
Clin.iobio: A Collaborative Diagnostic Workflow to Enable Team-Based Precision Genomics

{kind=link}
{kind=link}
Abstract
:1. Introduction
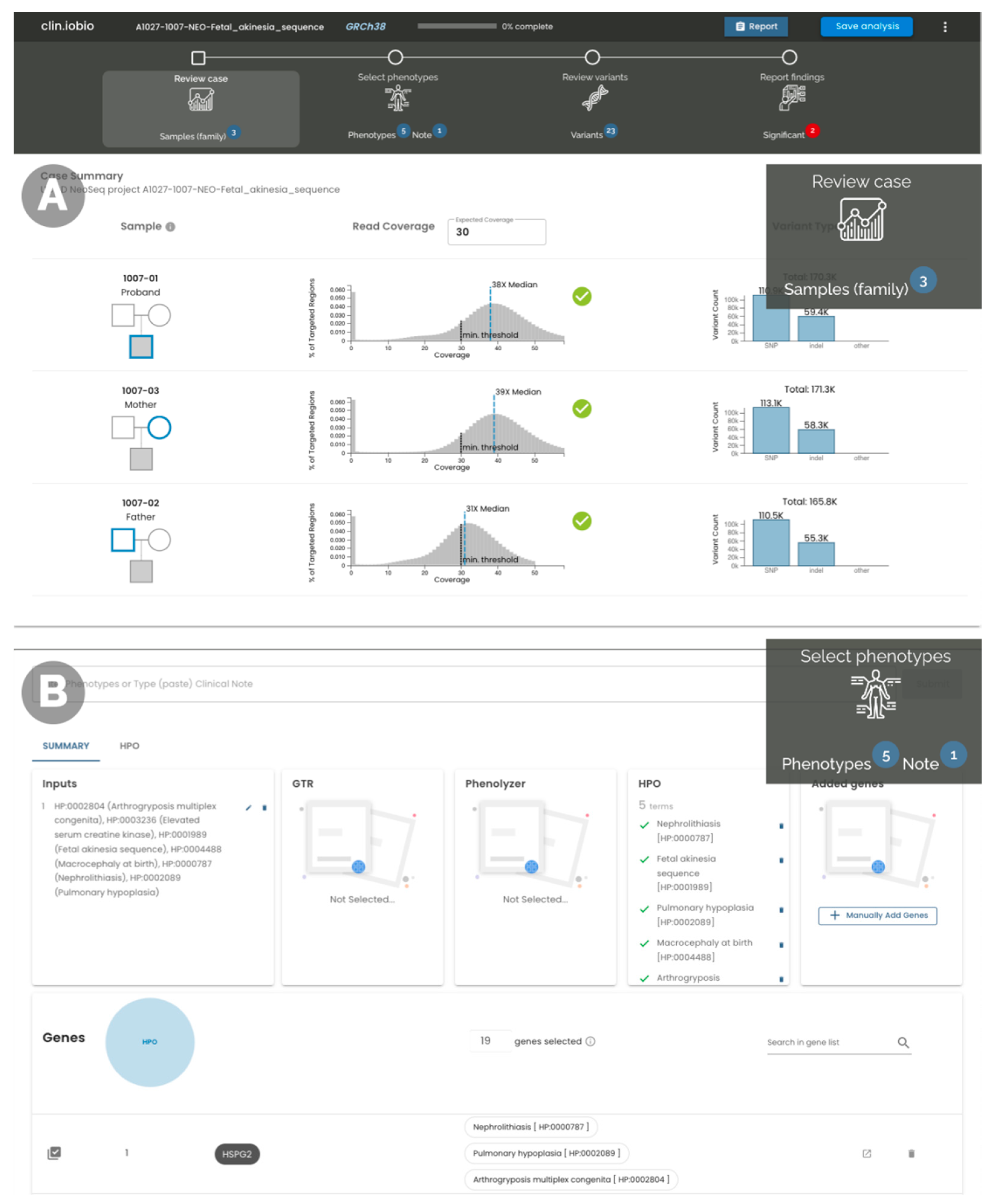
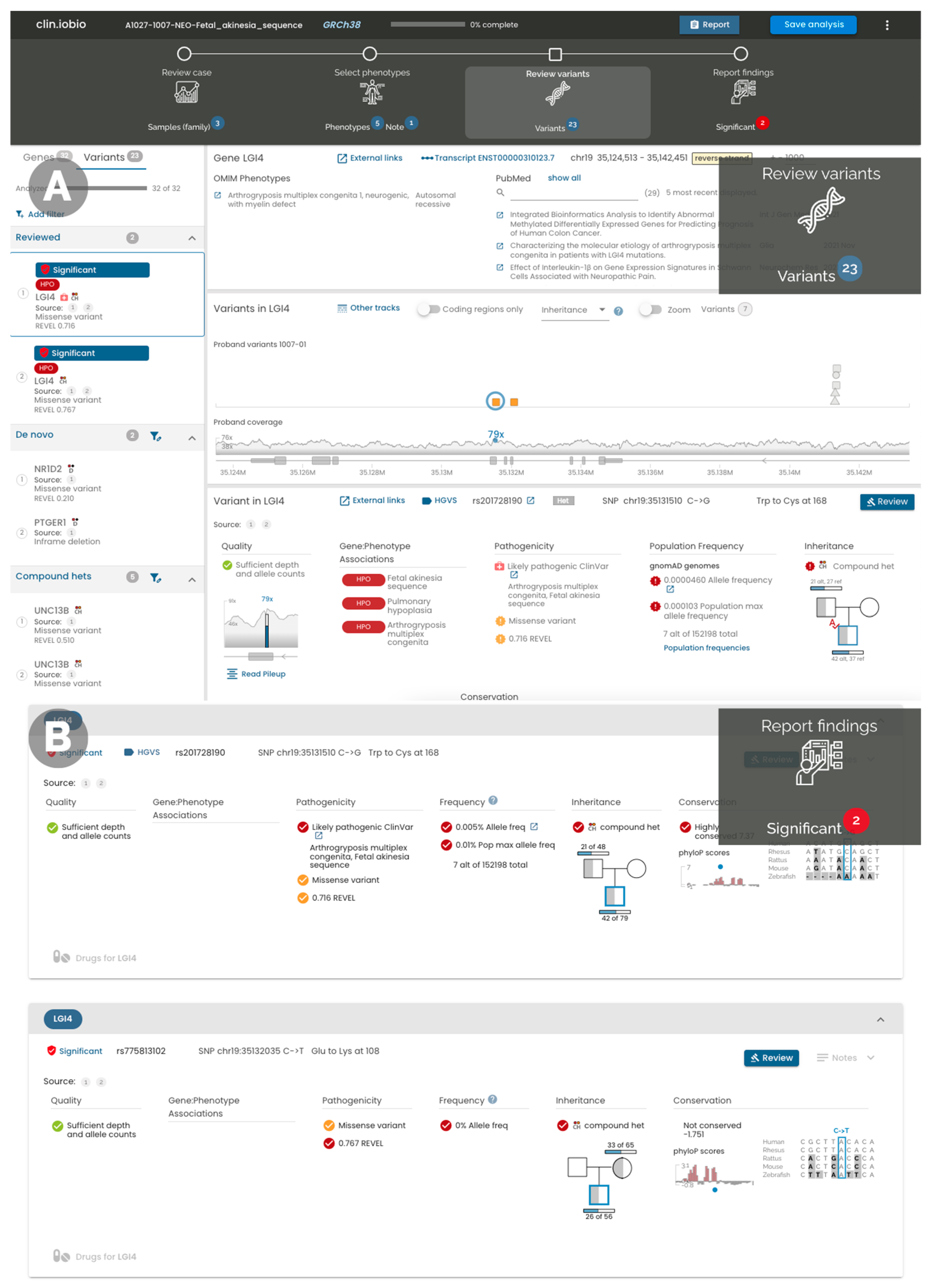
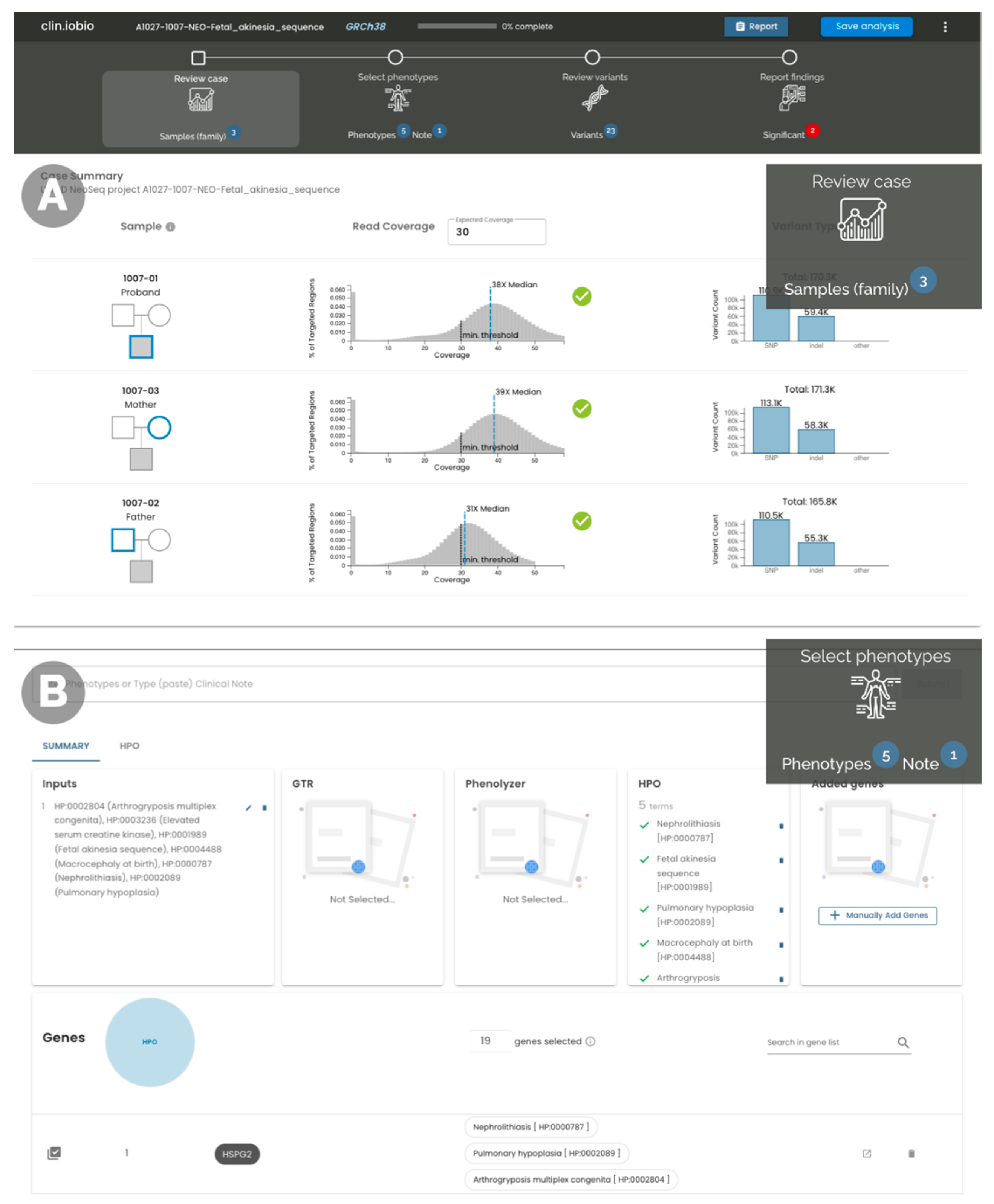
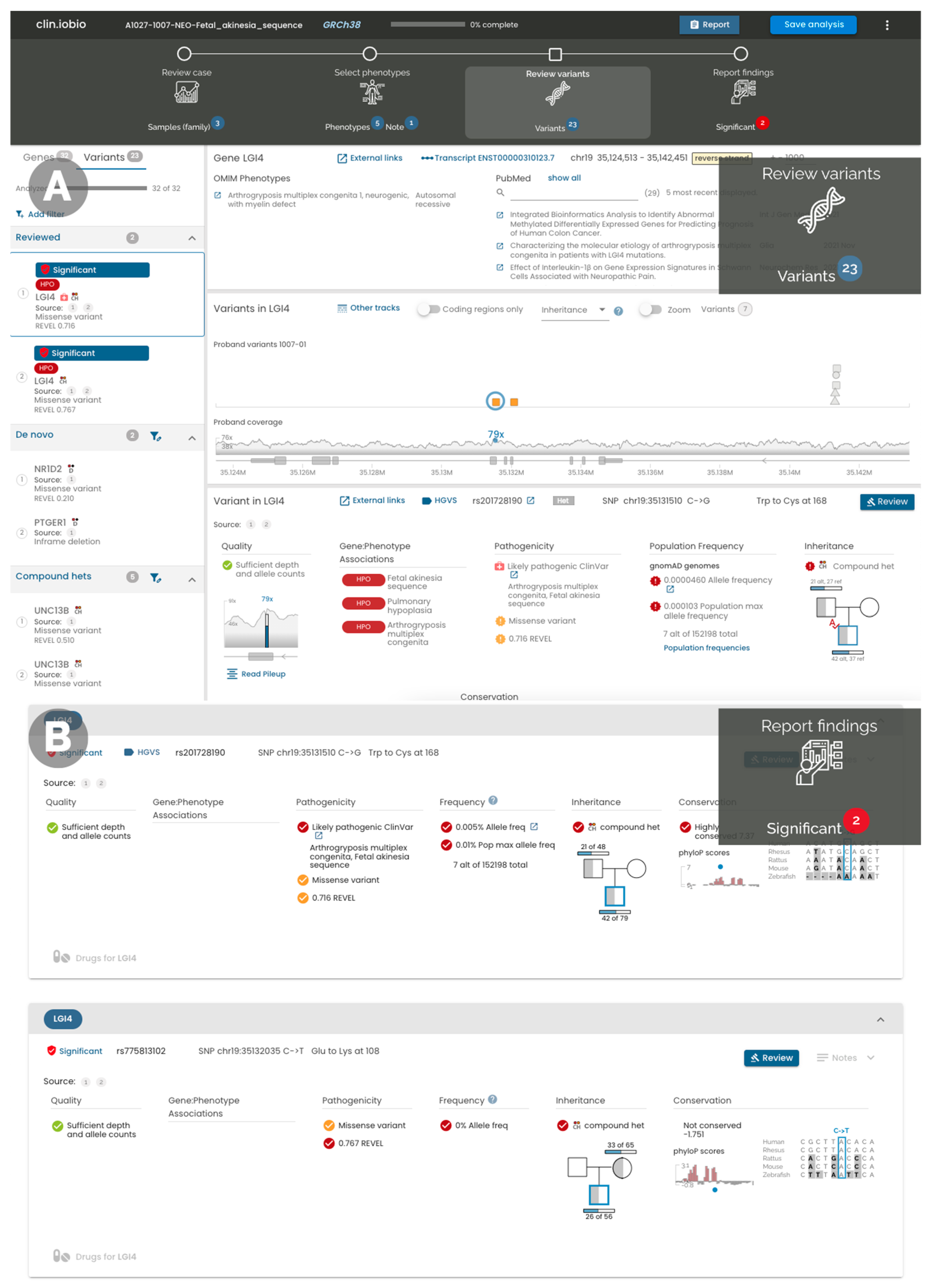
2. Results
3. Discussion
4. Materials and Methods
4.1. System Overview
4.2. File Input/Output
4.3. Sequencing Data Coverage and Alignment
4.4. IGV Integration
4.5. Variant Annotation
4.6. Gene–Disease Association
4.7. External Resources and Databases
4.8. Deployment, Usage and Availability
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Clark, M.M.; Stark, Z.; Farnaes, L.; Tan, T.Y.; White, S.M.; Dimmock, D.; Kingsmore, S.F. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom. Med. 2018, 3, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, I.A.; The BabySeq Project Team; Agrawal, P.B.; Ceyhan-Birsoy, O.; Christensen, K.D.; Fayer, S.; Frankel, L.A.; Genetti, C.A.; Krier, J.B.; LaMay, R.C.; et al. The BabySeq project: Implementing genomic sequencing in newborns. BMC Pediatr. 2018, 18, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanford, E.F.; Clark, M.M.; Farnaes, L.; Williams, M.R.; Perry, J.C.; Ingulli, E.G.; Sweeney, N.M.; Doshi, A.; Gold, J.J.; Briggs, B.; et al. Rapid Whole Genome Sequencing Has Clinical Utility in Children in the PICU. Pediatr. Crit. Care Med. 2019, 20, 1007–1020. [Google Scholar] [CrossRef]
- Farnaes, L.; Hildreth, A.; Sweeney, N.M.; Clark, M.M.; Chowdhury, S.; Nahas, S.; Cakici, J.A.; Benson, W.; Kaplan, R.H.; Kronick, R.; et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genom. Med. 2018, 3, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, J.S.; Agrawal, P.B.; Bailey, D.B., Jr.; Beggs, A.H.; Brenner, S.E.; Brower, A.M.; Cakici, J.A.; Ceyhan-Birsoy, O.; Chan, K.; Chen, F.; et al. Newborn Sequencing in Genomic Medicine and Public Health. Pediatrics 2017, 139, e20162252. [Google Scholar] [CrossRef] [Green Version]
- Robinson, P.N.; Köhler, S.; Bauer, S.; Seelow, D.; Horn, D.; Mundlos, S. The Human Phenotype Ontology: A Tool for Annotating and Analyzing Human Hereditary Disease. Am. J. Hum. Genet. 2008, 83, 610–615. [Google Scholar] [CrossRef] [Green Version]
- Solomon, B.D.; Muenke, M. When to suspect a genetic syndrome. Am. Fam. Physician 2012, 86, 826–833. [Google Scholar]
