
The Functionality of UDP-Glucuronosyltransferase Genetic Variants and their Association with Drug Responses and Human Diseases



Abstract
1. Introduction
2. UGT Isoforms and Genes
3. Expression of UGT Isoforms
4. The Role of UGTs in Xenobiotic Metabolism
5. Factors Affecting UGT Activity
6. The Clinical Impact of UGT1A Genotype on Drug Response and Toxicity
7. The Clinical Impact of the UGT2B7 Genotype on Drug Responses and Toxicity
8. The Role of UGTs in Endogenous Metabolism and Susceptibility to Human Diseases
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tukey, R.H.; Strassburg, C.P. Human UDP-glucuronosyltransferases: Metabolism, expression, and disease. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 581–616. [Google Scholar] [CrossRef]
- Miners, J.O.; Mackenzie, P.I.; Knights, K.M. The prediction of drug-glucuronidation parameters in humans: UDP-glucuronosyltransferase enzyme-selective substrate and inhibitor probes for reaction phenotyping and in vitro-in vivo extrapolation of drug clearance and drug-drug interaction potential. Drug Metab. Rev. 2010, 42, 196–208. [Google Scholar] [CrossRef]
- Rowland, A.; Miners, J.O.; Mackenzie, P.I. The UDP-glucuronosyltransferases: Their role in drug metabolism and detoxification. Int. J. Biochem. Cell Biol. 2013, 45, 1121–1132. [Google Scholar] [CrossRef]
- Perreault, M.; Wunsch, E.; Bialek, A.; Trottier, J.; Verreault, M.; Caron, P.; Poirier, G.G.; Milkiewicz, P.; Barbier, O. Urinary Elimination of Bile Acid Glucuronides under Severe Cholestatic Situations: Contribution of Hepatic and Renal Glucuronidation Reactions. Can. J. Gastroenterol. Hepatol. 2018, 2018, 8096314. [Google Scholar] [CrossRef]
- Klimas, R.; Mikus, G. Morphine-6-glucuronide is responsible for the analgesic effect after morphine administration: A quantitative review of morphine, morphine-6-glucuronide, and morphine-3-glucuronide. Br. J. Anaesth. 2014, 113, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Jarrar, Y.; Jarrar, Q.; Abu-Shalhoob, M.; Abed, A.; Sha’ban, E. Relative Expression of Mouse Udp-glucuronosyl Transferase 2b1 Gene in the Livers, Kidneys, and Hearts: The Influence of Nonsteroidal Anti-inflammatory Drug Treatment. Curr. Drug Metab. 2019, 20, 918–923. [Google Scholar] [CrossRef] [PubMed]
- Mays, D.C.; Dixon, K.F.; Balboa, A.; Pawluk, L.J.; Bauer, M.R.; Nawoot, S.; Gerber, N. A nonprimate animal model applicable to zidovudine pharmacokinetics in humans: Inhibition of glucuronidation and renal excretion of zidovudine by probenecid in rats. J. Pharmacol. Exp. Ther. 1991, 259, 1261–1270. [Google Scholar] [PubMed]
- Oguri, K.; Hanioka, N.; Yoshimura, H. Species differences in metabolism of codeine: Urinary excretion of codeine glucuronide, morphine-3-glucuronide and morphine-6-glucuronide in mice, rats, guinea pigs and rabbits. Xenobiotica 1990, 20, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Court, M.H. Feline drug metabolism and disposition: Pharmacokinetic evidence for species differences and molecular mechanisms. Vet. Clin. N. Am. Small Anim. Pract. 2013, 43, 1039–1054. [Google Scholar] [CrossRef]
- Miners, J.O.; Smith, P.A.; Sorich, M.J.; McKinnon, R.A.; Mackenzie, P.I. Predicting human drug glucuronidation parameters: Application of in vitro and in silico modeling approaches. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 1–25. [Google Scholar] [CrossRef]
