
Cerebrospinal Fluid Biomarker Profile in TDP-43-Related Genetic Frontotemporal Dementia


 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Patients
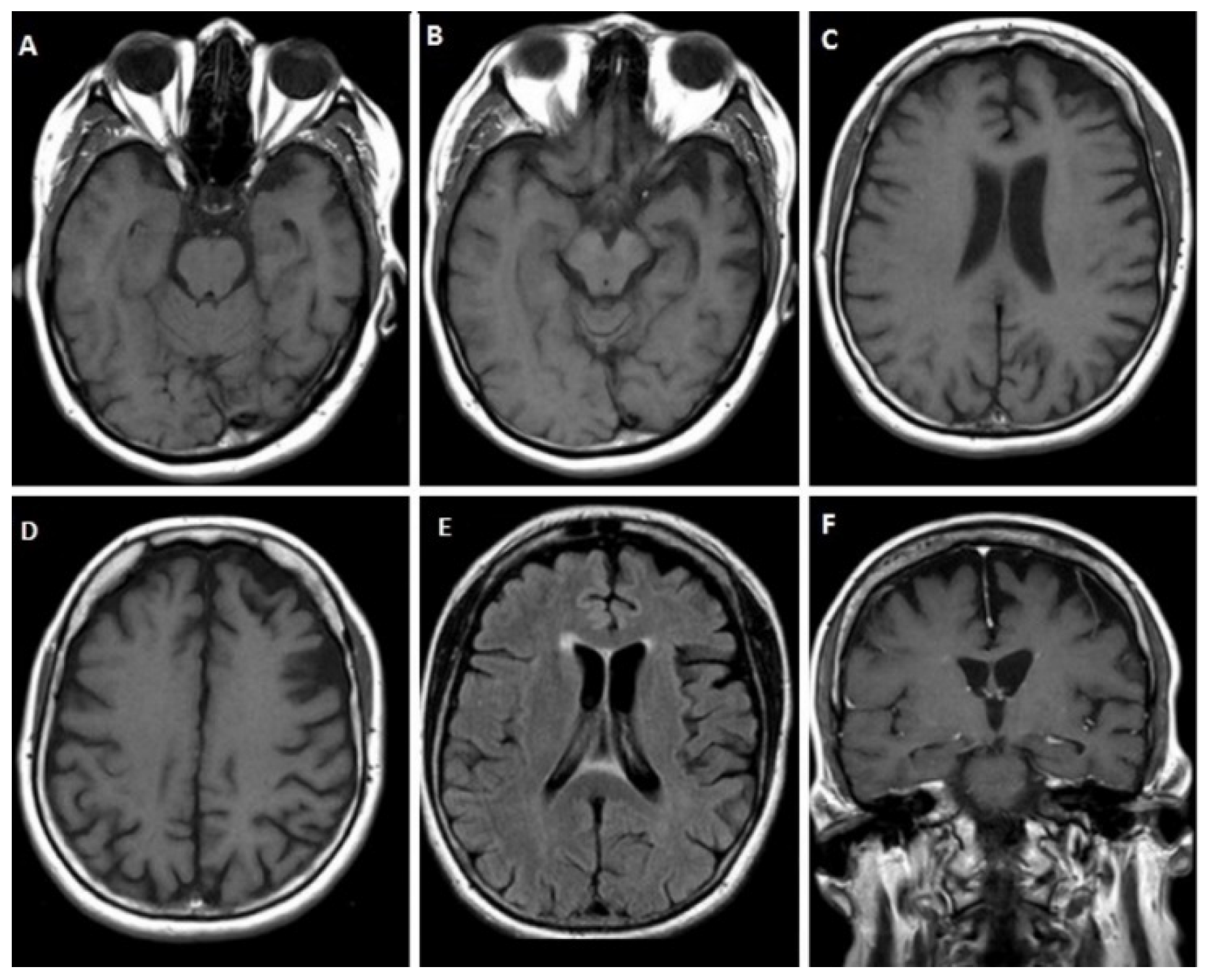
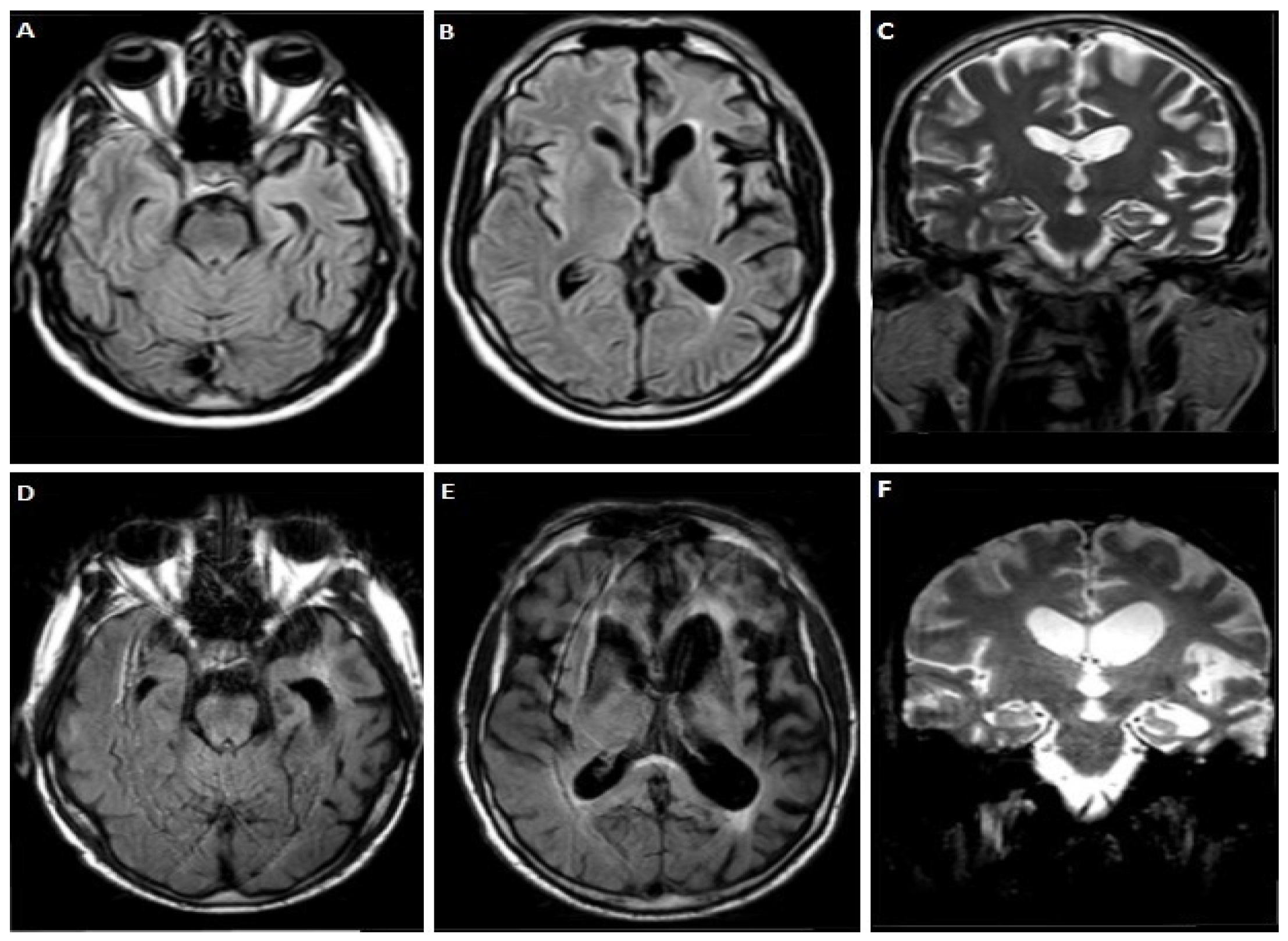
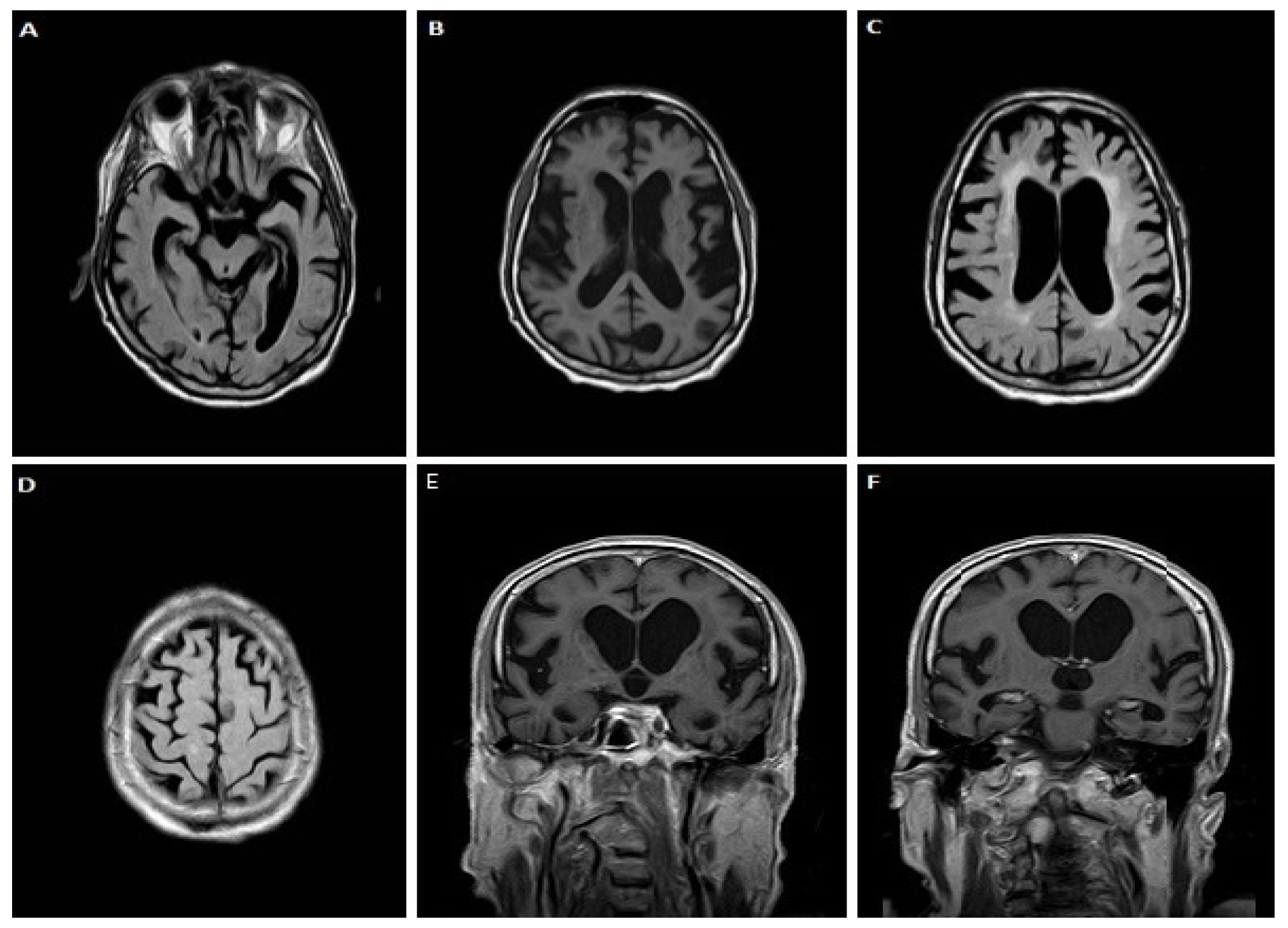
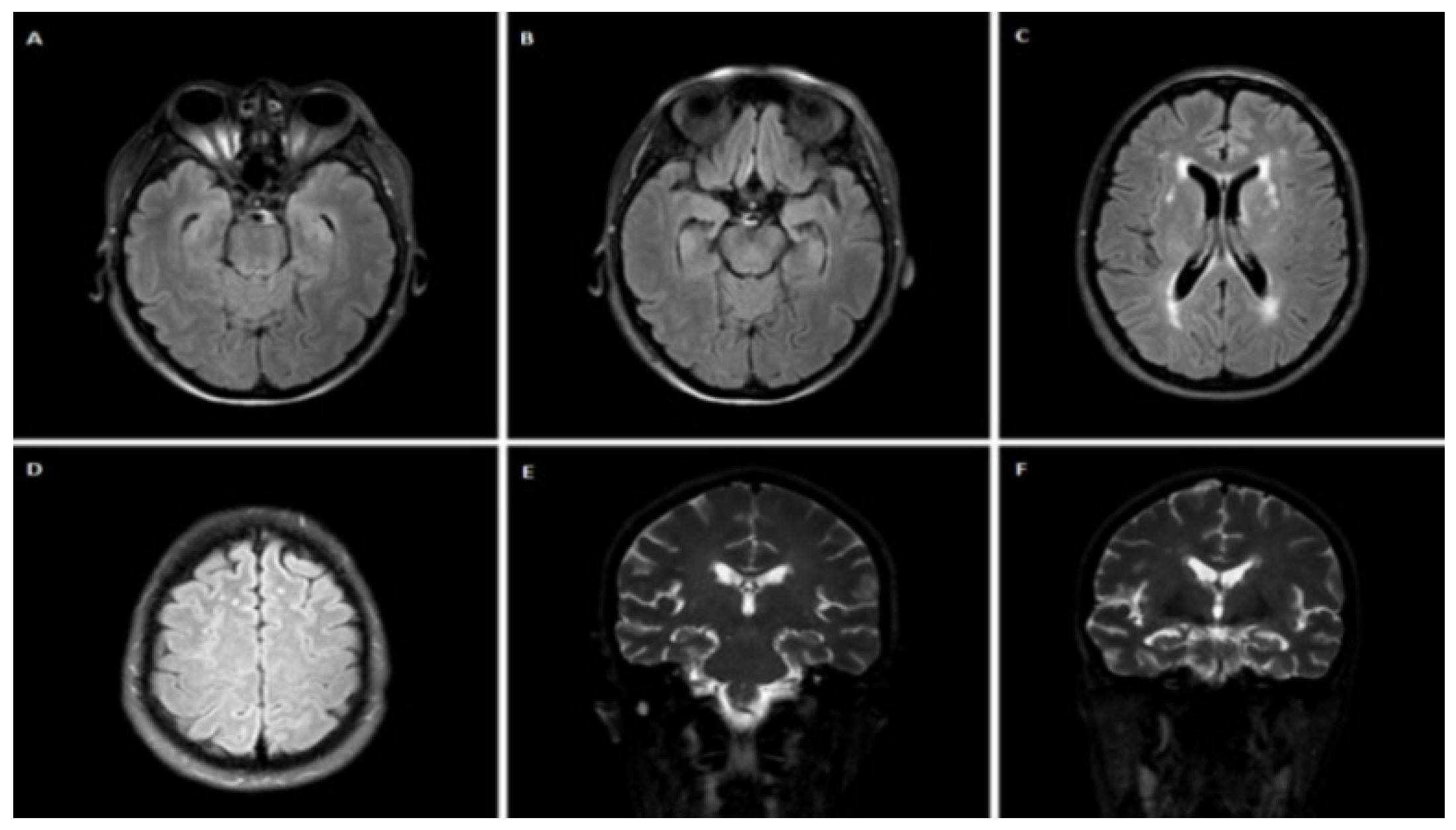
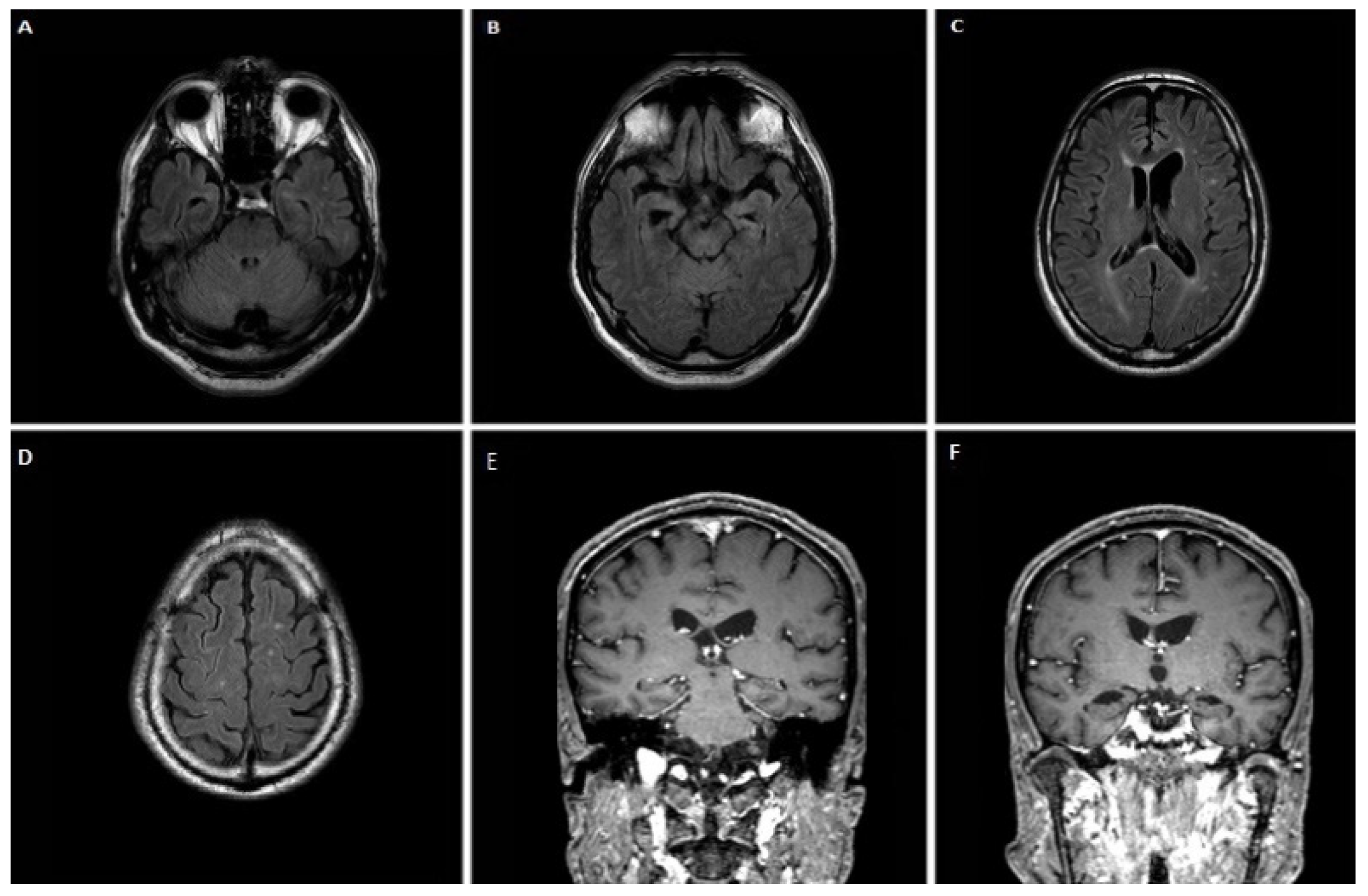
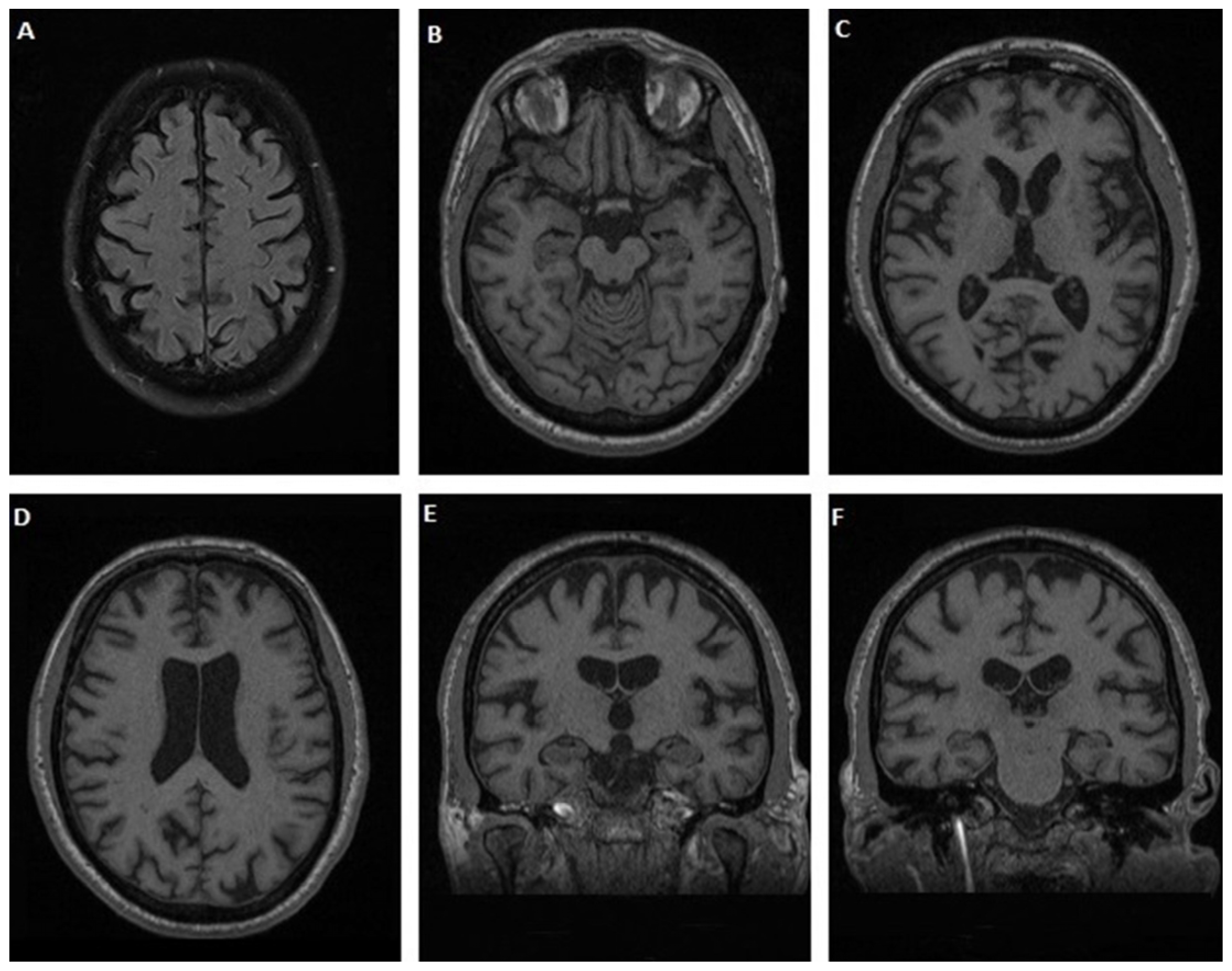
3.1.1. Case 1
3.1.2. Case 2
3.1.3. Case 3
3.1.4. Case 4
3.1.5. Case 5
3.1.6. Case 6
4. Discussion
5. Conclusions
- Established AD biomarkers are reliable indicators for the exclusion of AD pathology in genetic FTLD.
- A decreased τP-181/τΤ ratio may be useful as an indirect marker for non-tauopathic pathology.
- TDP-43 alone and combined with tau proteins, could be a useful tool for the diagnosis of genetic FTD patients with TDP-43 underlying pathology.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jo, M.; Lee, S.; Jeon, Y.M.; Kim, S.; Kwon, Y.; Kim, H.J. The role of TDP-43 propagation in neurodegenerative diseases: Integrating insights from clinical and experimental studies. Exp. Mol. Med. 2020, 52, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Bennion Callister, J.; Pickering-Brown, S.M. Pathogenesis/genetics of frontotemporal dementia and how it relates to ALS. Exp. Neurol. 2014, 262 (Pt B), 84–90. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Klotz, S.; Konig, T.; Erdler, M.; Ulram, A.; Nguyen, A.; Strobel, T.; Zimprich, A.; Stogmann, E.; Regelsberger, G.; Hoftberger, R.; et al. Co-incidental C9orf72 expansion mutation-related frontotemporal lobar degeneration pathology and sporadic Creutzfeldt-Jakob disease. Eur. J. Neurol. 2021, 28, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Gijselinck, I.; Van Broeckhoven, C.; Cruts, M. Granulin mutations associated with frontotemporal lobar degeneration and related disorders: An update. Hum. Mutat. 2008, 29, 1373–1386. [Google Scholar] [CrossRef]
