Abstract
The current coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus (SARS-CoV)-2, is affecting every aspect of global society, including public healthcare systems, medical care access, and the economy. Although the respiratory tract is primarily affected by SARS-CoV-2, emerging evidence suggests that the virus may also reach the central nervous system (CNS), leading to several neurological issues. In particular, people with a diagnosis of Alzheimer’s disease (AD) are a vulnerable group at high risk of contracting COVID-19, and develop more severe forms and worse outcomes, including death. Therefore, understanding shared links between COVID-19 and AD could aid the development of therapeutic strategies against both. Herein, we reviewed common risk factors and potential pathogenetic mechanisms that might contribute to the acceleration of neurodegenerative processes in AD patients infected by SARS-CoV-2.
1. Introduction
Coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), continues to spread rapidly across the globe, becoming a devastating pandemic infection with growing mortality rates [1]. Although SARS-CoV-2 predominantly affects the respiratory system, increasing evidence reports a close relationship between COVID-19 and central nervous system (CNS) disorders, with more than 30% of hospitalized COVID-19 patients exhibiting neurological manifestations [2]. In line with this observation, magnetic resonance imaging (MRI) showed brain structural changes associated with COVID-19 in both surviving patients and non-survivors [3,4], confirming SARS-CoV-2’s involvement within the CNS [5]. However, whether the neurological symptoms represent a direct consequence of SARS-CoV-2 infection of brain cells or a result of systemic illness remains to be clarified [6]. A viral infection of human neurons has been suggested by the presence of viral RNA and/or protein in the brains of COVID-19 patients with neurological manifestations [7,8,9]. A neurochemical study reported that patients with severe SARS-CoV-2 infections exhibit high plasma levels of neurofilament light chain protein (NfL) and glial fibrillary acidic protein (GFAP), known as biochemical indicators of neuronal injury and glial activation [10], further supporting a direct link between SARS-CoV-2 brain infection and neurological disturbances. Additionally, both human and animal models demonstrated that the virus can directly invade the olfactory bulb [11] without a primary lung involvement, by the interaction between the virus S1 spike protein and angiotensin-converting enzyme 2 (ACE2), which is widely expressed in the glial cells and neurons [12]. However, the distribution of ACE2 in the human brain regions and cell types is quite heterogeneous. It is highly expressed in the choroid plexus of the lateral ventricle and central glial substance, while low expression was detected in the hippocampus. Regarding the cell-type distribution, ACE2 was found both in excitatory and inhibitory neurons, as well as in non-neuronal cells such as the oligodendrocytes, astrocytes, and endothelial cells. This evidence supports the hypothesis that brain infection by SARS-CoV-2 may promote CNS symptoms in patients with COVID-19, and suggests new potential routes for viral entry and propagation into the cerebral tissue [13]. SARS-CoV-2 may also infect the brain through a disrupted blood-brain barrier (BBB) that is often compromised in the aging brain, and neurodegenerative disorders, mainly in Alzheimer’s disease (AD) [14].
AD represents the most common form of dementia in the elderly population world-wide, and it is clinically characterized by neuronal loss in the hippocampus and cortical areas, leading to memory deterioration, behavioral changes, and cognitive decline. Neuropathological hallmarks include the presence of intracellular neurofibrillary tangles (NFTs), as well as parenchymal and vascular amyloid β (Aβ) deposits [15]. In the progression of the neuropathological changes observed in AD pathogenesis, a central role is played by neuroinflammation, attributed to activated microglia cells and the release of several cytokines [16]. Intriguingly, severe outcomes after SARS-CoV-2 infection in elderly individuals are often associated with a cytokine storm producing an excessive inflammatory and immune response, which may in turn accelerate brain inflammatory neurodegeneration [17]. Moreover, some COVID-19 patients could develop cognitive deficits after the primary infection [18], which can be partially explained by the virus-related exacerbation of the underlying brain pathology in elderly people [14]. One important issue is whether or not COVID-19 actually infects neurons, enters into neurons, or replicates within them, leading to a lytic cycle. Some data reviewed in [19] point to neuronal infection. Moreover, a recent study reported that SARS-CoV-2 spike S1 protein could facilitate the spreading of aggregated tau via the secretion of extracellular vesicles (EV) or direct cell-to-cell contact [20]. As SARS-CoV-2 is able to infect human neurons and use the neuronal machinery to replicate [21], the virus could lend its glycoproteins to neurons and EV, thus perpetuating the pathology. In addition to neuronal cells, astrocytes can also be infected by SARS-CoV-2, causing metabolic alterations that impair neuronal viability, contributing to neurodegeneration [22].
Given the high prevalence of AD individuals affected by COVID-19, this review aimed to elucidate common underlying etiological and risk factors that may contribute to the exacerbation of the neurodegenerative processes in AD patients infected by SARS-CoV-2. Understanding the relationship between COVID-19 and AD could aid in the detection of potential biomarkers for the early identification of COVID-19 in patients with a high risk of developing AD, as well as the management and development of novel therapeutic approaches for both diseases. Figure 1 shows the possible association between AD and SARS-CoV-2 infection by summarizing shared risk factors and potential underlying mechanisms, which are described in the following paragraphs (Figure 1).
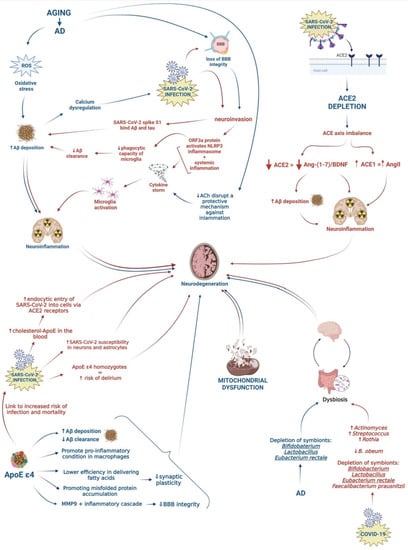
Figure 1.