- Pedersen, B.S.; Brown, J.M.; Dashnow, H.; Wallace, A.D.; Velinder, M.; Tristani-Firouzi, M.; Schiffman, J.D.; Tvrdik, T.; Mao, R.; Best, D.H.; et al. Effective variant filtering and expected candidate variant yield in studies of rare human disease. NPJ Genom. Med. 2021, 6, 60. [Google Scholar] [CrossRef]
- Smedley, D.; Jacobsen, J.O.B.; Jäger, M.; Köhler, S.; Holtgrewe, M.; Schubach, M.; Siragusa, E.; Zemojtel, T.; Buske, O.J.; Washington, N.L.; et al. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat. Protoc. 2015, 10, 2004–2015. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Huff, C.D.; Moore, B.; Flygare, S.; Reese, M.G.; Yandell, M. VAAST 2.0: Improved Variant Classification and Disease-Gene Identification Using a Conservation-Controlled Amino Acid Substitution Matrix. Genet. Epidemiol. 2013, 37, 622–634. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Di Sera, T.; Velinder, M.; Ward, A.; Qiao, Y.; Georges, S.; Miller, C.; Pitman, A.; Richards, W.; Ekawade, A.; Viskochil, D.; et al. Gene.iobio: An interactive web tool for versatile, clinically-driven variant interrogation and prioritization. Sci. Rep. 2021, 11, 20307. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Online Mendelian Inheritance in Man. Available online: https://omim.org/ (accessed on 11 November 2020).
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeukoSEQ: Whole Genome Sequencing as a First-Line Diagnostic Tool for Leukodystrophies. Available online: https://clinicaltrials.gov/ct2/show/NCT02699190 (accessed on 5 January 2021).
- Home—Genetic Testing Registry (GTR)—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gtr/ (accessed on 5 January 2021).