- Wong, T.H.; Pottier, C.; Hondius, D.C.; Meeter, L.H.H.; van Rooij, J.G.J.; Melhem, S.; Netherlands Brain, B.; van Minkelen, R.; van Duijn, C.M.; Rozemuller, A.J.M.; et al. Three VCP Mutations in Patients with Frontotemporal Dementia. J. Alzheimer’s Dis. JAD 2018, 65, 1139–1146. [Google Scholar] [CrossRef]
- Kacem, I.; Sghaier, I.; Ticozzi, N.; Mrabet, S.; Paverelli, S.; Nasri, A.; Ratti, A.; Ben Djebara, M.; Gargouri-Berrachid, A.; Silani, V.; et al. Expanding the phenotype of TARDBP mutation in a Tunisian family with clinical phenotype heterogeneity. Amyotroph. Lateral Scler. Front. Degener. 2022, 1–4. [Google Scholar] [CrossRef]
- Mattsson, N. CSF biomarkers in neurodegenerative diseases. Clin. Chem. Lab. Med. 2011, 49, 345–352. [Google Scholar] [CrossRef]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Dekosky, S.T.; Barberger-Gateau, P.; Cummings, J.; Delacourte, A.; Galasko, D.; Gauthier, S.; Jicha, G.; et al. Research criteria for the diagnosis of Alzheimer’s disease: Revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007, 6, 734–746. [Google Scholar] [CrossRef]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Biscetti, L.; Salvadori, N.; Farotti, L.; Cataldi, S.; Eusebi, P.; Paciotti, S.; Parnetti, L. The added value of Abeta42/Abeta40 in the CSF signature for routine diagnostics of Alzheimer’s disease. Clin. Chim. Acta Int. J. Clin. Chem. 2019, 494, 71–73. [Google Scholar] [CrossRef]
- Kasai, T.; Tokuda, T.; Ishigami, N.; Sasayama, H.; Foulds, P.; Mitchell, D.J.; Mann, D.M.; Allsop, D.; Nakagawa, M. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2009, 117, 55–62. [Google Scholar] [CrossRef]
