
LLM-PBC: Logic Learning Machine-Based Explainable Rules Accurately Stratify the Genetic Risk of Primary Biliary Cholangitis

 ,
,  , ,
, ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials, Methods, and Participants
2.1. Study Design and Participants
2.2. Patient Privacy and Ethical Issues
2.3. Data Preprocessing
2.3.1. Selection of Genetic Variants
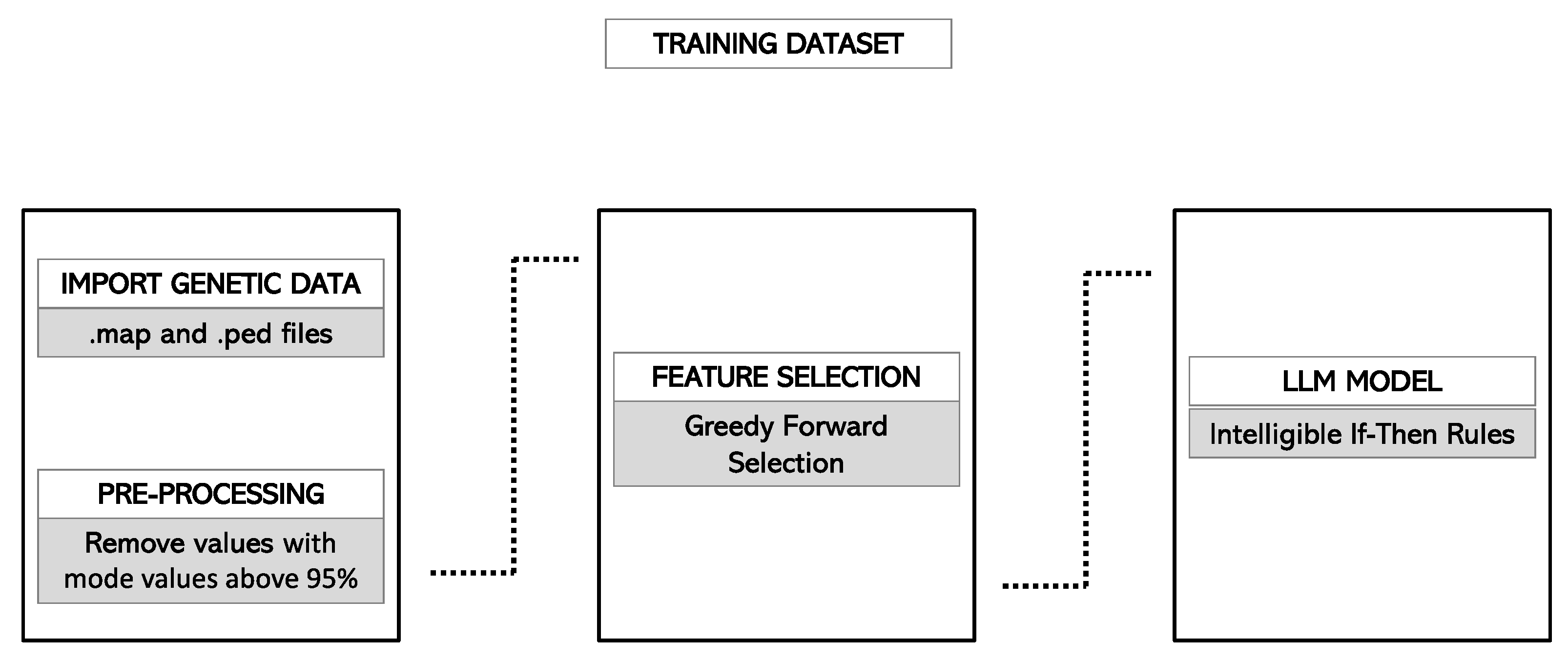
2.3.2. Import of PLINK Files in the Rulex Environment
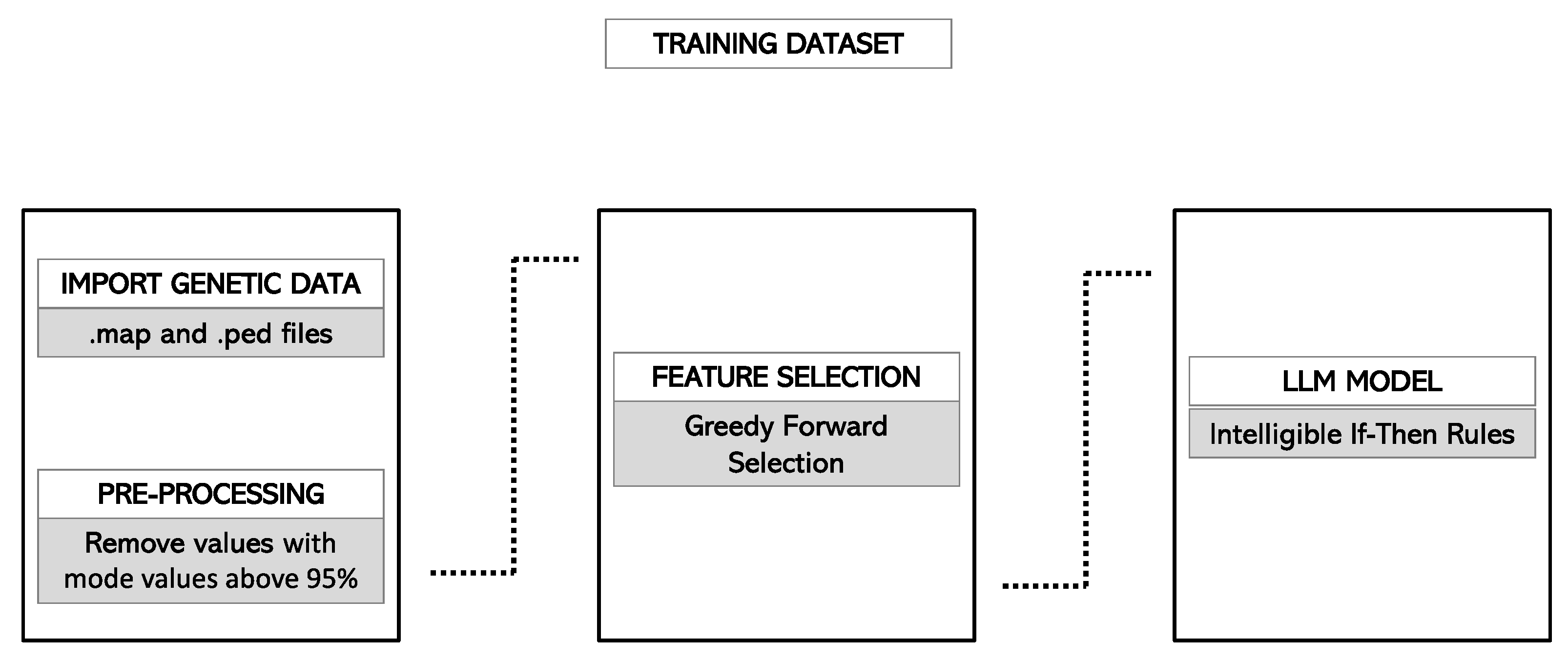
2.4. Model Development
2.4.1. Feature Selection
2.4.2. Internal Validation
2.4.3. Hyperparameter Tuning
- -
- correlation among features: three thresholds, t, based on Cramer’s V value were evaluated (0.7, 0.8 and 0.9);
- -
- number of pre-selected and selected features: several fixed combinations of thresholds for pre-selected features and selected features were evaluated (Supplementary Table S1);
- -
- max error (errmax): errmax represents the maximum level of error for each rule included in the ruleset. In other words, this corresponded to the maximum percentage of cases belonging to output classes different from the predicted one, which verified the rule.