- Yang, H.; Robinson, P.N.; Wang, K. Phenolyzer: Phenotype-based prioritization of candidate genes for human diseases. Nat. Methods 2015, 12, 841–843. [Google Scholar] [CrossRef]
- Ekawade, A.; Velinder, M.; Ward, A.; DiSera, T.; Miller, C.; Qiao, Y.; Marth, G. Genepanel.iobio—An easy to use web tool for generating disease- and phenotype-associated gene lists. BMC Med. Genom. 2019, 12, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, C.A.; Qiao, Y.; Di Sera, T.; D’Astous, B.; Marth, G.T. bam.iobio: A web-based, real-time, sequence alignment file inspector. Nat. Methods 2014, 11, 1189. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Tabix: Fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics 2011, 27, 718–719. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.; Abecasis, G.R.; Kang, H.M. Unified representation of genetic variants. Bioinformatics 2015, 31, 2202–2204. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.-F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.; et al. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2009, 20, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deisseroth, C.A.; Birgmeier, J.; Bodle, E.E.; Kohler, J.N.; Matalon, D.R.; Nazarenko, Y.; Genetti, C.A.; Brownstein, C.A.; Schmitz-Abe, K.; Schoch, K.; et al. ClinPhen extracts and prioritizes patient phenotypes directly from medical records to expedite genetic disease diagnosis. Genet. Med. 2019, 21, 1585–1593. [Google Scholar] [CrossRef]
- McKusick, V.A. Mendelian Inheritance in Man and Its Online Version, OMIM. Am. J. Hum. Genet. 2007, 80, 588–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayers, E. The E-utilities In-Depth: Parameters, Syntax and More. In Entrez Programming Utilities Help; National Center for Biotechnology Information: Bethesda, MD, USA, 2009. [Google Scholar]
- Wang, J.; Al-Ouran, R.; Hu, Y.; Kim, S.-Y.; Wan, Y.-W.; Wangler, M.; Yamamoto, S.; Chao, H.-T.; Comjean, A.; Mohr, S.; et al. MARRVEL: Integration of Human and Model Organism Genetic Resources to Facilitate Functional Annotation of the Human Genome. Am. J. Hum. Genet. 2017, 100, 843–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Firth, H.V.; Richards, S.M.; Bevan, P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. In Current Protocols in Bioinformatics; Bateman, A., Pearson, W.R., Stein, L.D., Stormo, G.D., Yates, J.R., III, Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2002; Volume 29, pp. 1.30.1–1.30.33. [Google Scholar]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Kalderimis, A.; Lyne, R.; Butano, D.; Contrino, S.; Lyne, M.; Heimbach, J.; Hu, F.; Smith, R.; Štěpán, R.; Sullivan, J.; et al. InterMine: Extensive web services for modern biology. Nucleic Acids Res. 2014, 42, W468–W472. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ward, A.; Velinder, M.; Di Sera, T.; Ekawade, A.; Malone Jenkins, S.; Moore, B.; Mao, R.; Bayrak-Toydemir, P.; Marth, G. Clin.iobio: A Collaborative Diagnostic Workflow to Enable Team-Based Precision Genomics. J. Pers. Med. 2022, 12, 73. https://doi.org/10.3390/jpm12010073
Ward A, Velinder M, Di Sera T, Ekawade A, Malone Jenkins S, Moore B, Mao R, Bayrak-Toydemir P, Marth G. Clin.iobio: A Collaborative Diagnostic Workflow to Enable Team-Based Precision Genomics. Journal of Personalized Medicine. 2022; 12(1):73. https://doi.org/10.3390/jpm12010073
Chicago/Turabian StyleWard, Alistair, Matt Velinder, Tonya Di Sera, Aditya Ekawade, Sabrina Malone Jenkins, Barry Moore, Rong Mao, Pinar Bayrak-Toydemir, and Gabor Marth. 2022. "Clin.iobio: A Collaborative Diagnostic Workflow to Enable Team-Based Precision Genomics" Journal of Personalized Medicine 12, no. 1: 73. https://doi.org/10.3390/jpm12010073
APA StyleWard, A., Velinder, M., Di Sera, T., Ekawade, A., Malone Jenkins, S., Moore, B., Mao, R., Bayrak-Toydemir, P., & Marth, G. (2022). Clin.iobio: A Collaborative Diagnostic Workflow to Enable Team-Based Precision Genomics. Journal of Personalized Medicine, 12(1), 73. https://doi.org/10.3390/jpm12010073