- Bourbouli, M.; Rentzos, M.; Bougea, A.; Zouvelou, V.; Constantinides, V.C.; Zaganas, I.; Evdokimidis, I.; Kapaki, E.; Paraskevas, G.P. Cerebrospinal Fluid TAR DNA-Binding Protein 43 Combined with Tau Proteins as a Candidate Biomarker for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Spectrum Disorders. Dement. Geriatr. Cogn. Disord. 2017, 44, 144–152. [Google Scholar] [CrossRef]
- Kojima, Y.; Kasai, T.; Noto, Y.I.; Ohmichi, T.; Tatebe, H.; Kitaoji, T.; Tsuji, Y.; Kitani-Morii, F.; Shinomoto, M.; Allsop, D.; et al. Amyotrophic lateral sclerosis: Correlations between fluid biomarkers of NfL, TDP-43, and tau, and clinical characteristics. PLoS ONE 2021, 16, e0260323. [Google Scholar] [CrossRef]
- Junttila, A.; Kuvaja, M.; Hartikainen, P.; Siloaho, M.; Helisalmi, S.; Moilanen, V.; Kiviharju, A.; Jansson, L.; Tienari, P.J.; Remes, A.M.; et al. Cerebrospinal Fluid TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis Patients with and without the C9ORF72 Hexanucleotide Expansion. Dement. Geriatr. Cogn. Disord. Extra 2016, 6, 142–149. [Google Scholar] [CrossRef]
- The Ronald and Nancy Reagan Research Institute of the Alzheimer’s Association and the National Institute on Aging Working Group. Consensus report of the Working Group on: “Molecular and Biochemical Markers of Alzheimer’s Disease”. Neurobiol. Aging 1998, 19, 109–116. [Google Scholar] [CrossRef]
- Borroni, B.; Benussi, A.; Archetti, S.; Galimberti, D.; Parnetti, L.; Nacmias, B.; Sorbi, S.; Scarpini, E.; Padovani, A. Csf p-tau181/tau ratio as biomarker for TDP pathology in frontotemporal dementia. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Paraskevas, G.P.; Kasselimis, D.; Kourtidou, E.; Constantinides, V.; Bougea, A.; Potagas, C.; Evdokimidis, I.; Kapaki, E. Cerebrospinal Fluid Biomarkers as a Diagnostic Tool of the Underlying Pathology of Primary Progressive Aphasia. J. Alzheimer’s Dis. JAD 2017, 55, 1453–1461. [Google Scholar] [CrossRef]
