
Targeting KRAS in Pancreatic Cancer


{kind=link}
Abstract
1. Introduction
2. Genomics of PDAC
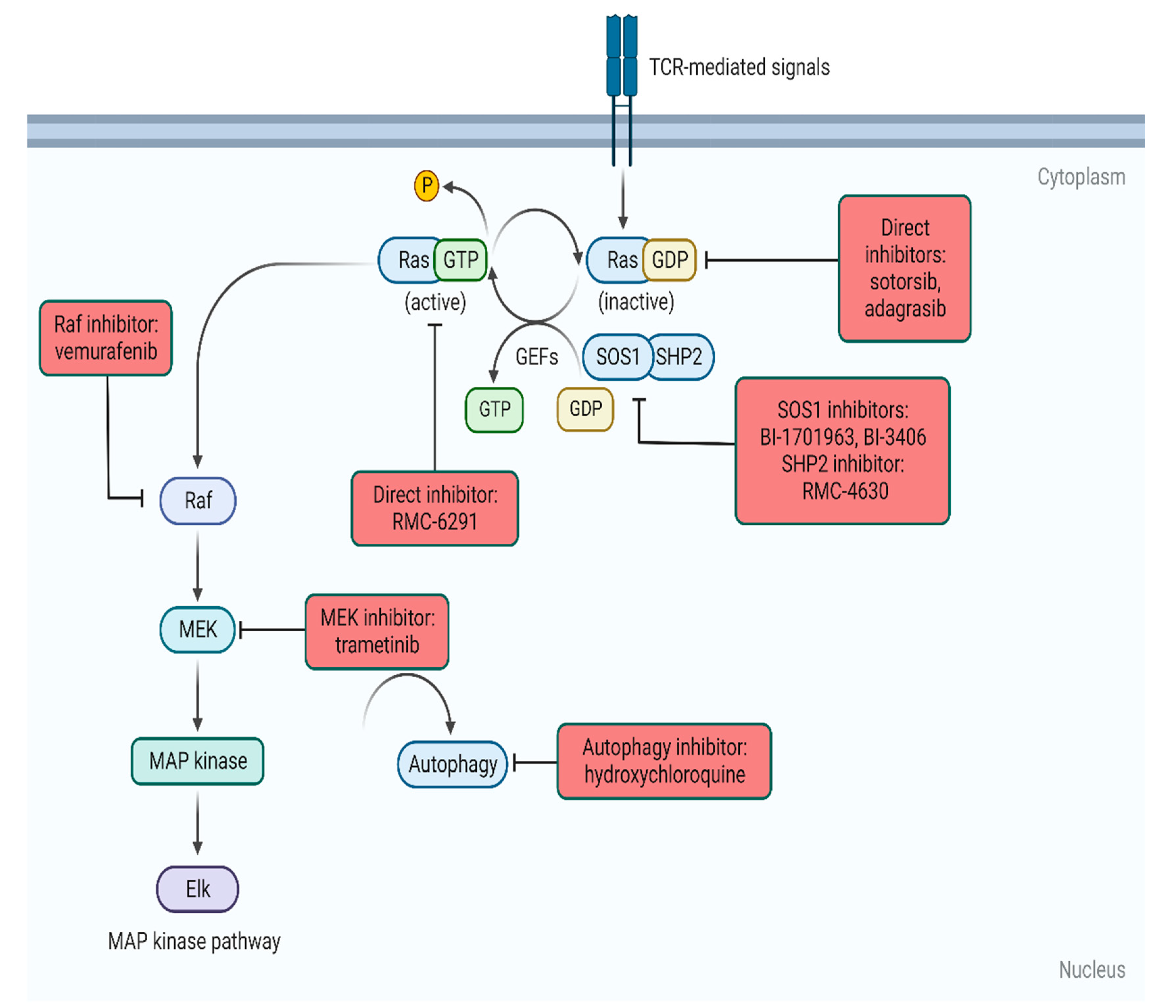
3. Methods of KRAS Inhibition
3.1. KRAS G12C-Specific
3.2. Other Allele-Specific ‘off’ Inhibitors
3.3. RAS ‘on’ Inhibitors
3.4. Adjuncts to RAS Inhibitors
3.5. Selective Downstream Inhibition
3.6. Autophagy
3.7. RNA Interference (RNAi) Approaches
3.8. Immune Approaches
3.8.1. Adoptive Cell Therapies
3.8.2. Vaccines
3.8.3. Immune Checkpoint Inhibitors
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Vander Heiden, M.G.; McCormick, F. The Metabolic Landscape of RAS-Driven Cancers from biology to therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Punekar, S.R.; Velcheti, V.; Neel, B.G.; Wong, K.K. The current state of the art and future trends in RAS-targeted cancer therapies. Nat. Rev. Clin. Oncol. 2022, 19, 637–655. [Google Scholar] [CrossRef] [PubMed]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 1–22. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef]
- Chung, V.; McDonough, S.; Philip, P.A.; Cardin, D.; Wang-Gillam, A.; Hui, L.; Tejani, M.A.; Seery, T.E.; Dy, I.A.; Al Baghdadi, T.; et al. Effect of Selumetinib and MK-2206 vs. Oxaliplatin and Fluorouracil in Patients with Metastatic Pancreatic Cancer after Prior Therapy: SWOG S1115 Study Randomized Clinical Trial. JAMA Oncol. 2017, 3, 516–522. [Google Scholar] [CrossRef]
- Bodoky, G.; Timcheva, C.; Spigel, D.R.; La Stella, P.J.; Ciuleanu, T.E.; Pover, G.; Tebbutt, N.C. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Investig. New Drugs 2012, 30, 1216–1223. [Google Scholar] [CrossRef]
- Aguirre, A.J.; Collisson, E.A. Advances in the Genetics and Biology of Pancreatic Cancer. Cancer J 2017, 23, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Kiel, C.; Matallanas, D.; Kolch, W. The Ins and Outs of RAS Effector Complexes. Biomolecules 2021, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Burkhart, D.L.; Haigis, K.M. Classification of KRAS-Activating Mutations and the Implications for Therapeutic Intervention. Cancer Discov. 2022, 12, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Stolze, B.; Reinhart, S.; Bulllinger, L.; Frohling, S.; Scholl, C. Comparative analysis of KRAS codon 12, 13, 18, 61, and 117 mutations using human MCF10A isogenic cell lines. Sci. Rep. 2015, 5, 8535. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted With Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253.e4. [Google Scholar] [CrossRef]
- Gentile, D.R.; Rathinaswamy, M.K.; Jenkins, M.L.; Moss, S.M.; Siempelkamp, B.D.; Renslo, A.R.; Burke, J.E.; Shokat, K.M. Ras Binder Induces a Modified Switch-II Pocket in GTP and GDP States. Cell Chem. Biol. 2017, 24, 1455–1466.e14. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef]