2.4.4. Final Model Selection and Rules Generation
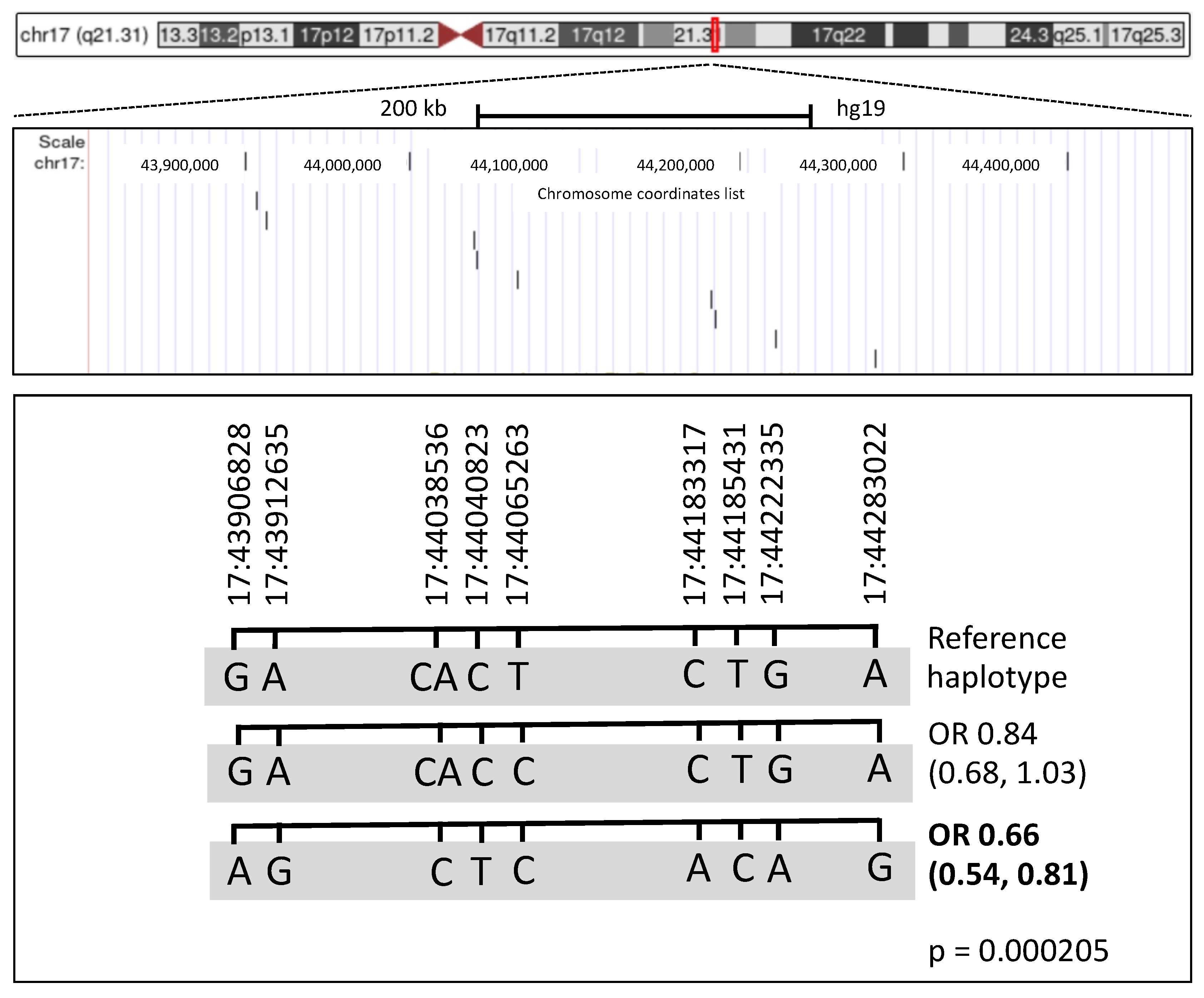
2.4.5. Haplotype Analysis
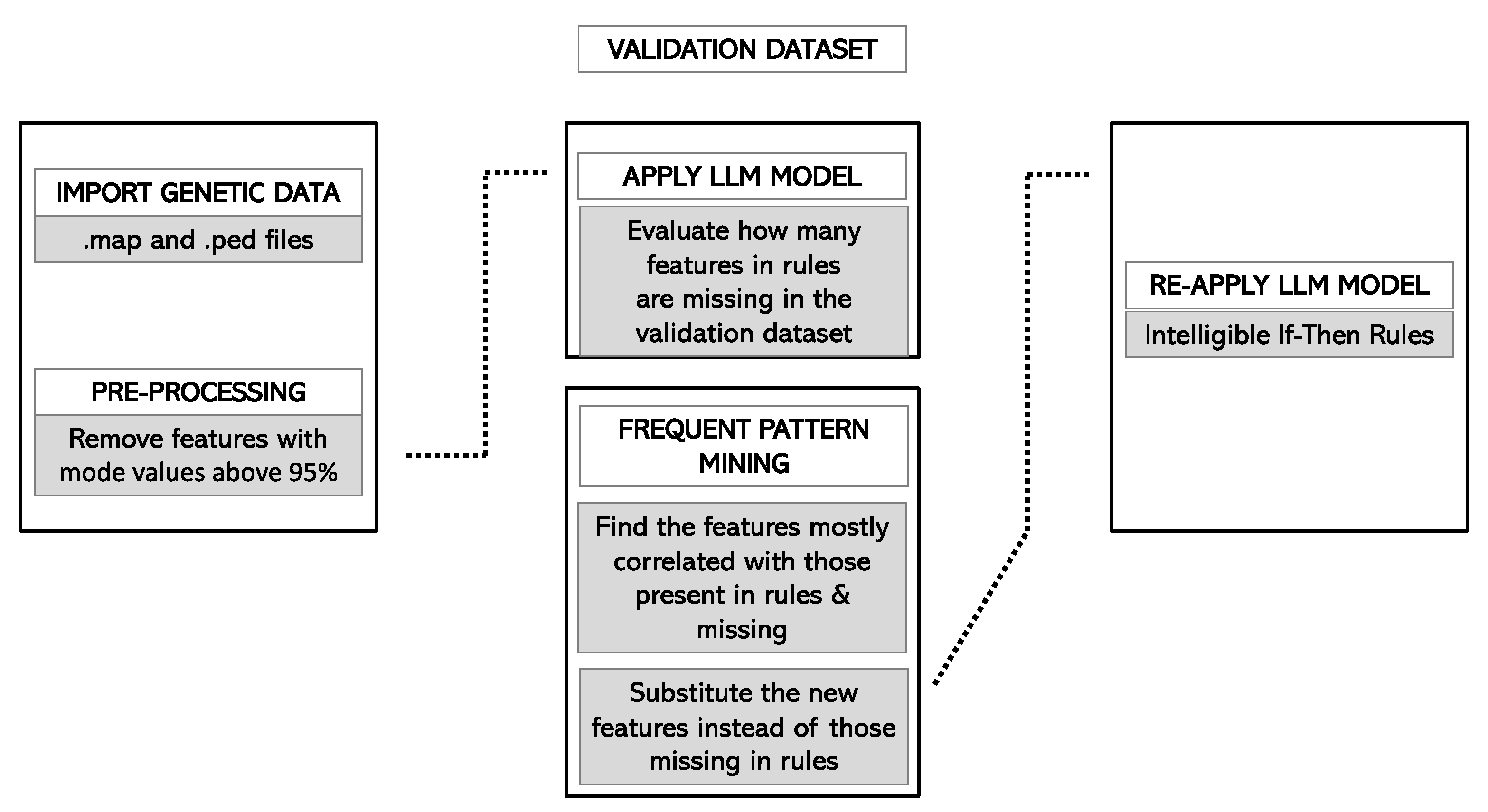
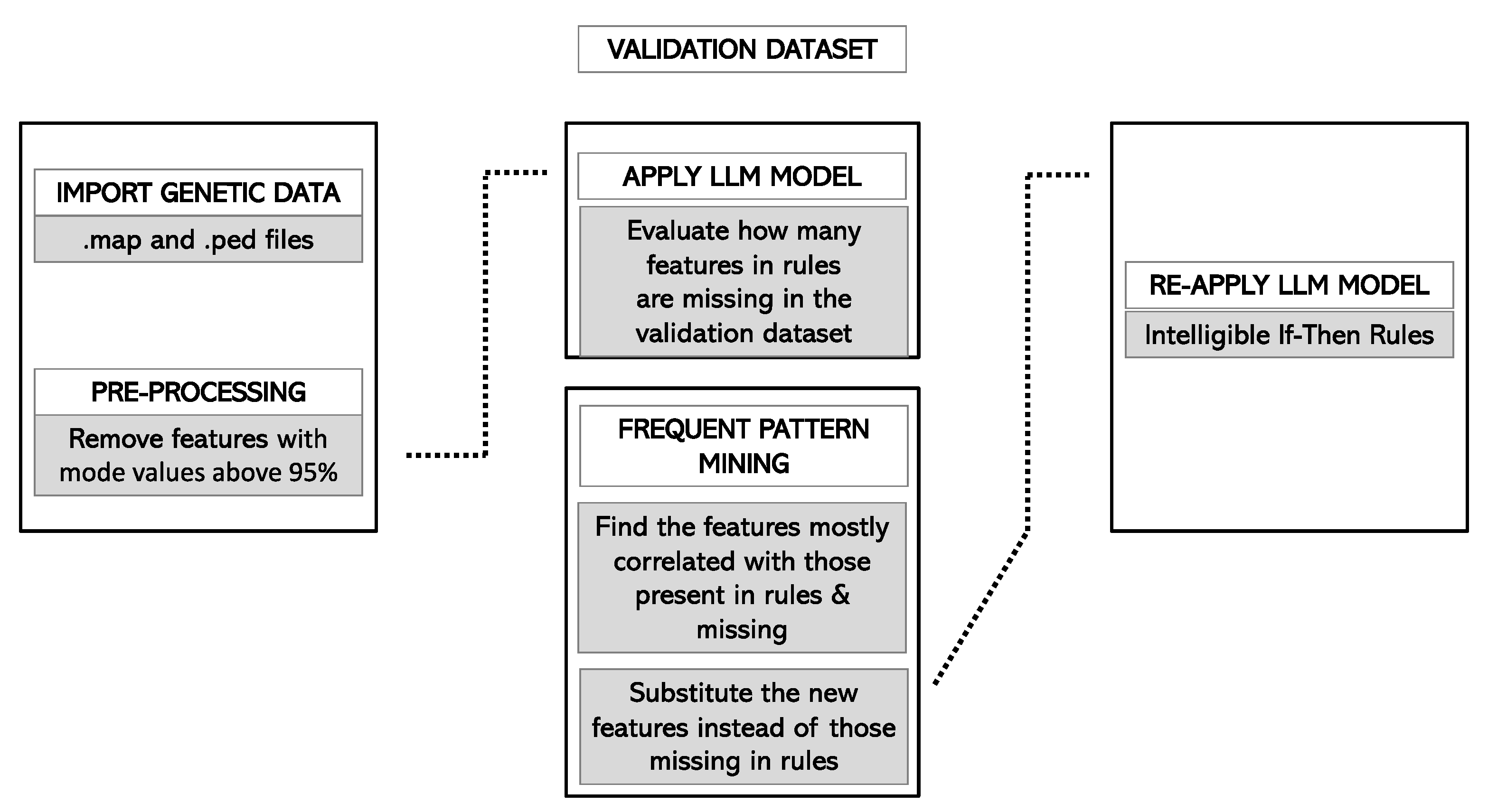
2.4.6. Validation of the Model
2.5. Statistical Analyses
3. Results
3.1. Description of the Training Cohort
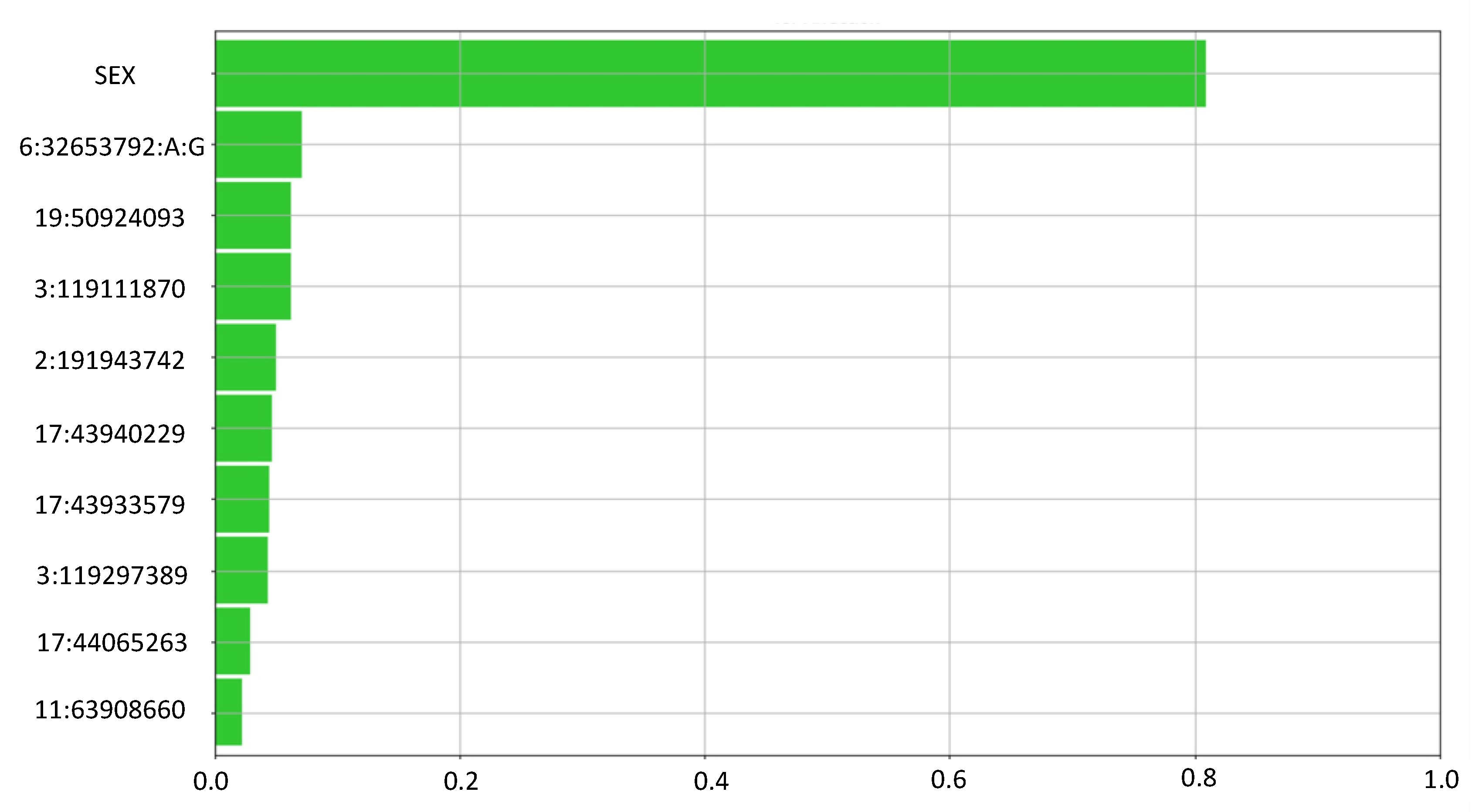
3.2. Feature Selection
3.3. Final Model
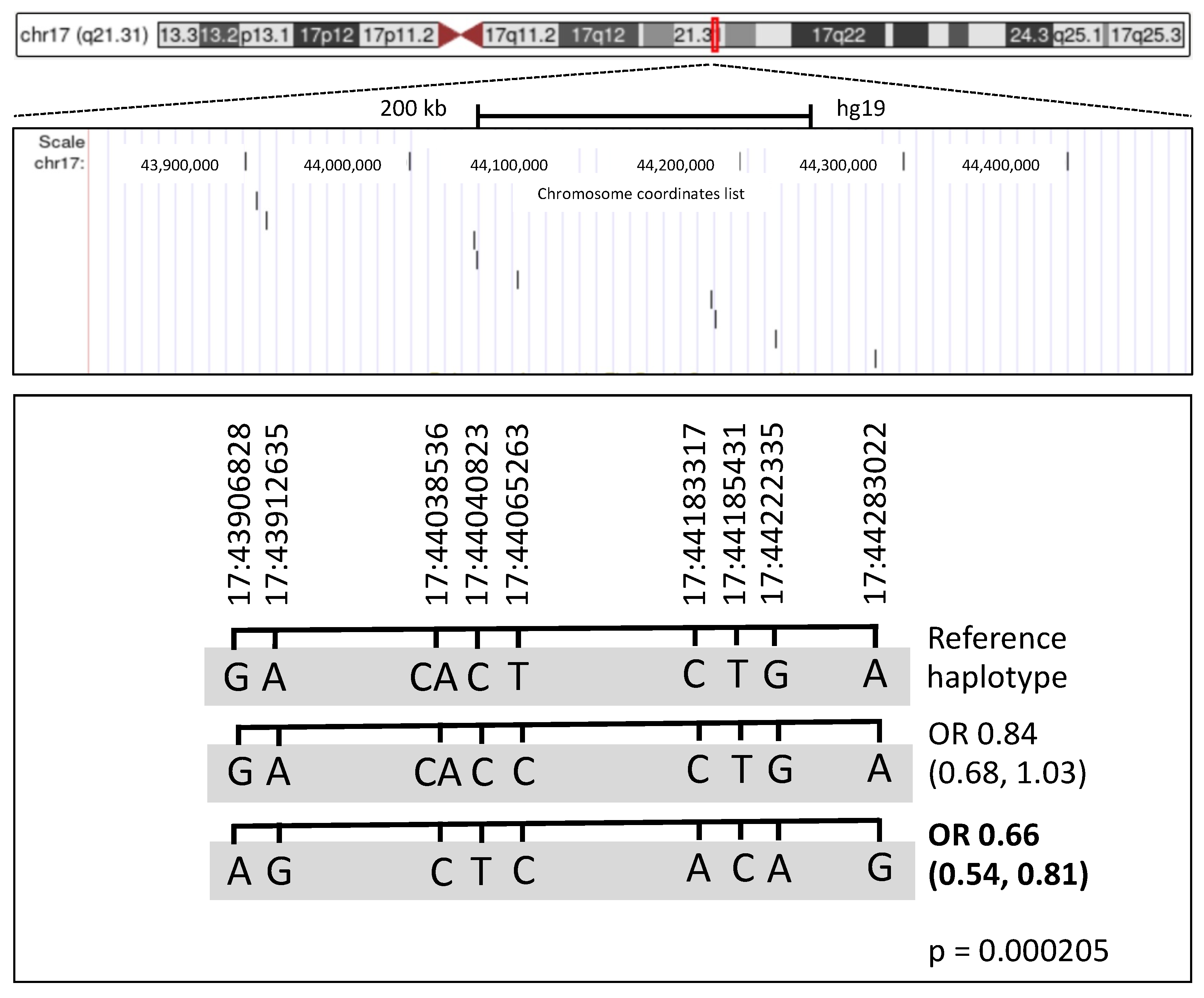
3.4. Haplotype Analysis
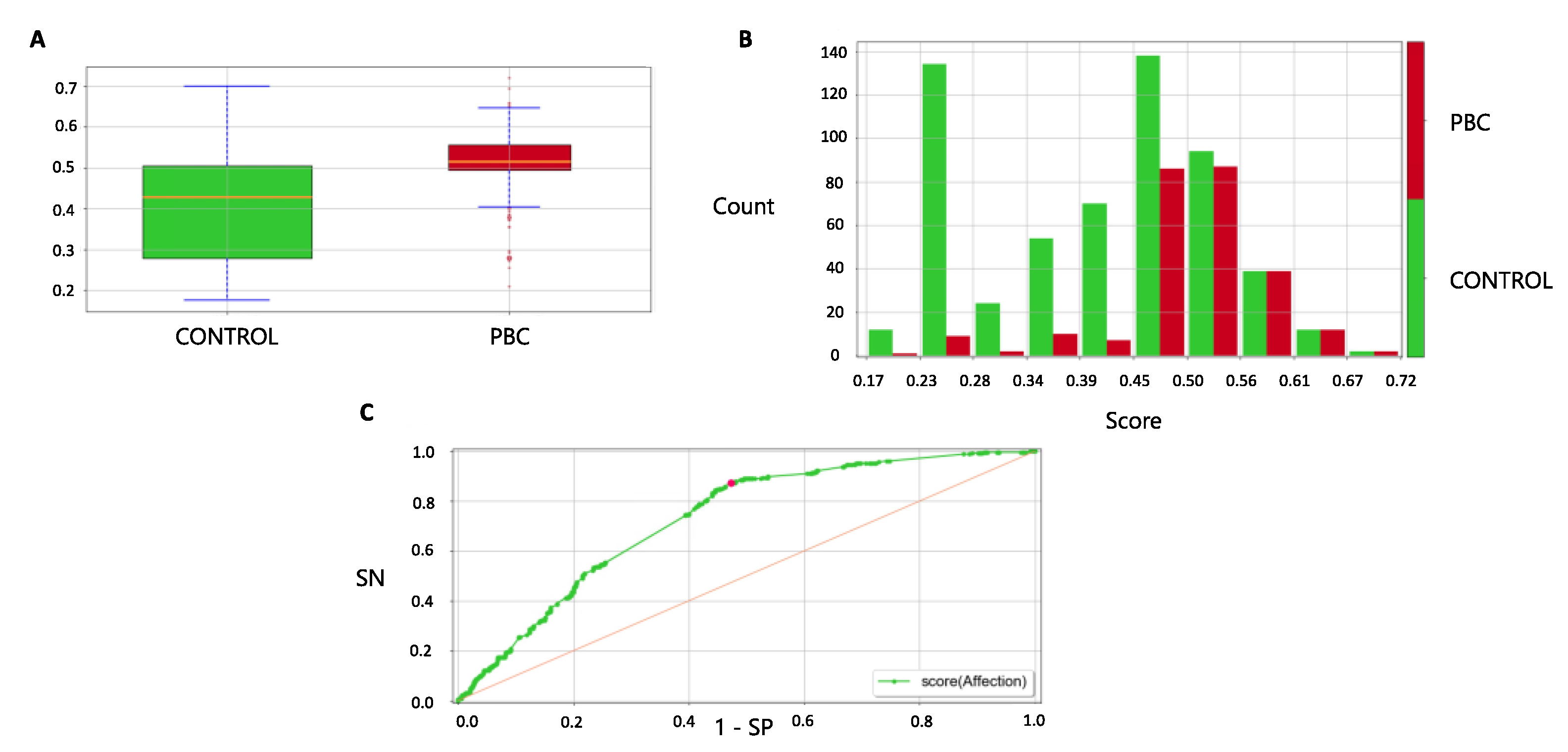
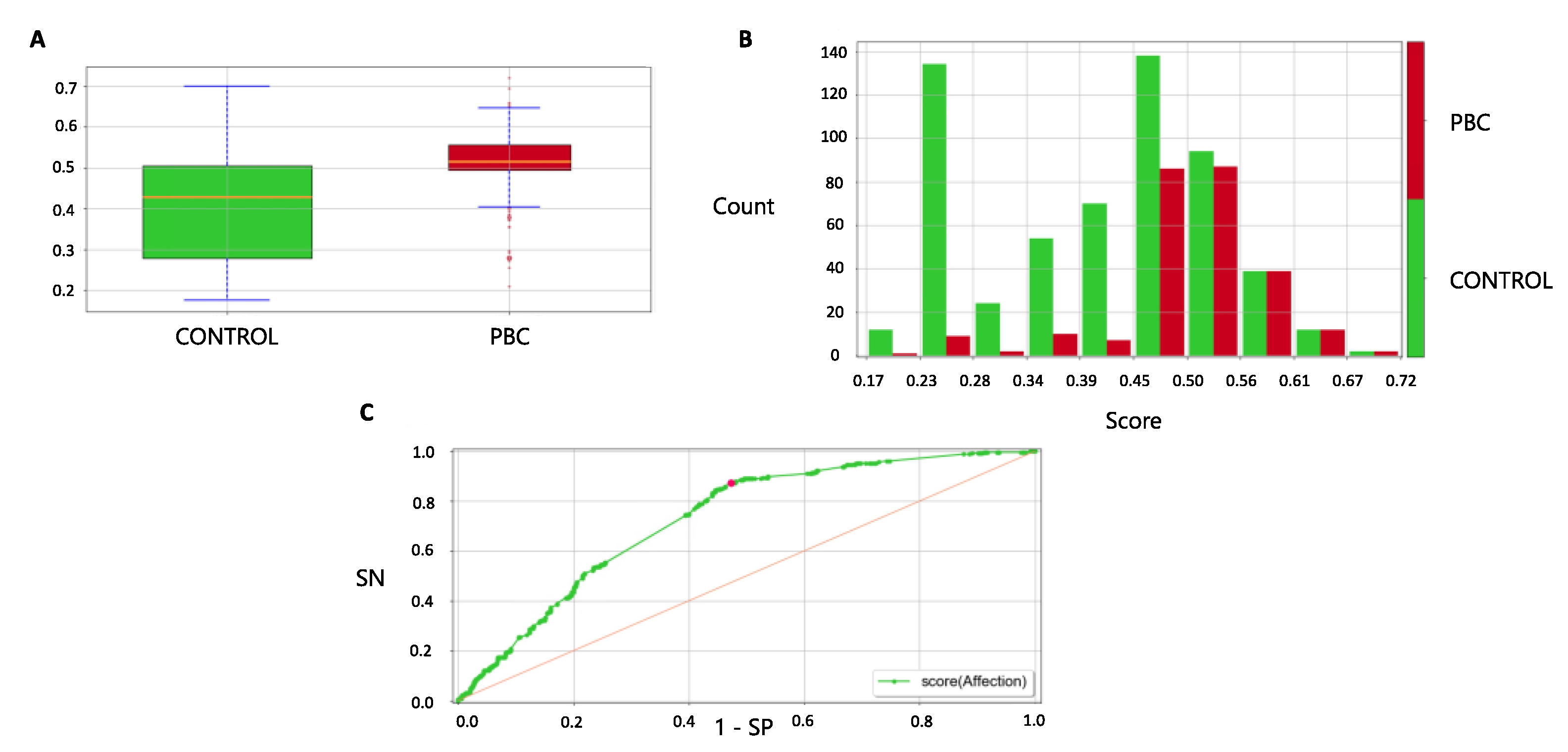
3.5. Forecast in the Validation Cohort
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torkamani, A.; Wineinger, N.E.; Topol, E.J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 2018, 19, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Topol, E.J. High-performance medicine: The convergence of human and artificial intelligence. Nat. Med. 2019, 25, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Gerussi, A.; Carbone, M.; Corpechot, C.; Schramm, C. The genetic architecture of primary biliary cholangitis. Eur. J. Med. Genet. 2021, 64, 104292. [Google Scholar] [CrossRef] [PubMed]
- Gulamhusein, A.F.; Hirschfield, G.M. Primary biliary cholangitis: Pathogenesis and therapeutic opportunities. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Villalón-García, I.; Álvarez-Córdoba, M.; Suárez-Rivero, J.M.; Povea-Cabello, S.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Sánchez-Alcázar, J.A. Precision Medicine in Rare Diseases. Diseases 2020, 8, 42. [Google Scholar] [CrossRef]
- Cordell, H.J.; Fryett, J.J.; Ueno, K.; Darlay, R.; Aiba, Y.; Hitomi, Y.; Kawashima, M.; Nishida, N.; Khor, S.-S.; Gervais, O.; et al. An international genome-wide meta-analysis of primary biliary cholangitis: Novel risk loci and candidate drugs. J. Hepatol. 2021, 75, 572–581. [Google Scholar] [CrossRef]