- Rascovsky, K.; Grossman, M. Clinical diagnostic criteria and classification controversies in frontotemporal lobar degeneration. Int. Rev. Psychiatry 2013, 25, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Gorno-Tempini, M.L.; Miller, B.L. Primary progressive aphasia as a model to study the neurobiology of language. Brain Lang. 2013, 127, 105. [Google Scholar] [CrossRef][Green Version]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Dubois, B.; Slachevsky, A.; Litvan, I.; Pillon, B. The FAB: A Frontal Assessment Battery at bedside. Neurology 2000, 55, 1621–1626. [Google Scholar] [CrossRef]
- Bourbouli, M.; Paraskevas, G.P.; Rentzos, M.; Mathioudakis, L.; Zouvelou, V.; Bougea, A.; Tychalas, A.; Kimiskidis, V.K.; Constantinides, V.; Zafeiris, S.; et al. Genotyping and Plasma/Cerebrospinal Fluid Profiling of a Cohort of Frontotemporal Dementia-Amyotrophic Lateral Sclerosis Patients. Brain Sci. 2021, 11, 1239. [Google Scholar] [CrossRef]
- Papadimas, G.K.; Paraskevas, G.P.; Zambelis, T.; Karagiaouris, C.; Bourbouli, M.; Bougea, A.; Walter, M.C.; Schumacher, N.U.; Krause, S.; Kapaki, E. The multifaceted clinical presentation of VCP-proteinopathy in a Greek family. Acta Myol. Myopathies Cardiomyopathies Off. J. Mediterr. Soc. Myol. 2017, 36, 203–206. [Google Scholar]
- Finch, N.; Baker, M.; Crook, R.; Swanson, K.; Kuntz, K.; Surtees, R.; Bisceglio, G.; Rovelet-Lecrux, A.; Boeve, B.; Petersen, R.C.; et al. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain J. Neurol. 2009, 132, 583–591. [Google Scholar] [CrossRef]