- Patricelli, M.P.; Janes, M.R.; Li, L.S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Smaill, J.B.; Liu, T.; Ding, K.; Lu, X. Small-Molecule Inhibitors Directly Targeting KRAS as Anticancer Therapeutics. J. Med. Chem. 2020, 63, 14404–14424. [Google Scholar] [CrossRef] [PubMed]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Blake, J.F.; Bouhana, K.; Briere, D.M.; Brown, K.D.; Burgess, L.E.; Burns, A.C.; Burkard, M.R.; et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRAS(G12C) Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 6679–6693. [Google Scholar] [CrossRef]
- Lanman, B.A.; Allen, J.R.; Allen, J.G.; Amegadzie, A.K.; Ashton, K.S.; Booker, S.K.; Chen, J.J.; Chen, N.; Frohn, M.J.; Goodman, G.; et al. Discovery of a Covalent Inhibitor of KRAS(G12C) (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Vasta, J.D.; Peacock, D.M.; Zheng, Q.; Walker, J.A.; Zhang, Z.; Zimprich, C.A.; Thomas, M.R.; Beck, M.T.; Binkowski, B.F.; Corona, C.R.; et al. KRAS is vulnerable to reversible switch-II pocket engagement in cells. Nat. Chem. Biol. 2022, 18, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Strickler, J.H.; Satake, H.; Hollebecque, A.; Sunakawa, Y.; Tomasini, P.; Bajor, D.L.; Schuler, M.H.; Yaeger, R.; George, T.J.; Garrido-Laguna, I.; et al. First data for sotorasib in patients with pancreatic cancer with KRAS p.G12C mutation: A phase I/II study evaluating efficacy and safety. J. Clin. Oncol. 2022, 40, 360490. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.S.; Spira, A.I.; Yaeger, R.; Buchschacher, G.L.; McRee, A.J.; Sabari, J.K.; Johnson, M.L.; Barve, M.A.; Hafez, N.; Velastegui, K. KRYSTAL-1: Updated activity and safety of adagrasib (MRTX849) in patients (Pts) with unresectable or metastatic pancreatic cancer (PDAC) and other gastrointestinal (GI) tumors harboring a KRASG12C mutation. J. Clin. Oncol. 2022, 40, 519. [Google Scholar] [CrossRef]
- Peng, S.-B.; Si, C.; Zhang, Y.; Van Horn, R.D.; Lin, X.; Gong, X.; Huber, L.; Donoho, G.; Curtis, C.; Strelow, J.M.; et al. Abstract 1259: Preclinical characterization of LY3537982, a novel, highly selective and potent KRAS-G12C inhibitor. Cancer Res. 2021, 81 (Suppl. 13), 1259. [Google Scholar] [CrossRef]
- Johnson, M.L.; de Langen, A.J.; Waterhouse, D.M.; Mazieres, J.; Dingemans, A.M.C.; Mountzios, G.; Pless, M.; Wolf, J.; Schuler, M.; Lena, H.; et al. LBA10 Sotorasib versus docetaxel for previously treated non-small cell lung cancer with KRAS G12C mutation: CodeBreaK 200 phase III study. Ann. Oncol. 2022, 33, S1417–S1418. [Google Scholar] [CrossRef]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J. Acquired resistance to KRASG12C inhibition in cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Koltun, E.S.; Rice, M.A.; Gustafson, W.C.; Wilds, D.; Jiang, J.; Lee, B.J.; Wang, Z.; Chang, S.; Flagella, M.; Mu, Y.; et al. Abstract 3597: Direct targeting of KRASG12X mutant cancers with RMC-6236, a first-in-class, RAS-selective, orally bioavailable, tri-complex RASMULTI(ON) inhibitor. Cancer Res. 2022, 82 (Suppl. 12), 3597. [Google Scholar] [CrossRef]
- Hofmann, M.H.; Gerlach, D.; Misale, S.; Petronczki, M.; Kraut, N. Expanding the Reach of Precision Oncology by Drugging All KRAS Mutants. Cancer Discov. 2022, 12, 924–937. [Google Scholar] [CrossRef] [PubMed]
