possible link to statin adverse reactions. Am. J. Pathol. 2014, 184, 2322–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, M.J.; Carrie, A. Mechanisms of statin-induced myopathy: A role for the ubiquitin-proteasome pathway? Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2441–2444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Trotman, L.C.; Koppie, T.; Alimonti, A.; Chen, Z.; Gao, Z.; Wang, J.; Erdjument-Bromage, H.; Tempst, P.; Cordon-Cardo, C.; et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 2007, 128, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Mygland, Å.; Ljøstad, U.; Krossnes, B.K. Persisting weakness after withdrawal of a statin. BMJ Case Rep. 2014, 2014, bcr2013203094. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, A.A.S.; da Silva, V.G.; Zanoteli, E.; Feder, D. Myopathy due to HMGCR antibodies in adult mimicking muscular dystrophy associated with cancer and statin exposure-narrative review of the literature-case report. Ther. Clin. Risk Manag. 2018, 14, 903–907. [Google Scholar] [CrossRef] [Green Version]
- Knoblauch, H.; Schoewel, V.; Rosada, A.; Spuler, S.; Kress, W. Another side to statin-related side effects. Ann. Int. Med. 2010, 152, 478–479. [Google Scholar] [CrossRef]
- Mohassel, P.; Landon-Cardinal, O.; Foley, A.R.; Donkervoort, S.; Pak, K.S.; Wahl, C.; Shebert, R.T.; Harper, A.; Fequiere, P.; Meriggioli, M.; et al. Anti-HMGCR myopathy may resemble limb-girdle muscular dystrophy. Neurol. Neuroimmunol. Neuroinflamm. 2018, 6, e523. [Google Scholar] [CrossRef] [Green Version]
- Ruaño, G.; Windemuth, A.; Wu, A.H.; Kane, J.P.; Malloy, M.J.; Pullinger, C.R.; Kocherla, M.; Bogaard, K.; Gordon, B.R.; Holford, T.R.; et al. Mechanisms of statin-induced myalgia assessed by physiogenomic associations. Atherosclerosis 2011, 218, 451–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulchandani, R.; Lyngdoh, T.; Kakkar, A.K. Statin use and safety concerns: An overview of the past, present, and the future. Expert Opin. Drug Saf. 2020, 19, 1011–1024. [Google Scholar] [CrossRef] [PubMed]
- Sahebzamani, F.M.; Munro, C.L.; Marroquin, O.C.; Diamond, D.M.; Keller, E.; Kip, K.E. Examination of the FDA warning for statins and cognitive dysfunction. J. Pharmacovigil. 2014, 2, 1000141. [Google Scholar] [CrossRef]
- Roy, S.; Weinstock, J.L.; Ishino, A.S.; Benites, J.F.; Pop, S.R.; Perez, C.D.; Gumbs, E.A.; Rosenbaum, J.A.; Roccato, M.K.; Shah, H.; et al. Association of cognitive impairment in patients on 3-Hydroxy-3-Methyl-Glutaryl-CoA reductase inhibitors. J. Clin. Med. Res. 2017, 9, 638–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahangari, N.; Doosti, M.; Ghayour Mobarhan, M.; Sahebkar, A.; Ferns, G.A.; Pasdar, A. Personalised medicine in hypercholesterolaemia: The role of pharmacogenetics in statin therapy. Ann. Med. 2020, 52, 462–470. [Google Scholar] [CrossRef]
- Barthold, D.; Joyce, G.; Brinton, R.D.; Wharton, W.; Kehoe, P.G.; Zissimopoulos, J. Association of combination statin and antihypertensive therapy with reduced Alzheimer’s disease and related dementia risk. PLoS ONE 2020, 15, e0229541. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, F.F.; Bertolucci, P.H.F.; Chen, E.S.; Smith, M.C. Pharmacogenetic analyses of therapeutic effects of lipophilic statins on cognitive and functional changes in Alzheimer’s disease. J. Alzheimer’s Dis. 2022, 87, 359–372. [Google Scholar] [CrossRef]
- Jamshidnejad-Tosaramandani, T.; Kashanian, S.; Al-Sabri, M.H.; Kročianová, D.; Clemensson, L.E.; Gentreau, M.; Schiöth, H.B. Statins and cognition: Modifying factors and possible underlying mechanisms. Front. Aging Neurosci. 2022, 14, 968039. [Google Scholar] [CrossRef]
- Rojas-Fernandez, C.H.; Cameron, J.C. Is statin-associated cognitive impairment clinically relevant? A narrative review and clinical recommendations. Ann. Pharmacother. 2012, 46, 549–557. [Google Scholar] [CrossRef]