- Ramos, E.M.; Koros, C.; Dokuru, D.R.; Van Berlo, V.; Kroupis, C.; Wojta, K.; Wang, Q.; Andronas, N.; Matsi, S.; Beratis, I.N.; et al. Frontotemporal dementia spectrum: First genetic screen in a Greek cohort. Neurobiol. Aging 2019, 75, 224.e1–224.e8. [Google Scholar] [CrossRef] [PubMed]
- Janelidze, S.; Zetterberg, H.; Mattsson, N.; Palmqvist, S.; Vanderstichele, H.; Lindberg, O.; van Westen, D.; Stomrud, E.; Minthon, L.; Blennow, K.; et al. CSF Abeta42/Abeta40 and Abeta42/Abeta38 ratios: Better diagnostic markers of Alzheimer disease. Ann. Clin. Transl. Neurol. 2016, 3, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Kapaki, E.; Paraskevas, G.P.; Papageorgiou, S.G.; Bonakis, A.; Kalfakis, N.; Zalonis, I.; Vassilopoulos, D. Diagnostic value of CSF biomarker profile in frontotemporal lobar degeneration. Alzheimer Dis. Assoc. Disord. 2008, 22, 47–53. [Google Scholar] [CrossRef]
- Paraskevas, G.P.; Kapaki, E.; Papageorgiou, S.G.; Kalfakis, N.; Andreadou, E.; Zalonis, I.; Vassilopoulos, D. CSF biomarker profile and diagnostic value in vascular dementia. Eur. J. Neurol. 2009, 16, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Lewczuk, P.; Riederer, P.; O’Bryant, S.E.; Verbeek, M.M.; Dubois, B.; Visser, P.J.; Jellinger, K.A.; Engelborghs, S.; Ramirez, A.; Parnetti, L.; et al. Cerebrospinal fluid and blood biomarkers for neurodegenerative dementias: An update of the Consensus of the Task Force on Biological Markers in Psychiatry of the World Federation of Societies of Biological Psychiatry. World J. Biol. Psychiatry Off. J. World Fed. Soc. Biol. Psychiatry 2018, 19, 244–328. [Google Scholar] [CrossRef]
- Paraskevas, G.P.; Bougea, A.; Constantinides, V.C.; Bourbouli, M.; Petropoulou, O.; Kapaki, E. In vivo Prevalence of Alzheimer Biomarkers in Dementia with Lewy Bodies. Dement. Geriatr. Cogn. Disord. 2019, 47, 289–296. [Google Scholar] [CrossRef]