- Kessler, D.; Gerlach, D.; Kraut, N.; McConnell, D.B. Targeting Son of Sevenless 1: The pacemaker of KRAS. Curr. Opin. Chem. Biol. 2021, 62, 109–118. [Google Scholar] [CrossRef]
- Buday, L.; Vas, V. Novel regulation of Ras proteins by direct tyrosine phosphorylation and dephosphorylation. Cancer Metastasis Rev. 2020, 39, 1067–1073. [Google Scholar] [CrossRef]
- Jeng, H.H.; Taylor, L.J.; Bar-Sagi, D. Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat. Commun. 2012, 3, 1168. [Google Scholar] [CrossRef]
- Gort, E.; Johnson, M.L.; Hwang, J.J.; Pant, S.; Dünzinger, U.; Riemann, K.; Kitzing, T.; Janne, P.A. A phase I, open-label, dose-escalation trial of BI 1701963 (SOS1::KRAS inhibitor) in patients with KRAS mutated solid tumours: A snapshot analysis. In Proceedings of the ESMO, Virtual, 16–21 September 2021; Available online: https://www.inoncology.com/sites/default/files/esmo_2021_1432.1_johnson_kras.pdf (accessed on 30 October 2022).
- Ou, S.I.; Koczywas, M.; Ulahannan, S.; Janne, P.; Pacheco, J.; Burris, H.; McCoach, C.; Wang, J.S.; Gordon, M.; Haura, E.; et al. A12 The SHP2 Inhibitor RMC-4630 in Patients with KRAS-Mutant Non-Small Cell Lung Cancer: Preliminary Evaluation of a First-in-Man Phase 1 Clinical Trial. J. Thorac. Oncol. 2020, 15, S15–S16. [Google Scholar] [CrossRef]
- Liu, C.; Lu, H.; Wang, H.; Loo, A.; Zhang, X.; Yang, G.; Kowal, C.; Delach, S.; Wang, Y.; Goldoni, S. Combinations with Allosteric SHP2 Inhibitor TNO155 to Block Receptor Tyrosine Kinase SignalingCombinations with SHP2 Inhibitor to Block RTK Signaling. Clin. Cancer Res. 2021, 27, 342–354. [Google Scholar] [CrossRef]
- Theard, P.L.; Sheffels, E.; Sealover, N.E.; Linke, A.J.; Pratico, D.J.; Kortum, R.L. Marked synergy by vertical inhibition of EGFR signaling in NSCLC spheroids shows SOS1 is a therapeutic target in EGFR-mutated cancer. Elife 2020, 9, e58204. [Google Scholar] [CrossRef] [PubMed]
- Janne, P.A.; van den Heuvel, M.M.; Barlesi, F.; Cobo, M.; Mazieres, J.; Crino, L.; Orlov, S.; Blackhall, F.; Wolf, J.; Garrido, P.; et al. Selumetinib Plus Docetaxel Compared With Docetaxel Alone and Progression-Free Survival in Patients With KRAS-Mutant Advanced Non-Small Cell Lung Cancer: The SELECT-1 Randomized Clinical Trial. JAMA 2017, 317, 1844–1853. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Somer, B.G.; Park, J.O.; Li, C.-P.; Scheulen, M.E.; Kasubhai, S.M.; Oh, D.-Y.; Liu, Y.; Redhu, S.; Steplewski, K. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 2014, 50, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Rauch, N.; Rukhlenko, O.S.; Kolch, W.; Kholodenko, B.N. MAPK kinase signalling dynamics regulate cell fate decisions and drug resistance. Curr. Opin. Struct. Biol. 2016, 41, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Jambrina, P.G.; Rauch, N.; Pilkington, R.; Rybakova, K.; Nguyen, L.K.; Kholodenko, B.N.; Buchete, N.V.; Kolch, W.; Rosta, E. Phosphorylation of RAF Kinase Dimers Drives Conformational Changes that Facilitate Transactivation. Angew. Chem. Int. Ed. Engl. 2016, 55, 983–986. [Google Scholar] [CrossRef]