Schematic representation of possible pathogenetic mechanisms leading to neurodegeneration. Pathways activated by COVID-19 and AD are represented by red and blue arrows, respectively.
3. Conclusions and Future Directions
The ongoing COVID-19 outbreak in late 2019 has caused a global pandemic with serious public health concerns. Apart from the well-known consequences to the respiratory system, increasing evidence reports that SARS-CoV-2 can invade the CNS, leading to severe neurological sequelae [126,127]. Once penetrated into the brain, the virus can cause neurodegeneration, demyelination, and cellular senescence, thus accelerating brain aging and potentially exacerbating the underlying neurodegenerative pathology [14].
Regarding AD, several mechanisms have been suggested to explain SARS-CoV-2-mediated neurological damage (Table 1), though its effects at the molecular and mechanistic levels remain only hypothetical or speculative, due to the absence of reliable post-mortem data on Aβ and NFTs in SARS-CoV-2-infected patients. AD and COVID-19 share many risk factors and pathogenetic mechanisms that may also partially explain the high incidence and mortality rate in people with AD. On the other hand, patients affected by AD could be more susceptible to contracting COVID-19. Preventive strategies to contain the SARS-CoV-2 spread, such as isolation or quarantine, negatively affect AD patients, increasing the risk of cognitive impairment due to a lack of social interaction [128]. Moreover, people living with dementia may be not able to follow recommendations from government authorities, such as sanitizing hands, covering the mouth and nose when coughing, and maintaining social distancing, partially due to their general cognitive impairment and short-term memory loss [129]. These patients are also more susceptible to circulating SARS-CoV-2, as they are frequently exposed to the virus during hospital care or required institutionalization and often suffer from pre-existing comorbidities, such as hypertension, diabetes mellitus and cerebrovascular diseases [130]. In this regard, the majority of patients with COVID-19 display severe coagulopathies, such as thrombotic microangiopathy and disseminated intravascular coagulation (DIC), probably due to hypoxic conditions or proinflammatory cytokines produced by infected cells [131]. Therefore, it is possible that microembolic events may contribute to cerebrovascular disease, which in turn can contribute to worsen AD pathology. However, it remains to be determined whether SARS-CoV-2 infection exacerbates cognitive decline in AD patients or triggers dementia in infected people, although many elements described here support this hypothesis. Additionally, it should be noted that some of pathogenetic mechanisms reported in this review are common to other neurodegenerative disorders, such as Parkinson’s disease and amyotrophic lateral sclerosis. In fact, novel research articles are continuing to report interesting results on other aspects. Among them, an overlap has been found between genetic risk factors for AD and severe COVID-19, such as single-nucleotide polymorphisms (SNPs) in oligoadenylate synthetase 1 (OAS1) [132] and bridging integrator 1 (BIN1) genes [133]. Single cell sequencing studies performed on brains of COVID-19 patients revealed that astrocyte and microglia cells show some pathological features shared with those observed in neurodegenerative disorders [134].

Table 1.
Summary of shared biological links between AD and COVID-19.
Although few studies addressing the relationship between COVID-19 and AD are currently available given the recent onset of the pandemic, their numerous shared links strengthen the necessity to assess neurological symptoms and implement preventive strategies to mitigate the risk of developing AD in SARS-CoV-2-infected people. Longitudinal follow-up studies of COVID-19 patients are needed to evaluate the long-term neurological effects of SARS-CoV-2 infection. Furthermore, large-scale retrospective analysis, in combination with preclinical studies, will be useful to fully understand the implications of SARS-CoV-2 infection for the development and progression of AD. Finally, these studies should be also implemented with cognitive impairment evaluation, blood and neuroimaging biomarkers evaluating inflammation, oxidative damage, or metabolic alterations, in order to assess the pathogenetic pathways shared by AD and COVID-19 that we reviewed.