- Tan, R.H.; Yang, Y.; Kim, W.S.; Dobson-Stone, C.; Kwok, J.B.; Kiernan, M.C.; Halliday, G.M. Distinct TDP-43 inclusion morphologies in frontotemporal lobar degeneration with and without amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2017, 5, 76. [Google Scholar] [CrossRef]
- Sellami, L.; Saracino, D.; Le Ber, I. Genetic forms of frontotemporal lobar degeneration: Current diagnostic approach and new directions in therapeutic strategies. Rev. Neurol. 2020, 176, 571–581. [Google Scholar] [CrossRef]
- de Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M.; et al. TDP-43 proteinopathies: A new wave of neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 2021, 92, 86–95. [Google Scholar] [CrossRef]
- Foulds, P.; McAuley, E.; Gibbons, L.; Davidson, Y.; Pickering-Brown, S.M.; Neary, D.; Snowden, J.S.; Allsop, D.; Mann, D.M. TDP-43 protein in plasma may index TDP-43 brain pathology in Alzheimer’s disease and frontotemporal lobar degeneration. Acta Neuropathol. 2008, 116, 141–146. [Google Scholar] [CrossRef]
- Babic Leko, M.; Zupunski, V.; Kirincich, J.; Smilovic, D.; Hortobagyi, T.; Hof, P.R.; Simic, G. Molecular Mechanisms of Neurodegeneration Related to C9orf72 Hexanucleotide Repeat Expansion. Behav. Neurol. 2019, 2019, 2909168. [Google Scholar] [CrossRef] [PubMed]
- Freibaum, B.D.; Taylor, J.P. The Role of Dipeptide Repeats in C9ORF72-Related ALS-FTD. Front. Mol. Neurosci. 2017, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Arzberger, T.; Grasser, F.A.; Gijselinck, I.; May, S.; Rentzsch, K.; Weng, S.M.; Schludi, M.H.; van der Zee, J.; Cruts, M.; et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013, 126, 881–893. [Google Scholar] [CrossRef]