- Mackenzie, P.I.; Bock, K.W.; Burchell, B.; Guillemette, C.; Ikushiro, S.; Iyanagi, T.; Miners, J.O.; Owens, I.S.; Nebert, D.W. Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily. Pharmacogenet Genomics 2005, 15, 677–685. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, P.I.; Rogers, A.; Elliot, D.J.; Chau, N.; Hulin, J.A.; Miners, J.O.; Meech, R. The novel UDP glycosyltransferase 3A2: Cloning, catalytic properties, and tissue distribution. Mol. Pharmacol. 2011, 79, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Kiang, T.K.; Ensom, M.H.; Chang, T.K. UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharmacol. Ther. 2005, 106, 97–132. [Google Scholar] [CrossRef] [PubMed]
- Court, M.H.; Zhang, X.; Ding, X.; Yee, K.K.; Hesse, L.M.; Finel, M. Quantitative distribution of mRNAs encoding the 19 human UDP-glucuronosyltransferase enzymes in 26 adult and 3 fetal tissues. Xenobiotica 2012, 42, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Izukawa, T.; Nakajima, M.; Fujiwara, R.; Yamanaka, H.; Fukami, T.; Takamiya, M.; Aoki, Y.; Ikushiro, S.; Sakaki, T.; Yokoi, T. Quantitative analysis of UDP-glucuronosyltransferase (UGT) 1A and UGT2B expression levels in human livers. Drug Metab. Dispos. 2009, 37, 1759–1768. [Google Scholar] [CrossRef]
- Gaganis, P.; Miners, J.O.; Knights, K.M. Glucuronidation of fenamates: Kinetic studies using human kidney cortical microsomes and recombinant UDP-glucuronosyltransferase (UGT) 1A9 and 2B7. Biochem. Pharmacol. 2007, 73, 1683–1691. [Google Scholar] [CrossRef]
- Ohno, S.; Nakajin, S. Determination of mRNA expression of human UDP-glucuronosyltransferases and application for localization in various human tissues by real-time reverse transcriptase-polymerase chain reaction. Drug Metab. Dispos. 2009, 37, 32–40. [Google Scholar] [CrossRef]
- Radominska-Pandya, A.; Czernik, P.J.; Little, J.M.; Battaglia, E.; Mackenzie, P.I. Structural and functional studies of UDP-glucuronosyltransferases. Drug Metab. Rev. 1999, 31, 817–899. [Google Scholar] [CrossRef]
- Tien, E.S.; Negishi, M. Nuclear receptors CAR and PXR in the regulation of hepatic metabolism. Xenobiotica 2006, 36, 1152–1163. [Google Scholar] [CrossRef]
- Mackenzie, P.I.; Hu, D.G.; Gardner-Stephen, D.A. The regulation of UDP-glucuronosyltransferase genes by tissue-specific and ligand-activated transcription factors. Drug Metab. Rev. 2010, 42, 99–109. [Google Scholar] [CrossRef]
- Sugatani, J.; Nishitani, S.; Yamakawa, K.; Yoshinari, K.; Sueyoshi, T.; Negishi, M.; Miwa, M. Transcriptional regulation of human UGT1A1 gene expression: Activated glucocorticoid receptor enhances constitutive androstane receptor/pregnane X receptor-mediated UDP-glucuronosyltransferase 1A1 regulation with glucocorticoid receptor-interacting protein 1. Mol. Pharmacol. 2005, 67, 845–855. [Google Scholar] [PubMed]
- Gardner-Stephen, D.; Heydel, J.M.; Goyal, A.; Lu, Y.; Xie, W.; Lindblom, T.; Mackenzie, P.; Radominska-Pandya, A. Human PXR variants and their differential effects on the regulation of human UDP-glucuronosyltransferase gene expression. Drug Metab. Dispos. 2004, 32, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Barbier, O.; Duran-Sandoval, D.; Pineda-Torra, I.; Kosykh, V.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptor alpha induces hepatic expression of the human bile acid glucuronidating UDP-glucuronosyltransferase 2B4 enzyme. J. Biol. Chem. 2003, 278, 32852–32860. [Google Scholar] [CrossRef]