Author Contributions
C.V. carried out the literature review, conceptualized and wrote the manuscript; E.R. contributed to carry out the literature review and designed the figure; M.L. edited the manuscript; R.C. carried out the literature review and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Park, M.; Cook, A.R.; Lim, J.T.; Sun, Y.; Dickens, B.L. A Systematic Review of COVID-19 Epidemiology Based on Current Evidence. J. Clin. Med. 2020, 9, 967. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic Manifestations of Hospitalized Patients with Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Coolen, T.; Lolli, V.; Sadeghi, N.; Rovai, A.; Trotta, N.; Taccone, F.S.; Creteur, J.; Henrard, S.; Goffard, J.C.; Dewitte, O.; et al. Early postmortem brain MRI findings in COVID-19 non-survivors. Neurology 2020, 95, e2016–e2027. [Google Scholar] [CrossRef]
- Chougar, L.; Shor, N.; Weiss, N.; Galanaud, D.; Leclercq, D.; Mathon, B.; Belkacem, S.; Ströer, S.; Burrel, S.; Boutolleau, D.; et al. Retrospective Observational Study of Brain MRI Findings in Patients with Acute SARS-CoV-2 Infection and Neurologic Manifestations. Radiology 2020, 297, E313–E323. [Google Scholar] [CrossRef]
- Vespignani, H.; Colas, D.; Lavin, B.S.; Soufflet, C.; Maillard, L.; Pourcher, V.; Paccoud, O.; Medjebar, S.; Frouin, P.Y. Report on Electroencephalographic Findings in Critically Ill Patients with COVID-19. Ann. Neurol. 2020, 88, 626–630. [Google Scholar] [CrossRef]
- Ellul, M.A.; Benjamin, L.; Singh, B.; Lant, S.; Michael, B.D.; Easton, A.; Kneen, R.; Defres, S.; Sejvar, J.; Solomon, T. Neurological associations of COVID-19. Lancet Neurol. 2020, 19, 767–783. [Google Scholar] [CrossRef]
- Moriguchi, T.; Harii, N.; Goto, J.; Harada, D.; Sugawara, H.; Takamino, J.; Ueno, M.; Sakata, H.; Kondo, K.; Myose, N.; et al. A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int. J. Infect. Dis. 2020, 94, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Paniz-Mondolfi, A.; Bryce, C.; Grimes, Z.; Gordon, R.E.; Reidy, J.; Lednicky, J.; Sordillo, E.M.; Fowkes, M. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J. Med. Virol. 2020, 92, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Solomon, I.H.; Normandin, E.; Bhattacharyya, S.; Mukerji, S.S.; Keller, K.; Ali, A.S.; Adams, G.; Hornick, J.L.; Padera, R.F.; Sabeti, P. Neuropathological Features of Covid-19. N. Engl. J. Med. 2020, 383, 989–992. [Google Scholar] [CrossRef]
- Kanberg, N.; Ashton, N.J.; Andersson, L.M.; Yilmaz, A.; Lindh, M.; Nilsson, S.; Price, R.W.; Blennow, K.; Zetterberg, H.; Gisslén, M. Neurochemical evidence of astrocytic and neuronal injury commonly found in COVID-19. Neurology 2020, 95, e1754–e1759. [Google Scholar] [CrossRef]
- Li, Y.C.; Bai, W.Z.; Hashikawa, T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J. Med. Virol. 2020, 92, 552–555. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef]
- Chen, R.; Wang, K.; Yu, J.; Howard, D.; French, L.; Chen, Z.; Wen, C.; Xu, Z. The Spatial and Cell-Type Distribution of SARS-CoV-2 Receptor ACE2 in the Human and Mouse Brains. Front. Neurol. 2020, 11, 573095. [Google Scholar] [CrossRef]
- Hascup, E.R.; Hascup, K.N. Does SARS-CoV-2 infection cause chronic neurological complications? Geroscience 2020, 42, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Villa, C.; Lavitrano, M.; Salvatore, E.; Combi, R. Molecular and Imaging Biomarkers in Alzheimer’s Disease: A Focus on Recent Insights. J. Pers. Med. 2020, 10, 61. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Heneka, M.T.; Golenbock, D.; Latz, E.; Morgan, D.; Brown, R. Immediate and long-term consequences of COVID-19 infections for the development of neurological disease. Alzheimers Res. Ther. 2020, 12, 69. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Lou, J.J.; Movassaghi, M.; Gordy, D.; Olson, M.G.; Zhang, T.; Khurana, M.S.; Chen, Z.; Perez-Rosendahl, M.; Thammachantha, S.; Singer, E.J.; et al. Neuropathology of COVID-19 (neuro-COVID): Clinicopathological update. Free Neuropathol. 2021, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Hossinger, A.; Heumüller, S.E.; Hornberger, A.; Buravlova, O.; Konstantoulea, K.; Müller, S.A.; Paulsen, L.; Rousseau, F.; Schymkowitz, J.; et al. Highly efficient intercellular spreading of protein misfolding mediated by viral ligand-receptor interactions. Nat. Commun. 2021, 12, 5739. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.Z.; Chu, H.; Han, S.; Shuai, H.; Deng, J.; Hu, Y.F.; Gong, H.R.; Lee, A.C.; Zou, Z.; Yau, T.; et al. SARS-CoV-2 infects human neural progenitor cells and brain organoids. Cell Res. 2020, 30, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Crunfli, F.; Carregari, V.C.; Veras, F.P.; Vendramini, P.H.; Fragnani Valença, A.G.; Leão Marcelo Antunes, A.S.; Brandão-Teles, C.; da Silva Zuccoli, G.; Reis-de-Oliveira, G.; Silva-Costa, L.C.; et al. SARS-CoV-2 infects brain astrocytes of COVID-19 patients and impairs neuronal viability. MedRxiv 2020. [Google Scholar] [CrossRef]