- Zu, T.; Liu, Y.; Banez-Coronel, M.; Reid, T.; Pletnikova, O.; Lewis, J.; Miller, T.M.; Harms, M.B.; Falchook, A.E.; Subramony, S.H.; et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2013, 110, E4968–E4977. [Google Scholar] [CrossRef] [PubMed]
- Ash, P.E.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.L.; Dejesus-Hernandez, M.; van Blitterswijk, M.M.; Jansen-West, K.; Paul, J.W., 3rd; Rademakers, R.; et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Trojanowski, J.Q.; Abner, E.L.; Al-Janabi, O.M.; Jicha, G.A.; Schmitt, F.A.; Smith, C.D.; Fardo, D.W.; Wang, W.X.; Kryscio, R.J.; et al. “New Old Pathologies”: AD, PART, and Cerebral Age-Related TDP-43 with Sclerosis (CARTS). J. Neuropathol. Exp. Neurol. 2016, 75, 482–498. [Google Scholar] [CrossRef]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain J. Neurol. 2019, 142, 1503–1527. [Google Scholar] [CrossRef] [PubMed]
- Brunnstrom, H.; Englund, E. Clinicopathological concordance in dementia diagnostics. Am. J. Geriatr. Psychiatry Off. J. Am. Assoc. Geriatr. Psychiatry 2009, 17, 664–670. [Google Scholar] [CrossRef]
- Delaby, C.; Alcolea, D.; Carmona-Iragui, M.; Illan-Gala, I.; Morenas-Rodriguez, E.; Barroeta, I.; Altuna, M.; Estelles, T.; Santos-Santos, M.; Turon-Sans, J.; et al. Differential levels of Neurofilament Light protein in cerebrospinal fluid in patients with a wide range of neurodegenerative disorders. Sci. Rep. 2020, 10, 9161. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CSF Biomarker | Cut-off Values * |
|---|---|
| total tau protein (τT) | <376 pg/mL |
| tau phosphorylated at threonine-181 (τP-181) | <57 pg/mL |
| amyloid-β peptide with 42 amino acids (Aβ42) | >580 pg/mL |
| Aβ42/Aβ40 | >0.067 |
| τP-181/τT | >0.165 |
| TDP-43 | <3.73 pg/mL |
| TDP-43 × τΤ/τP-181 | <20.5 |