- Choi, S.W.; Mak, T.S.H.; O’Reilly, P.F. Tutorial: A guide to performing polygenic risk score analyses. Nat. Protoc. 2020, 3, 11–13. [Google Scholar] [CrossRef]
- Ho, D.S.W.; Schierding, W.; Wake, M.; Saffery, R.; O’Sullivan, J. Machine Learning SNP Based Prediction for Precision Medicine. Front. Genet. 2019, 10, 267. [Google Scholar] [CrossRef]
- Libbrecht, M.W.; Noble, W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef]
- de los Campos, G.; Vazquez, A.I.; Hsu, S.; Lello, L. Complex-Trait Prediction in the Era of Big Data. Trends Genet. 2018, 34, 746–754. [Google Scholar] [CrossRef]
- Lindor, K.D.; Gershwin, M.E.; Poupon, R.; Kaplan, M.; Bergasa, N.V.; Heathcote, E.J. Primary biliary cirrhosis. Hepatology 2009, 50, 291–308. [Google Scholar] [CrossRef] [PubMed]
- de Hond, A.A.H.; Leeuwenberg, A.M.; Hooft, L.; Kant, I.M.J.; Nijman, S.W.J.; van Os, H.J.A.; Aardoom, J.J.; Debray, T.P.A.; Schuit, E.; van Smeden, M.; et al. Guidelines and quality criteria for artificial intelligence-based prediction models in healthcare: A scoping review. NPJ Digit. Med. 2022, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Gunning, D.; Stefik, M.; Choi, J.; Miller, T.; Stumpf, S.; Yang, G.-Z. XAI—Explainable artificial intelligence. Sci. Robot. 2019, 4, eaay7120. [Google Scholar] [CrossRef] [PubMed]
- Price, W.N. Big data and black-box medical algorithms. Sci. Transl. Med. 2018, 10, eaao5333. [Google Scholar] [CrossRef]
- Azodi, C.B.; Tang, J.; Shiu, S.-H. Opening the Black Box: Interpretable Machine Learning for Geneticists. Trends Genet. 2020, 36, 442–455. [Google Scholar] [CrossRef]
- Cangelosi, D.; Blengio, F.; Versteeg, R.; Eggert, A.; Garaventa, A.; Gambini, C.; Conte, M.; Eva, A.; Muselli, M.; Varesio, L. Logic Learning Machine creates explicit and stable rules stratifying neuroblastoma patients. BMC Bioinform. 2013, 14, S12. [Google Scholar] [CrossRef]