- Trevisan, K.; Cristina-Pereira, R.; Silva-Amaral, D.; Aversi-Ferreira, T.A. Theories of Aging and the Prevalence of Alzheimer’s Disease. Biomed. Res. Int. 2019, 2019, 9171424. [Google Scholar] [CrossRef]
- Chen, Y.; Klein, S.L.; Garibaldi, B.T.; Li, H.; Wu, C.; Osevala, N.M.; Li, T.; Margolick, J.B.; Pawelec, G.; Leng, S.X. Aging in COVID-19: Vulnerability, immunity and intervention. Ageing Res. Rev. 2021, 65, 101205. [Google Scholar] [CrossRef]
- Wu, J.T.; Leung, K.; Bushman, M.; Kishore, N.; Niehus, R.; de Salazar, P.M.; Cowling, B.J.; Lipsitch, M.; Leung, G.M. Estimating clinical severity of COVID-19 from the transmission dynamics in Wuhan, China. Nat. Med. 2020, 26, 506–510. [Google Scholar] [CrossRef]
- Onder, G.; Rezza, G.; Brusaferro, S. Case-Fatality Rate and Characteristics of Patients Dying in Relation to COVID-19 in Italy. JAMA 2020, 323, 1775–1776. [Google Scholar] [CrossRef]
- Xia, X.; Jiang, Q.; McDermott, J.; Han, J.J. Aging and Alzheimer’s disease: Comparison and associations from molecular to system level. Aging Cell 2018, 17, e12802. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Islam, K.; Rahman, S.; Alamin, M. Neurobiochemical Cross-talk Between COVID-19 and Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 1017–1023. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Liu, Q.; Shi, Y.; Cai, J.; Duan, Y.; Wang, R.; Zhang, H.; Ruan, Q.; Li, J.; Zhao, L.; Ping, Y.; et al. Pathological changes in the lungs and lymphatic organs of 12 COVID-19 autopsy cases. Natl. Sci. Rev. 2020, 7, 1868–1878. [Google Scholar] [CrossRef] [PubMed]
- Duarte, C.; Akkaoui, J.; Ho, A.; Garcia, C.; Yamada, C.; Movila, A. Age-dependent effects of the recombinant spike protein/SARS-CoV-2 on the M-CSF- and IL-34-differentiated macrophages in vitro. Biochem. Biophys. Res. Commun. 2021, 546, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS ONE 2010, 5, e9505. [Google Scholar] [CrossRef]
- Idrees, D.; Kumar, V. SARS-CoV-2 spike protein interactions with amyloidogenic proteins: Potential clues to neurodegeneration. Biochem. Biophys. Res. Commun. 2021, 554, 94–98. [Google Scholar] [CrossRef]
- Green, K.N.; LaFerla, F.M. Linking calcium to Abeta and Alzheimer’s disease. Neuron 2008, 59, 190–194. [Google Scholar] [CrossRef]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cao, R.; Zhong, W. Host Calcium Channels and Pumps in Viral Infections. Cells 2019, 9, 94. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.I.; de Oliveira, E.M. Brain renin angiotensin in disease. J. Mol. Med. 2008, 86, 715–722. [Google Scholar] [CrossRef]
- Abiodun, O.A.; Ola, M.S. Role of brain renin angiotensin system in neurodegeneration: An update. Saudi J. Biol. Sci. 2020, 27, 905–912. [Google Scholar] [CrossRef]
- Kaur, P.; Muthuraman, A.; Kaur, M. The implications of angiotensin-converting enzymes and their modulators in neurodegenerative disorders: Current and future perspectives. ACS Chem. Neurosci. 2015, 6, 508–521. [Google Scholar] [CrossRef] [PubMed]
- Rabie, M.A.; Abd El Fattah, M.A.; Nassar, N.N.; El-Abhar, H.S.; Abdallah, D.M. Angiotensin 1–7 ameliorates 6-hydroxydopamine lesions in hemiparkinsonian rats through activation of MAS receptor/PI3K/Akt/BDNF pathway and inhibition of angiotensin II type-1 receptor/NF-κB axis. Biochem. Pharmacol. 2018, 151, 126–134. [Google Scholar] [CrossRef]
- Motaghinejad, M.; Gholami, M. Possible Neurological and Mental Outcomes of COVID-19 Infection: A Hypothetical Role of ACE-2\Mas\BDNF Signaling Pathway. Int. J. Prev. Med. 2020, 11, 84. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Iwanami, J.; Min, L.J.; Tsukuda, K.; Nakaoka, H.; Bai, H.Y.; Shan, B.S.; Kan-No, H.; Kukida, M.; Chisaka, T.; et al. Deficiency of angiotensin-converting enzyme 2 causes deterioration of cognitive function. NPJ Aging Mech. Dis. 2016, 2, 16024. [Google Scholar] [CrossRef]
- Evans, C.E.; Miners, J.S.; Piva, G.; Willis, C.L.; Heard, D.M.; Kidd, E.J.; Good, M.A.; Kehoe, P.G. ACE2 activation protects against cognitive decline and reduces amyloid pathology in the Tg2576 mouse model of Alzheimer’s disease. Acta Neuropathol. 2020, 139, 485–502. [Google Scholar] [CrossRef]
- Kamel, A.S.; Abdelkader, N.F.; Abd El-Rahman, S.S.; Emara, M.; Zaki, H.F.; Khattab, M.M. Stimulation of ACE2/ANG(1–7)/Mas Axis by Diminazene Ameliorates Alzheimer’s Disease in the D-Galactose-Ovariectomized Rat Model: Role of PI3K/Akt Pathway. Mol. Neurobiol. 2018, 55, 8188–8202. [Google Scholar] [CrossRef]