| Case | τT (pg/mL) | τP-181 (pg/mL) | Aβ42 (pg/mL) | Aβ42/Aβ40 | τP-181/τT | TDP-43 (pg/mL) | Formula (f) |
|---|---|---|---|---|---|---|---|
| 1 | 511.6 | 28.7 | 504.2 | 0.10 | 0.056 | 4.46 | 79.5 |
| 2 | 565.1 | 41.9 | 640 | 0.11 | 0.074 | 5.92 | 79.9 |
| 3 | 316.2 | 27.2 | 501.5 | 0.14 | 0.086 | 5.74 | 66.7 |
| 4 | 250.3 | 35.5 | 935.9 | - | 0.140 | 2.48 | 17.5 |
| 5 | 356.8 | 32.8 | 431 | 0.10 | 0.091 | 5.27 | 57.3 |
| 6 | 328.1 | 32.1 | 397.1 | 0.17 | 0.098 | 5.74 | 57.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapaki, E.; Boufidou, F.; Bourbouli, M.; Pyrgelis, E.-S.; Constantinides, V.C.; Anastassopoulou, C.; Paraskevas, G.P. Cerebrospinal Fluid Biomarker Profile in TDP-43-Related Genetic Frontotemporal Dementia. J. Pers. Med. 2022, 12, 1747. https://doi.org/10.3390/jpm12101747
Kapaki E, Boufidou F, Bourbouli M, Pyrgelis E-S, Constantinides VC, Anastassopoulou C, Paraskevas GP. Cerebrospinal Fluid Biomarker Profile in TDP-43-Related Genetic Frontotemporal Dementia. Journal of Personalized Medicine. 2022; 12(10):1747. https://doi.org/10.3390/jpm12101747
Chicago/Turabian StyleKapaki, Elisabeth, Foteini Boufidou, Mara Bourbouli, Efstratios-Stylianos Pyrgelis, Vasilios C. Constantinides, Cleo Anastassopoulou, and George P. Paraskevas. 2022. "Cerebrospinal Fluid Biomarker Profile in TDP-43-Related Genetic Frontotemporal Dementia" Journal of Personalized Medicine 12, no. 10: 1747. https://doi.org/10.3390/jpm12101747
APA StyleKapaki, E., Boufidou, F., Bourbouli, M., Pyrgelis, E.-S., Constantinides, V. C., Anastassopoulou, C., & Paraskevas, G. P. (2022). Cerebrospinal Fluid Biomarker Profile in TDP-43-Related Genetic Frontotemporal Dementia. Journal of Personalized Medicine, 12(10), 1747. https://doi.org/10.3390/jpm12101747