- Cangelosi, D.; Muselli, M.; Parodi, S.; Blengio, F.; Becherini, P.; Versteeg, R.; Conte, M.; Varesio, L. Use of Attribute Driven Incremental Discretization and Logic Learning Machine to build a prognostic classifier for neuroblastoma patients. BMC Bioinform. 2014, 15, S4. [Google Scholar] [CrossRef]
- Cangelosi, D.; Morini, M.; Zanardi, N.; Sementa, A.R.; Muselli, M.; Conte, M.; Garaventa, A.; Pfeffer, U.; Bosco, M.C.; Varesio, L.; et al. Hypoxia predicts poor prognosis in neuroblastoma patients and associates with biological mechanisms involved in telomerase activation and tumor microenvironment reprogramming. Cancers 2020, 12, 2343. [Google Scholar] [CrossRef]
- Mordenti, M.; Ferrari, E.; Pedrini, E.; Fabbri, N.; Campanacci, L.; Muselli, M.; Sangiorgi, L. Validation of a new multiple osteochondromas classification through Switching Neural Networks. Am. J. Med. Genet. A 2013, 161, 556–560. [Google Scholar] [CrossRef]
- Parodi, S.; Filiberti, R.; Marroni, P.; Libener, R.; Ivaldi, G.P.; Mussap, M.; Ferrari, E.; Manneschi, C.; Montani, E.; Muselli, M. Differential diagnosis of pleural mesothelioma using Logic Learning Machine. BMC Bioinform. 2015, 16, S3. [Google Scholar] [CrossRef] [PubMed]
- Parodi, S.; Dosi, C.; Zambon, A.; Ferrari, E.; Muselli, M. Identifying Environmental and Social Factors Predisposing to Pathological Gambling Combining Standard Logistic Regression and Logic Learning Machine. J. Gambl. Stud. 2017, 33, 1121–1137. [Google Scholar] [CrossRef] [PubMed]
- Verda, D.; Parodi, S.; Ferrari, E.; Muselli, M. Analyzing gene expression data for pediatric and adult cancer diagnosis using logic learning machine and standard supervised methods. BMC Bioinform. 2019, 20, 390. [Google Scholar] [CrossRef] [PubMed]