- Duan, R.; Xue, X.; Zhang, Q.Q.; Wang, S.Y.; Gong, P.Y.; E, Y.; Jiang, T.; Zhang, Y.D. ACE2 activator diminazene aceturate ameliorates Alzheimer’s disease-like neuropathology and rescues cognitive impairment in SAMP8 mice. Aging (Albany NY) 2020, 12, 14819–14829. [Google Scholar] [CrossRef]
- Fu, X.; Lin, R.; Qiu, Y.; Yu, P.; Lei, B. Overexpression of Angiotensin-Converting Enzyme 2 Ameliorates Amyloid β-Induced Inflammatory Response in Human Primary Retinal Pigment Epithelium. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3018–3028. [Google Scholar] [CrossRef][Green Version]
- Kehoe, P.G.; Wong, S.; Al Mulhim, N.; Palmer, L.E.; Miners, J.S. Angiotensin-converting enzyme 2 is reduced in Alzheimer’s disease in association with increasing amyloid-β and tau pathology. Alzheimers Res. Ther. 2016, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Tan, L.; Gao, Q.; Lu, H.; Zhu, X.C.; Zhou, J.S.; Zhang, Y.D. Plasma Angiotensin-(1-7) is a Potential Biomarker for Alzheimer’s Disease. Curr. Neurovasc. Res. 2016, 13, 96–99. [Google Scholar] [CrossRef]
- Jiang, T.; Zhang, Y.D.; Zhou, J.S.; Zhu, X.C.; Tian, Y.Y.; Zhao, H.D.; Lu, H.; Gao, Q.; Tan, L.; Yu, J.T. Angiotensin-(1-7) is Reduced and Inversely Correlates with Tau Hyperphosphorylation in Animal Models of Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 2489–2497. [Google Scholar] [CrossRef] [PubMed]
- Miners, S.; Kehoe, P.G.; Love, S. Cognitive impact of COVID-19: Looking beyond the short term. Alzheimers Res. Ther. 2020, 12, 170. [Google Scholar] [CrossRef] [PubMed]
- Monteonofrio, L.; Florio, M.C.; AlGhatrif, M.; Lakatta, E.G.; Capogrossi, M.C. Aging- and gender-related modulation of RAAS: Potential implications in COVID-19 disease. Vasc. Biol. 2021, 3, R1–R14. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Korwek, K.M.; Trotter, J.H.; Ladu, M.J.; Sullivan, P.M.; Weeber, E.J. ApoE isoform-dependent changes in hippocampal synaptic function. Mol. Neurodegener. 2009, 4, 21. [Google Scholar] [CrossRef]
- Drouet, B.; Fifre, A.; Pinçon-Raymond, M.; Vandekerckhove, J.; Rosseneu, M.; Guéant, J.L.; Chambaz, J.; Pillot, T. ApoE protects cortical neurones against neurotoxicity induced by the non-fibrillar C-terminal domain of the amyloid-beta peptide. J. Neurochem. 2001, 76, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Polazzi, E.; Mengoni, I.; Peña-Altamira, E.; Massenzio, F.; Virgili, M.; Petralla, S.; Monti, B. Neuronal Regulation of Neuroprotective Microglial Apolipoprotein E Secretion in Rat In Vitro Models of Brain Pathophysiology. J. Neuropathol. Exp. Neurol. 2015, 74, 818–834. [Google Scholar] [CrossRef]
- Shi, Y.; Manis, M.; Long, J.; Wang, K.; Sullivan, P.M.; Remolina Serrano, J.; Hoyle, R.; Holtzman, D.M. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J. Exp. Med. 2019, 216, 2546–2561. [Google Scholar] [CrossRef]
- Fernandez, C.G.; Hamby, M.E.; McReynolds, M.L.; Ray, W.J. The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 14. [Google Scholar] [CrossRef]
- Bertram, L.; Tanzi, R.E. Thirty years of Alzheimer’s disease genetics: The implications of systematic meta-analyses. Nat. Rev. Neurosci. 2008, 9, 768–778. [Google Scholar] [CrossRef]
- Riedel, B.C.; Thompson, P.M.; Brinton, R.D. Age, APOE and sex: Triad of risk of Alzheimer’s disease. J. Steroid. Biochem. Mol. Biol. 2016, 160, 134–147. [Google Scholar] [CrossRef]
- Kloske, C.M.; Wilcock, D.M. The Important Interface Between Apolipoprotein E and Neuroinflammation in Alzheimer’s Disease. Front. Immunol. 2020, 11, 754. [Google Scholar] [CrossRef]
- Kuo, C.L.; Pilling, L.C.; Atkins, J.L.; Masoli, J.A.H.; Delgado, J.; Kuchel, G.A.; Melzer, D. APOE e4 Genotype Predicts Severe COVID-19 in the UK Biobank Community Cohort. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 2231–2232. [Google Scholar] [CrossRef]
- Wang, H.; Yuan, Z.; Pavel, M.A.; Hansen, S.B. The role of high cholesterol in age-related COVID19 lethality. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, M.; Garcia, G.; Tian, E.; Cui, Q.; Chen, X.; Sun, G.; Wang, J.; Arumugaswami, V.; Shi, Y. ApoE-Isoform-Dependent SARS-CoV-2 Neurotropism and Cellular Response. Cell Stem Cell 2021, 28, 331–342. [Google Scholar] [CrossRef]
- Mao, X.Y.; Jin, W.L. The COVID-19 Pandemic: Consideration for Brain Infection. Neuroscience 2020, 437, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Atkins, J.L.; Masoli, J.A.H.; Delgado, J.; Pilling, L.C.; Kuo, C.L.; Kuchel, G.A.; Melzer, D. Preexisting Comorbidities Predicting COVID-19 and Mortality in the UK Biobank Community Cohort. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 2224–2230. [Google Scholar] [CrossRef]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: A meta-analysis. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Cameron, B.; Landreth, G.E. Inflammation, microglia, and Alzheimer’s disease. Neurobiol. Dis. 2010, 37, 503–509. [Google Scholar] [CrossRef]