- Rukhlenko, O.S.; Khorsand, F.; Krstic, A.; Rozanc, J.; Alexopoulos, L.G.; Rauch, N.; Erickson, K.E.; Hlavacek, W.S.; Posner, R.G.; Gómez-Coca, S.; et al. Dissecting RAF Inhibitor Resistance by Structure-based Modeling Reveals Ways to Overcome Oncogenic RAS Signaling. Cell Syst. 2018, 7, 161–179.e114. [Google Scholar] [CrossRef]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF-->MEK-->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A.; et al. Effect of Gemcitabine and nab-Paclitaxel With or Without Hydroxychloroquine on Patients With Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef]
- Xavier, C.B.; Marchetti, K.R.; Castria, T.B.; Jardim, D.L.; Fernandes, G.S. Trametinib and Hydroxychloroquine (HCQ) combination treatment in KRAS-mutated advanced pancreatic adenocarcinoma: Detailed description of two cases. J. Gastrointest. Cancer 2021, 52, 374–380. [Google Scholar] [CrossRef]
- Yuan, T.L.; Fellmann, C.; Lee, C.-S.; Ritchie, C.D.; Thapar, V.; Lee, L.C.; Hsu, D.J.; Grace, D.; Carver, J.O.; Zuber, J. Development of siRNA Payloads to Target KRAS-Mutant CancerRNAi Therapy for KRAS-Mutant Cancer. Cancer Discov. 2014, 4, 1182–1197. [Google Scholar] [CrossRef]
- Brummelkamp, T.R.; Bernards, R.; Agami, R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell 2002, 2, 243–247. [Google Scholar] [CrossRef]
- Ross, S.J.; Revenko, A.S.; Hanson, L.L.; Ellston, R.; Staniszewska, A.; Whalley, N.; Pandey, S.K.; Revill, M.; Rooney, C.; Buckett, L.K. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci. Transl. Med. 2017, 9, eaal5253. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; David, E.B.; Raskin, S.; et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560–24570. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Goff, S.L.; Dudley, M.E.; Citrin, D.E.; Somerville, R.P.; Wunderlich, J.R.; Danforth, D.N.; Zlott, D.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S. Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J. Clin. Oncol. 2016, 34, 2389. [Google Scholar] [CrossRef]
- Wang, J.N.; Gu, T.; Hu, Y.; Huang, H. Novel cellular immunotherapies for hematological malignancies: Recent updates from the 2021 ASH annual meeting. Exp Hematol Oncol 2022, 11, 61. [Google Scholar] [CrossRef]
- Wagner, J.; Wickman, E.; DeRenzo, C.; Gottschalk, S. CAR T cell therapy for solid tumors: Bright future or dark reality? Mol. Ther. 2020, 28, 2320–2339. [Google Scholar] [CrossRef]
- Beatty, G.L.; O’Hara, M.H.; Nelson, A.M.; McGarvey, M.; Torigian, D.A.; Lacey, S.F.; Melenhorst, J.J.; Levine, B.; Plesa, G.; June, C.H. Safety and Antitumor Activity of Chimeric Antigen Receptor Modified T Cells in Patients with Chemotherapy Refractory Metastatic Pancreatic Cancer; American Society of Clinical Oncology, 2015; Available online: https://ascopubs.org/doi/abs/10.1200/jco.2015.33.15_suppl.3007 (accessed on 30 October 2022).