- Skotko, B.G.; Macklin, E.A.; Muselli, M.; Voelz, L.; McDonough, M.E.; Davidson, E.; Allareddy, V.; Jayaratne, Y.S.N.; Bruun, R.; Ching, N.; et al. A predictive model for obstructive sleep apnea and Down syndrome. Am. J. Med. Genet. Part A 2017, 173, 889–896. [Google Scholar] [CrossRef]
- Caruana, R.; Freitag, D. Greedy Attribute Selection. In Machine Learning Proceedings 1994; Elsevier: Amsterdam, The Netherlands, 1994; pp. 28–36. [Google Scholar]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Omiecinski, E.R. Alternative Interest Measures for Mining Associations in Databases. IEEE Trans. Knowl. Data Eng. 2003, 15, 57–69. [Google Scholar] [CrossRef]
- Bommert, A.; Sun, X.; Bischl, B.; Rahnenführer, J.; Lang, M. Benchmark for filter methods for feature selection in high-dimensional classification data. Comput. Stat. Data Anal. 2020, 143, 106839. [Google Scholar] [CrossRef]
- Jones, D.E.J.; Watt, F.E.; Metcalf, J.V.; Bassendine, M.F.; James, O.F.W. Familial primary biliary cirrhosis reassessed: A geographically-based population study. J. Hepatol. 1999, 30, 402–407. [Google Scholar] [CrossRef]
- Hartl, J.; Kroll, C.; Stahl, F.; Lohse, A.W.; Schramm, C.; Sebode, M. Risk of Primary Biliary Cholangitis Relatives: A Prospective Cohort Study. In Proceedings of the International Liver Congress (ILC) 2021, 23–26 June 2021. [Google Scholar]
- McGee, S. Simplifying likelihood ratios. J. Gen. Intern. Med. 2002, 17, 646–649. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef]
- Wei, W.H.; Hemani, G.; Haley, C.S. Detecting epistasis in human complex traits. Nat. Rev. Genet. 2014, 15, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Paulus, J.K.; Kent, D.M. Predictably unequal: Understanding and addressing concerns that algorithmic clinical prediction may increase health disparities. NPJ Digit. Med. 2020, 3, 99. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Huss, M.; Abid, A.; Mohammadi, P.; Torkamani, A.; Telenti, A. A primer on deep learning in genomics. Nat. Genet. 2019, 51, 12–18. [Google Scholar] [CrossRef]