- Kim, Y.S.; Lee, K.J.; Kim, H. Serum tumour necrosis factor-α and interleukin-6 levels in Alzheimer’s disease and mild cognitive impairment. Psychogeriatrics 2017, 17, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Cojocaru, I.M.; Cojocaru, M.; Miu, G.; Sapira, V. Study of interleukin-6 production in Alzheimer’s disease. Rom. J. Intern. Med. 2011, 49, 55–58. [Google Scholar]
- Pugh, C.R.; Nguyen, K.T.; Gonyea, J.L.; Fleshner, M.; Wakins, L.R.; Maier, S.F.; Rudy, J.W. Role of interleukin-1 beta in impairment of contextual fear conditioning caused by social isolation. Behav. Brain Res. 1999, 106, 109–118. [Google Scholar] [CrossRef]
- Depino, A.M.; Alonso, M.; Ferrari, C.; del Rey, A.; Anthony, D.; Besedovsky, H.; Medina, J.H.; Pitossi, F. Learning modulation by endogenous hippocampal IL-1: Blockade of endogenous IL-1 facilitates memory formation. Hippocampus 2004, 14, 526–535. [Google Scholar] [CrossRef]
- Bialuk, I.; Taranta, A.; Winnicka, M.M. IL-6 deficiency alters spatial memory in 4- and 24-month-old mice. Neurobiol. Learn. Mem. 2018, 155, 21–29. [Google Scholar] [CrossRef]
- Naughton, S.X.; Raval, U.; Pasinetti, G.M. Potential Novel Role of COVID-19 in Alzheimer’s Disease and Preventative Mitigation Strategies. J. Alzheimers Dis. 2020, 76, 21–25. [Google Scholar] [CrossRef]
- Benameur, K.; Agarwal, A.; Auld, S.C.; Butters, M.P.; Webster, A.S.; Ozturk, T.; Howell, J.C.; Bassit, L.C.; Velasquez, A.; Schinazi, R.F.; et al. Encephalopathy and Encephalitis Associated with Cerebrospinal Fluid Cytokine Alterations and Coronavirus Disease, Atlanta, Georgia, USA, 2020. Emerg. Infect. Dis. 2020, 26, 2016–2021. [Google Scholar] [CrossRef]
- Farhadian, S.; Glick, L.R.; Vogels, C.B.F.; Thomas, J.; Chiarella, J.; Casanovas-Massana, A.; Zhou, J.; Odio, C.; Vijayakumar, P.; Geng, B.; et al. Acute encephalopathy with elevated CSF inflammatory markers as the initial presentation of COVID-19. BMC Neurol. 2020, 20, 248. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, B.; Qu, Y.; Chen, Y.; Xiong, J.; Feng, Y.; Men, D.; Huang, Q.; Liu, Y.; Yang, B.; et al. Detectable Serum Severe Acute Respiratory Syndrome Coronavirus 2 Viral Load (RNAemia) Is Closely Correlated with Drastically Elevated Interleukin 6 Level in Critically Ill Patients With Coronavirus Disease 2019. Clin. Infect. Dis. 2020, 71, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, S.; Moosaie, F.; Aarabi, M.H. Understanding the Immunologic Characteristics of Neurologic Manifestations of SARS-CoV-2 and Potential Immunological Mechanisms. Mol. Neurobiol. 2020, 57, 5263–5275. [Google Scholar] [CrossRef]
- Cauchois, R.; Koubi, M.; Delarbre, D.; Manet, C.; Carvelli, J.; Blasco, V.B.; Jean, R.; Fouche, L.; Bornet, C.; Pauly, V.; et al. Early IL-1 receptor blockade in severe inflammatory respiratory failure complicating COVID-19. Proc. Natl. Acad. Sci. USA. 2020, 117, 18951–18953. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.L.; Yuen, K.S.; Castaño-Rodriguez, C.; Ye, Z.W.; Yeung, M.L.; Fung, S.Y.; Yuan, S.; Chan, C.P.; Yuen, K.Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef] [PubMed]
- Lara, P.C.; Macías-Verde, D.; Burgos-Burgos, J. Age-induced NLRP3 Inflammasome Over-activation Increases Lethality of SARS-CoV-2 Pneumonia in Elderly Patients. Aging Dis. 2020, 11, 756–762. [Google Scholar] [CrossRef]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Aβ clearance through NLRP3 inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef]
- Reynolds, J.L.; Mahajan, S.D. SARS-COV2 Alters Blood Brain Barrier Integrity Contributing to Neuro-Inflammation. J. Neuroimmune Pharmacol. 2021, 16, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Hanley, B.; Naresh, K.N.; Roufosse, C.; Nicholson, A.G.; Weir, J.; Cooke, G.S.; Thursz, M.; Manousou, P.; Corbett, R.; Goldin, R.; et al. Histopathological findings and viral tropism in UK patients with severe fatal COVID-19: A post-mortem study. Lancet Microbe 2020, 1, e245–e253. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Wang, H.; Qin, R.; Zhang, J.; Chen, Y. Possible immunity, inflammation, and oxidative stress mechanisms of Alzheimer’s disease in COVID-19 patients. Clin. Neurol. Neurosurg. 2021, 201, 106414. [Google Scholar] [CrossRef]
- Hoover, D.B. Cholinergic modulation of the immune system presents new approaches for treating inflammation. Pharmacol. Ther. 2017, 179, 1–16. [Google Scholar] [CrossRef]
- Geula, C.; Nagykery, N.; Nicholas, A.; Wu, C.K. Cholinergic neuronal and axonal abnormalities are present early in aging and in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2008, 67, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Stanciu, G.D.; Luca, A.; Rusu, R.N.; Bild, V.; Beschea Chiriac, S.I.; Solcan, C.; Bild, W.; Ababei, D.C. Alzheimer’s Disease Pharmacotherapy in Relation to Cholinergic System Involvement. Biomolecules 2019, 10, 40. [Google Scholar] [CrossRef]