- Chmielewski, M.; Hahn, O.; Rappl, G.; Nowak, M.; Schmidt-Wolf, I.H.; Hombach, A.A.; Abken, H. T cells that target carcinoembryonic antigen eradicate orthotopic pancreatic carcinomas without inducing autoimmune colitis in mice. Gastroenterology 2012, 143, 1095–1107. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Arbelaez, C.A.; Estrada, J.; Gessner, M.A.; Glaus, C.; Morales, A.B.; Mohn, D.; Phee, H.; Lipford, J.R.; Johnston, J.A. A nanoparticle vaccine that targets neoantigen peptides to lymphoid tissues elicits robust antitumor T cell responses. NPJ Vaccines 2020, 5, 1–14. [Google Scholar] [CrossRef]
- Liu, H.; Moynihan, K.D.; Zheng, Y.; Szeto, G.L.; Li, A.V.; Huang, B.; Van Egeren, D.S.; Park, C.; Irvine, D.J. Structure-based programming of lymph-node targeting in molecular vaccines. Nature 2014, 507, 519–522. [Google Scholar] [CrossRef]
- Moynihan, K.D.; Opel, C.F.; Szeto, G.L.; Tzeng, A.; Zhu, E.F.; Engreitz, J.M.; Williams, R.T.; Rakhra, K.; Zhang, M.H.; Rothschilds, A.M.; et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat. Med. 2016, 22, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Pant, S.; Furqan, M.; Abdul-Karim, R.M.; Chung, V.; Devoe, C.E.; Johnson, M.L.; Leal, A.D.; Park, H.; Wainberg, Z.A.; Welkowsky, E.; et al. First-in-human phase 1 trial of ELI-002 immunotherapy as treatment for subjects with Kirsten rat sarcoma (KRAS)-mutated pancreatic ductal adenocarcinoma and other solid tumors. J. Clin. Oncol. 2022, 40, TPS2701. [Google Scholar] [CrossRef]
- Chaft, J.E.; Litvak, A.; Arcila, M.E.; Patel, P.; D’Angelo, S.P.; Krug, L.M.; Rusch, V.; Mattson, A.; Coeshott, C.; Park, B. Phase II study of the GI-4000 KRAS vaccine after curative therapy in patients with stage I-III lung adenocarcinoma harboring a KRAS G12C, G12D, or G12V mutation. Clin. Lung Cancer 2014, 15, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Hochster, H.S.; Kim, E.J.; George, B.; Kaylan, A.; Chiorean, E.G.; Waterhouse, D.M.; Guiterrez, M.; Parikh, A.; Jain, R. Open-label, Phase I Study of Nivolumab Combined with nab-Paclitaxel Plus Gemcitabine in Advanced Pancreatic CancerNivo Plus nab-Pac and Gem in Advanced Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4814–4822. [Google Scholar] [CrossRef]
- Saka, D.; Gökalp, M.; Piyade, B.; Cevik, N.C.; Arik Sever, E.; Unutmaz, D.; Ceyhan, G.O.; Demir, I.E.; Asimgil, H. Mechanisms of T-cell exhaustion in pancreatic cancer. Cancers 2020, 12, 2274. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N. The clinical KRAS (G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cowzer, D.; Zameer, M.; Conroy, M.; Kolch, W.; Duffy, A.G. Targeting KRAS in Pancreatic Cancer. J. Pers. Med. 2022, 12, 1870. https://doi.org/10.3390/jpm12111870
Cowzer D, Zameer M, Conroy M, Kolch W, Duffy AG. Targeting KRAS in Pancreatic Cancer. Journal of Personalized Medicine. 2022; 12(11):1870. https://doi.org/10.3390/jpm12111870
Chicago/Turabian StyleCowzer, Darren, Mohammed Zameer, Michael Conroy, Walter Kolch, and Austin G. Duffy. 2022. "Targeting KRAS in Pancreatic Cancer" Journal of Personalized Medicine 12, no. 11: 1870. https://doi.org/10.3390/jpm12111870
APA StyleCowzer, D., Zameer, M., Conroy, M., Kolch, W., & Duffy, A. G. (2022). Targeting KRAS in Pancreatic Cancer. Journal of Personalized Medicine, 12(11), 1870. https://doi.org/10.3390/jpm12111870