- Wiens, J.; Saria, S.; Sendak, M.; Ghassemi, M.; Liu, V.X.; Doshi-Velez, F.; Jung, K.; Heller, K.; Kale, D.; Saeed, M.; et al. Do no harm: A roadmap for responsible machine learning for health care. Nat. Med. 2019, 25, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Asselta, R.; Paraboschi, E.M.; Gerussi, A.; Cordell, H.J.; Mells, G.F.; Sandford, R.N.; Jones, D.E.; Nakamura, M.; Ueno, K.; Hitomi, Y.; et al. X Chromosome Contribution to the Genetic Architecture of Primary Biliary Cholangitis. Gastroenterology 2021, 160, 2483–2495. [Google Scholar] [CrossRef] [PubMed]
- Kuo, R.J.; Zulvia, F.E. The gradient evolution algorithm: A new metaheuristic. Inf. Sci. 2015, 316, 246–265. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr | Gene | Start | End |
|---|---|---|---|
| 1 | MMEL1 | 2,273,723 | 2,773,723 |
| 1 | IL12RB2 | 67,570,194 | 68,070,194 |
| 1 | CD58 | 116,815,083 | 117,315,083 |
| 1 | FCRL3 | 157,420,290 | 157,920,290 |
| 1 | DENND1B | 197,530,966 | 198,030,966 |
| 1 | CACNA1S | 200,769,059 | 201,269,059 |
| 2 | DNMT3A | 25,264,333 | 25,764,333 |
| 2 | TMEM163 | 135,091,200 | 135,591,200 |
| 2 | STAT4 | 191,693,742 | 192,193,742 |
| 3 | PLCL2 | 16,711,265 | 17,211,265 |
| 3 | RARB | 25,133,587 | 25,633,587 |
| 3 | TIMMDC1 | 118,969,934 | 119,469,934 |
| 3 | IL12A-AS1 | 159,410,283 | 159,910,283 |
| 4 | NFKB1 | 103,290,780 | 103,790,780 |
| 4 | TET2 | 105,878,954 | 106,378,954 |
| 5 | IL7R | 35,631,130 | 36,131,130 |
| 5 | LOC285626 | 158,509,900 | 159,009,900 |
| 6 | OLIG3 | 137,723,068 | 138,223,068 |
| 7 | ITGB8 | 20,128,801 | 20,628,801 |
| 7 | ELMO1 | 37,132,465 | 37,632,465 |
| 7 | TNPO3 | 128,367,466 | 128,867,466 |
| 7 | ZC3HAV1 | 138,479,543 | 138,979,543 |