- Villa, C.; Ferini-Strambi, L.; Combi, R. The Synergistic Relationship between Alzheimer’s Disease and Sleep Disorders: An Update. J. Alzheimers Dis. 2015, 46, 571–580. [Google Scholar] [CrossRef]
- Chowdhury, P.; Pathak, P. Neuroprotective immunity by essential nutrient “Choline” for the prevention of SARS CoV2 infections: An in silico study by molecular dynamics approach. Chem. Phys. Lett. 2020, 761, 138057. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, E.M.; Ivanova, E.; Grechko, A.V.; Wu, W.K.; Starodubova, A.V.; Orekhov, A.N. Involvement of Oxidative Stress and the Innate Immune System in SARS-CoV-2 Infection. Diseases 2021, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, R.; Cecchini, A.L. SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med. Hypotheses 2020, 143, 110102. [Google Scholar] [CrossRef]
- Beltrán-García, J.; Osca-Verdegal, R.; Pallardó, F.V.; Ferreres, J.; Rodríguez, M.; Mulet, S.; Sanchis-Gomar, F.; Carbonell, N.; García-Giménez, J.L. Oxidative Stress and Inflammation in COVID-19-Associated Sepsis: The Potential Role of Anti-Oxidant Therapy in Avoiding Disease Progression. Antioxidants 2020, 9, 936. [Google Scholar] [CrossRef] [PubMed]
- Karkhanei, B.; Ghane, E.T.; Mehri, F. Evaluation of oxidative stress level: Total antioxidant capacity, total oxidant status and glutathione activity in patients with Covid-19. New Microbes New Infect. 2021, 42, 100897. [Google Scholar] [CrossRef]
- Singh, K.K.; Chaubey, G.; Chen, J.Y.; Suravajhala, P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am. J. Physiol. Cell Physiol. 2020, 319, C258–C267. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Y.; Xiao, Z.G.; Zou, C.; Zhang, X.; Wang, Z.; Wu, D.; Yu, S.; Chan, F.L. Nuclear receptor ERRα and transcription factor ERG form a reciprocal loop in the regulation of TMPRSS2:ERG fusion gene in prostate cancer. Oncogene 2018, 37, 6259–6274. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Saleh, J.; Peyssonnaux, C.; Singh, K.K.; Edeas, M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion 2020, 54, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.; Cevenini, E.; Nasi, M.; De Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 2014, 44, 1552–1562. [Google Scholar] [CrossRef]
- Banchini, F.; Cattaneo, G.M.; Capelli, P. Serum ferritin levels in inflammation: A retrospective comparative analysis between COVID-19 and emergency surgical non-COVID-19 patients. World J. Emerg. Surg. 2021, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef]
- Ayton, S.; Faux, N.G.; Bush, A.I. Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat. Commun. 2015, 6, 6760. [Google Scholar] [CrossRef]
- Wu, K.E.; Fazal, F.M.; Parker, K.R.; Zou, J.; Chang, H.Y. RNA-GPS Predicts SARS-CoV-2 RNA Residency to Host Mitochondria and Nucleolus. Cell Syst. 2020, 11, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Zhang, F.; Lui, G.C.Y.; Yeoh, Y.K.; Li, A.Y.L.; Zhan, H.; Wan, Y.; Chung, A.C.K.; Cheung, C.P.; Chen, N.; et al. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology 2020, 159, 944–955. [Google Scholar] [CrossRef]
- Wu, Y.; Cheng, X.; Jiang, G.; Tang, H.; Ming, S.; Tang, L.; Lu, J.; Guo, C.; Shan, H.; Huang, X. Altered oral and gut microbiota and its association with SARS-CoV-2 viral load in COVID-19 patients during hospitalization. NPJ Biofilms Microbiomes 2021, 7, 61. [Google Scholar] [CrossRef]
- Gu, S.; Chen, Y.; Wu, Z.; Gao, H.; Lv, L.; Guo, F.; Zhang, X.; Luo, R.; Huang, C.; Lu, H.; et al. Alterations of the Gut Microbiota in Patients With Coronavirus Disease 2019 or H1N1 Influenza. Clin. Infect. Dis. 2020, 71, 2669–2678. [Google Scholar] [CrossRef]
- Yeoh, Y.K.; Zuo, T.; Lui, G.C.; Zhang, F.; Liu, Q.; Li, A.Y.; Chung, A.C.; Cheung, C.P.; Tso, E.Y.; Fung, K.S.; et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 2021, 70, 698–706. [Google Scholar] [CrossRef]
- Prasad, R.; Patton, M.J.; Floyd, J.L.; Vieira, C.P.; Fortmann, S.; DuPont, M.; Harbour, A.; Jeremy, C.S.; Wright, J.; Lamendella, R.; et al. Plasma microbiome in COVID-19 subjects: An indicator of gut barrier defects and dysbiosis. Biorxiv 2021. [Google Scholar] [CrossRef]
- Xu, W.; Luo, Z.; Alekseyenko, A.V.; Martin, L.; Wan, Z.; Ling, B.; Qin, Z.; Heath, S.L.; Maas, K.; Cong, X.; et al. Distinct systemic microbiome and microbial translocation are associated with plasma level of anti-CD4 autoantibody in HIV infection. Sci. Rep. 2018, 8, 12863. [Google Scholar] [CrossRef]