- Kim, M.-Y.; Jung, M.; Noh, Y.; Shin, S.; Hong, C.H.; Lee, S.; Jung, Y.-S. Impact of statin use on dementia incidence in elderly men and women with ischemic heart disease. Biomedicines 2020, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Faubion, S.S.; Kapoor, E.; Moyer, A.M.; Hodis, H.N.; Miller, V.M. Statin therapy: Does sex matter? Menopause 2019, 26, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Alisha, K.; Tripti, S. Repurposing statins as a potential ligand for estrogen receptor alpha via molecular docking. Res. J. Pharm. Tech. 2021, 14, 3757–3762. [Google Scholar] [CrossRef]
- Joosten, H.; Visser, S.T.; van Eersel, M.E.; Gansevoort, R.T.; Bilo, H.J.; Slaets, J.P.; Izaks, G.J. Statin use and cognitive function: Population-based observational study with long-term follow-up. PLoS ONE 2014, 9, e115755. [Google Scholar] [CrossRef] [PubMed]
- Swiger, K.J.; Manalac, R.J.; Blumenthal, R.S.; Blaha, M.J.; Martin, S.S. Statins and cognition: A systematic review and meta-analysis of short- and long-term cognitive effects. Mayo Clin. Proc. 2013, 88, 1213–1221. [Google Scholar] [CrossRef]
- Rost, N.S.; Brodtmann, A.; Pase, M.P.; van Veluw, S.J.; Biffi, A.; Duering, M.; Hinman, J.D.; Dichgans, M. Post-stroke cognitive impairment and dementia. Circ. Res. 2022, 130, 1252–1271. [Google Scholar] [CrossRef]
- Davis, K.A.; Bishara, D.; Perera, G.; Molokhia, M.; Rajendran, L.; Stewart, R.J. Benefits and harms of statins in people with dementia: A systematic review and meta-analysis. J. Am. Geriatr. Soc. 2020, 68, 650–658. [Google Scholar] [CrossRef]
- Alsehli, A.M.; Olivo, G.; Clemensson, L.E.; Williams, M.J.; Schiöth, H.B. The cognitive effects of statins are modified by age. Sci. Rep. 2020, 10, 6187. [Google Scholar] [CrossRef] [Green Version]
- Jurcau, A.; Simion, A. Cognition, statins, and cholesterol in elderly ischemic stroke patients: A neurologist’s perspective. Medicina 2021, 57, 616. [Google Scholar] [CrossRef]
- Bosch, J.; Donnell, M.; Swaminathan, B.; Lonn, E.M.; Sharma, M.; Dagenais, G.; Diaz, R.; Khunti, K.; Lewis, B.S.; Avezum, A.; et al. Effects of blood pressure and lipid lowering on cognition. Neurology 2019, 92, e1435. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Mudher, A. Alzheimer’s disease and type 2 diabetes: A critical assessment of the shared pathological traits. Front. Neurosci. 2018, 12, 383. [Google Scholar] [CrossRef] [Green Version]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.-Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Hyman, D.; Ayyala, S.; Bakhshi, A.; Kim, S.H.; Anoruo, N.; Weinstock, J.; Balogun, A.; D’Souza, M.; Filatova, N.; et al. Cognitive function assessment in patients on moderate-or high-intensity statin therapy. J. Clin. Med. Res. 2020, 12, 255–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poly, T.N.; Islam, M.M.; Walther, B.A.; Yang, H.-C.; Wu, C.-C.; Lin, M.-C.; Li, Y.-C. Association between use of statin and risk of dementia: A meta-analysis of observational studies. Neuroepidemiology 2020, 54, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.-L.; Hsu, C.-C.; Chen, Y.-M.; Yu, H.-K.; Hu, G.-C. Statin use and the risk of dementia in patients with stroke: A nationwide population-based cohort study. J. Stroke Cerebrovasc. Dis. 2018, 27, 3001–3007. [Google Scholar] [CrossRef]
- Sinyavskaya, L.; Gauthier, S.; Renoux, C.; Dell’Aniello, S.; Suissa, S.; Brassard, P. Comparative effect of statins on the risk of incident Alzheimer disease. Neurology 2018, 90, e179–e187. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, R.P.; Wiviott, S.D.; Blazing, M.A.; De Ferrari, G.M.; Park, J.G.; Murphy, S.A.; White, J.A.; Tershakovec, A.M.; Cannon, C.P.; Braunwald, E. Long-term safety and efficacy of achieving very low levels of low-density lipoprotein cholesterol: A prespecified analysis of the IMPROVE-IT Trial. JAMA Cardiol. 2017, 2, 547–555. [Google Scholar] [CrossRef]