| 9 | HEMGN | 100,491,912 | 100,991,912 |
| 10 | WDFY4 | 49,775,396 | 50,275,396 |
| 11 | DEAF1 | 396,986 | 896,986 |
| 11 | CCDC88B, | 63,860,422 | 64,360,422 |
| 11 | POU2AF1 | 110,989,365 | 111,489,365 |
| 11 | DDX6 | 118,490,104 | 118,990,104 |
| 12 | TNFRSF1A | 6,190,009 | 6,690,009 |
| 12 | ATXN2 | 111,657,431 | 112,157,431 |
| 13 | LINC02341 | 42,805,002 | 43,305,002 |
| 13 | DLEU1 | 50,561,220 | 51,061,220 |
| 14 | RAD51B | 68,499,927 | 68,999,927 |
| 14 | RIN3 | 92,864,787 | 93,364,787 |
| 14 | EXOC3L4 | 103,314,807 | 103,814,807 |
| 16 | CLEC16A | 10,924,365 | 11,424,365 |
| 16 | IL4R | 27,153,469 | 27,653,469 |
| 16 | DPEP2 | 67,786,939 | 68,286,939 |
| 16 | LOC105371388 | 85,769,271 | 86,269,271 |
| 17 | ZPBP2 | 37,794,893 | 38,294,893 |
| 17 | KANSL1 | 43,899,348 | 44,399,348 |
| 18 | CD226 | 67,276,026 | 67,776,026 |
| 19 | TYK2 | 10,225,652 | 10,725,652 |
| 19 | MAST3 | 17,985,882 | 18,485,882 |
| 19 | SPIB | 50,676,742 | 51,176,742 |
| 22 | RPL3 | 39,490,078 | 39,990,078 |
| Total | 46 | 23,000 kb | |
| Id Rule | 4 |
|---|---|
| Number of conditions | 13 |
| Output attribute | Affection |
| Output value | Case |
| Covering % | 19.14 |
| Error % | 3.88 |
| Condition 1 | 14:92932650_2 = “C” |
| Condition 2 | 17:43906828_1 = “G” |
| Condition 3 | 17:43912635_1 = “A” |
| Condition 4 | 17:44038536_1 = “CA” |
| Condition 5 | 17:44040823_1 = “C” |
| Condition 6 | 17:44065263_1 = “T” |
| Condition 7 | 17:44183317_1 = “C” |
| Condition 8 | 17:44185431_1 = “T” |
| Condition 9 | 17:44222335_1 = “G” |
| Condition 10 | 17:44283022_1 = “A” |
| Condition 11 | 3:119111870_1 = “T” |
| Condition 12 | 7:128705730_1 = “T” |
| Condition 13 | Sex = F |
| Id Rule | 11 |
|---|---|
| Number of conditions | 7 |
| Output attribute | Affection |
| Output value | Case |
| Covering % | 12,162162 |
| Error % | 4,661487 |