- Luo, Z.; Li, M.; Wu, Y.; Meng, Z.; Martin, L.; Zhang, L.; Ogunrinde, E.; Zhou, Z.; Qin, S.; Wan, Z.; et al. Systemic translocation of Staphylococcus drives autoantibody production in HIV disease. Microbiome 2019, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Dinh, D.M.; Volpe, G.E.; Duffalo, C.; Bhalchandra, S.; Tai, A.K.; Kane, A.V.; Wanke, C.A.; Ward, H.D. Intestinal microbiota, microbial translocation, and systemic inflammation in chronic HIV infection. J. Infect. Dis. 2015, 211, 19–27. [Google Scholar] [CrossRef]
- Villanueva-Millán, M.J.; Pérez-Matute, P.; Recio-Fernández, E.; Lezana Rosales, J.M.; Oteo, J.A. Differential effects of antiretrovirals on microbial translocation and gut microbiota composition of HIV-infected patients. J. Int. AIDS Soc. 2017, 20, 21526. [Google Scholar] [CrossRef] [PubMed]
- Manosso, L.M.; Arent, C.O.; Borba, L.A.; Ceretta, L.B.; Quevedo, J.; Réus, G.Z. Microbiota-Gut-Brain Communication in the SARS-CoV-2 Infection. Cells 2021, 10, 1993. [Google Scholar] [CrossRef]
- Cattaneo, A.; Cattane, N.; Galluzzi, S.; Provasi, S.; Lopizzo, N.; Festari, C.; Ferrari, C.; Guerra, U.P.; Paghera, B.; Muscio, C.; et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 2017, 49, 60–68. [Google Scholar] [CrossRef]
- Zhuang, Z.Q.; Shen, L.L.; Li, W.W.; Fu, X.; Zeng, F.; Gui, L.; Lü, Y.; Cai, M.; Zhu, C.; Tan, Y.L.; et al. Gut Microbiota is Altered in Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2018, 63, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Haran, J.P.; Bhattarai, S.K.; Foley, S.E.; Dutta, P.; Ward, D.V.; Bucci, V.; McCormick, B.A. Alzheimer’s Disease Microbiome Is Associated with Dysregulation of the Anti-Inflammatory P-Glycoprotein Pathway. MBio 2019, 10, e00632-19. [Google Scholar] [CrossRef]
- Lundmark, K.; Westermark, G.T.; Olsén, A.; Westermark, P. Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism. Proc. Natl. Acad. Sci. USA 2005, 102, 6098–6102. [Google Scholar] [CrossRef] [PubMed]
- Mezö, C.; Dokalis, N.; Mossad, O.; Staszewski, O.; Neuber, J.; Yilmaz, B.; Schnepf, D.; de Agüero, M.G.; Ganal-Vonarburg, S.C.; Macpherson, A.J.; et al. Different effects of constitutive and induced microbiota modulation on microglia in a mouse model of Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 119. [Google Scholar] [CrossRef] [PubMed]
- Leblhuber, F.; Steiner, K.; Schuetz, B.; Fuchs, D.; Gostner, J.M. Probiotic Supplementation in Patients with Alzheimer’s Dementia—An Explorative Intervention Study. Curr. Alzheimer Res. 2018, 15, 1106–1113. [Google Scholar] [CrossRef]
- Dolatshahi, M.; Sabahi, M.; Aarabi, M.H. Pathophysiological Clues to How the Emergent SARS-CoV-2 Can Potentially Increase the Susceptibility to Neurodegeneration. Mol. Neurobiol. 2021, 58, 2379–2394. [Google Scholar] [CrossRef] [PubMed]
- Hassett, C.E.; Frontera, J.A. Neurologic aspects of coronavirus disease of 2019 infection. Curr. Opin. Infect. Dis. 2021, 34, 217–227. [Google Scholar] [CrossRef]
- de Erausquin, G.A.; Snyder, H.; Carrillo, M.; Hosseini, A.A.; Brugha, T.S.; Seshadri, S. The chronic neuropsychiatric sequelae of COVID-19: The need for a prospective study of viral impact on brain functioning. Alzheimers Dement. 2021, 17, 1056–1065. [Google Scholar] [CrossRef]
- Padala, K.P.; Parkes, C.M.; Padala, P.R. Neuropsychological and Functional Impact of COVID-19 on Mild Cognitive Impairment. Am. J. Alzheimers Dis Other Dement. 2020, 35, 1533317520960875. [Google Scholar] [CrossRef]
- Brown, E.E.; Kumar, S.; Rajji, T.K.; Pollock, B.G.; Mulsant, B.H. Anticipating and Mitigating the Impact of the COVID-19 Pandemic on Alzheimer’s Disease and Related Dementias. Am. J. Geriatr. Psychiatry 2020, 28, 712–721. [Google Scholar] [CrossRef]
- Wang, J.H.; Wu, Y.J.; Tee, B.L.; Lo, R.Y. Medical Comorbidity in Alzheimer’s Disease: A Nested Case-Control Study. J. Alzheimers Dis. 2018, 63, 773–781. [Google Scholar] [CrossRef]
- Pyne, J.D.; Brickman, A.M. The Impact of the COVID-19 Pandemic on Dementia Risk: Potential Pathways to Cognitive Decline. Neurodegener. Dis. 2021, 21, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Magusali, N.; Graham, A.C.; Piers, T.M.; Panichnantakul, P.; Yaman, U.; Shoai, M.; Reynolds, R.H.; Botia, J.A.; Brookes, K.J.; Guetta-Baranes, T.; et al. A genetic link between risk for Alzheimer’s disease and severe COVID-19 outcomes via the OAS1 gene. Brain 2021, 144, 3727–3741. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, S.; Rheinstein, P.H. BIN1 rs744373 SNP and COVID-19 mortality. World Acad. Sci. J. 2021, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.C.; Kern, F.; Losada, P.M.; Agam, M.R.; Maat, C.A.; Schmartz, G.P.; Fehlmann, T.; Stein, J.A.; Schaum, N.; Lee, D.P.; et al. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 2021, 595, 565–571. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).