- Giugliano, R.P.; Mach, F.; Zavitz, K.; Kurtz, C.; Im, K.; Kanevsky, E.; Schneider, J.; Wang, H.; Keech, A.; Pedersen, T.R.; et al. EBBINGHAUS Investigators. Cognitive function in a randomized trial of evolocumab. N. Engl. J. Med. 2017, 377, 633–643. [Google Scholar] [CrossRef]
- Adams, H.P., Jr.; Bendixen, B.H.; Kappelle, L.J.; Biller, J.; Love, B.B.; Gordon, D.L.; Marsh, E.E., 3rd. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993, 24, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Adelhoefer, S.; Uddin, S.M.I.; Osei, A.D.; Obisesan, O.H.; Blaha, M.J.; Dzaye, O. Coronary artery calcium scoring: New insights into clinical interpretation-lessons from the CAC Consortium. Radiol. Cardiothorac. Imaging 2020, 2, e200281. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Statin | Dose | Solubility | Liver Metabolization | Half-Life (Hours) |
|---|---|---|---|---|
| Lovastatin | 10–80 mg | lipophilic | CYP450 3A4 | 2 |
| Fluvastatin | 20–80 mg | lipophilic | CYP450 2C9 | 4.7 |
| Simvastatin | 5–40 mg | lipophilic | CYP4503A4 | 1–2 |
| Atorvastatin | 10–80 mg | lipophilic | CYP450 3A4 | 14 |
| Pitavastatin | 1–4 mg | lipophilic | CYP450 2C9 | 96 |
| Rosuvastatin | 5–40 mg | hydrophilic | CYP450 2C9 and 2C19 | 19 |
| Pravastatin | 20–80 mg | hydrophilic | sulphation | 1–2 |
| SRM Classification | Phenotype | Definition |
|---|---|---|
| SRM 0 | Asymptomatic | Elevations of <4× upper normal limit in serum creatine kinase (CK) |
| SRM 1 | Myalgia, tolerable | Muscle aches, cramps and/or weakness with no elevation of CK |
| SRM 2 | Myalgia, intolerable | Muscle aches, cramps and/or weakness with < 4× upper normal limit of CK |
| SRM 3 | Myopathy | CK levels > 4× but < 10× upper normal limit of CK |
| SRM 4 | Severe myopathy | CK levels >10× but < 50× upper normal limit |
| SRM 5 | Rhabdomyolysis | Either CK > 10× upper normal limit, muscle symptoms and renal impairment, or CK > 50× upper normal limit |
| SRM 6 | Autoimmune-mediated, necrotizing myositis | HMGCR antibodies, HMGCR expression in muscle biopsy, incomplete resolution after statin discontinuation |
| Solubility | Statin | Effects of Statin Use on Cognitive Function | Number of Patients | Reference |
|---|---|---|---|---|
| Lipophilic | Simvastatin |
| 4724 pairs of patients 9,162,509 total number 10,888 334,861 | [154] [153] [140] [155] |
| Atorvastatin |
| 4724 pairs of patients 9,162,509 total number 45,753 | [154] [153] [140] | |
| Lovastatin |
| Total of 9,162,509 567 | [153] [140] | |
| Fluvastatin |
| Total of 9,162,509 patients 4724 pairs of patients | [153] [154] | |
| Hydrophilic | Rosuvastatin |
| 4724 pairs of patients 62,387 9,162,509 8251 | [154] [136] [153] [140] |
| Pravastatin |
| 9,162,509 2524 77,795 | [153] [140] [136] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andronie-Cioară, F.L.; Jurcău, A.; Jurcău, M.C.; Nistor-Cseppentö, D.C.; Simion, A. Cholesterol Management in Neurology: Time for Revised Strategies? J. Pers. Med. 2022, 12, 1981. https://doi.org/10.3390/jpm12121981
Andronie-Cioară FL, Jurcău A, Jurcău MC, Nistor-Cseppentö DC, Simion A. Cholesterol Management in Neurology: Time for Revised Strategies? Journal of Personalized Medicine. 2022; 12(12):1981. https://doi.org/10.3390/jpm12121981
Chicago/Turabian StyleAndronie-Cioară, Felicia Liana, Anamaria Jurcău, Maria Carolina Jurcău, Delia Carmen Nistor-Cseppentö, and Aurel Simion. 2022. "Cholesterol Management in Neurology: Time for Revised Strategies?" Journal of Personalized Medicine 12, no. 12: 1981. https://doi.org/10.3390/jpm12121981
APA StyleAndronie-Cioară, F. L., Jurcău, A., Jurcău, M. C., Nistor-Cseppentö, D. C., & Simion, A. (2022). Cholesterol Management in Neurology: Time for Revised Strategies? Journal of Personalized Medicine, 12(12), 1981. https://doi.org/10.3390/jpm12121981