| Condition 1 | 17:38020058_2 = “AC” |
| Condition 2 | 17:38049589_2 = “T” |
| Condition 3 | 17:38070071_2 = “C” |
| Condition 4 | 17:43933579_1 = “C” |
| Condition 5 | 2:135188248_1 = “A” |
| Condition 6 | 2:25332696_2 = “C” |
| Condition 7 | 3:159726324_1 = “A” |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerussi, A.; Verda, D.; Cappadona, C.; Cristoferi, L.; Bernasconi, D.P.; Bottaro, S.; Carbone, M.; Muselli, M.; Invernizzi, P.; Asselta, R.; et al. LLM-PBC: Logic Learning Machine-Based Explainable Rules Accurately Stratify the Genetic Risk of Primary Biliary Cholangitis. J. Pers. Med. 2022, 12, 1587. https://doi.org/10.3390/jpm12101587
Gerussi A, Verda D, Cappadona C, Cristoferi L, Bernasconi DP, Bottaro S, Carbone M, Muselli M, Invernizzi P, Asselta R, et al. LLM-PBC: Logic Learning Machine-Based Explainable Rules Accurately Stratify the Genetic Risk of Primary Biliary Cholangitis. Journal of Personalized Medicine. 2022; 12(10):1587. https://doi.org/10.3390/jpm12101587
Chicago/Turabian StyleGerussi, Alessio, Damiano Verda, Claudio Cappadona, Laura Cristoferi, Davide Paolo Bernasconi, Sandro Bottaro, Marco Carbone, Marco Muselli, Pietro Invernizzi, Rosanna Asselta, and et al. 2022. "LLM-PBC: Logic Learning Machine-Based Explainable Rules Accurately Stratify the Genetic Risk of Primary Biliary Cholangitis" Journal of Personalized Medicine 12, no. 10: 1587. https://doi.org/10.3390/jpm12101587
APA StyleGerussi, A., Verda, D., Cappadona, C., Cristoferi, L., Bernasconi, D. P., Bottaro, S., Carbone, M., Muselli, M., Invernizzi, P., Asselta, R., & on behalf of The Italian PBC Genetics Study Group. (2022). LLM-PBC: Logic Learning Machine-Based Explainable Rules Accurately Stratify the Genetic Risk of Primary Biliary Cholangitis. Journal of Personalized Medicine, 12(10), 1587. https://doi.org/10.3390/jpm12101587