- Lu, Y.; Heydel, J.M.; Li, X.; Bratton, S.; Lindblom, T.; Radominska-Pandya, A. Lithocholic acid decreases expression of UGT2B7 in Caco-2 cells: A potential role for a negative farnesoid X receptor response element. Drug Metab. Dispos. 2005, 33, 937–946. [Google Scholar] [CrossRef]
- Verreault, M.; Senekeo-Effenberger, K.; Trottier, J.; Bonzo, J.A.; Belanger, J.; Kaeding, J.; Staels, B.; Caron, P.; Tukey, R.H.; Barbier, O. The liver X-receptor alpha controls hepatic expression of the human bile acid-glucuronidating UGT1A3 enzyme in human cells and transgenic mice. Hepatology 2006, 44, 368–378. [Google Scholar] [CrossRef]
- Senekeo-Effenberger, K.; Chen, S.; Brace-Sinnokrak, E.; Bonzo, J.A.; Yueh, M.F.; Argikar, U.; Kaeding, J.; Trottier, J.; Remmel, R.P.; Ritter, J.K.; et al. Expression of the human UGT1 locus in transgenic mice by 4-chloro-6-(2,3-xylidino)-2-pyrimidinylthioacetic acid (WY-14643) and implications on drug metabolism through peroxisome proliferator-activated receptor alpha activation. Drug Metab. Dispos. 2007, 35, 419–427. [Google Scholar] [CrossRef]
- Lankisch, T.O.; Gillman, T.C.; Erichsen, T.J.; Ehmer, U.; Kalthoff, S.; Freiberg, N.; Munzel, P.A.; Manns, M.P.; Strassburg, C.P. Aryl hydrocarbon receptor-mediated regulation of the human estrogen and bile acid UDP-glucuronosyltransferase 1A3 gene. Arch. Toxicol. 2008, 82, 573–582. [Google Scholar] [CrossRef]
- Hu, D.G.; Mackenzie, P.I. Estrogen receptor alpha, fos-related antigen-2, and c-Jun coordinately regulate human UDP glucuronosyltransferase 2B15 and 2B17 expression in response to 17beta-estradiol in MCF-7 cells. Mol. Pharmacol. 2009, 76, 425–439. [Google Scholar] [CrossRef]
- Ohno, A.; Saito, Y.; Hanioka, N.; Jinno, H.; Saeki, M.; Ando, M.; Ozawa, S.; Sawada, J. Involvement of human hepatic UGT1A1, UGT2B4, and UGT2B7 in the glucuronidation of carvedilol. Drug Metab. Dispos. 2004, 32, 235–239. [Google Scholar] [CrossRef]
- Watanabe, Y.; Nakajima, M.; Ohashi, N.; Kume, T.; Yokoi, T. Glucuronidation of etoposide in human liver microsomes is specifically catalyzed by UDP-glucuronosyltransferase 1A1. Drug Metab. Dispos. 2003, 31, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tracy, T.S.; Remmel, R.P. Correlation between bilirubin glucuronidation and estradiol-3-gluronidation in the presence of model UDP-glucuronosyltransferase 1A1 substrates/inhibitors. Drug Metab. Dispos. 2011, 39, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Nguyen, N.; Peterkin, V.; Yang, Y.S.; Hotz, K.; La Placa, D.B.; Chen, S.; Tukey, R.H.; Stevens, J.C. A humanized UGT1 mouse model expressing the UGT1A1*28 allele for assessing drug clearance by UGT1A1-dependent glucuronidation. Drug Metab. Dispos. 2010, 38, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Saka, H.; Asai, G.; Sugiura, S.; Shimokata, K.; Kamataki, T. UGT1A1 genotypes and glucuronidation of SN-38, the active metabolite of irinotecan. Ann. Oncol. 1998, 9, 845–847. [Google Scholar] [CrossRef]
- Ghosal, A.; Hapangama, N.; Yuan, Y.; Achanfuo-Yeboah, J.; Iannucci, R.; Chowdhury, S.; Alton, K.; Patrick, J.E.; Zbaida, S. Identification of human UDP-glucuronosyltransferase enzyme(s) responsible for the glucuronidation of ezetimibe (Zetia). Drug Metab. Dispos. 2004, 32, 314–320. [Google Scholar] [CrossRef]
- Gill, K.L.; Houston, J.B.; Galetin, A. Characterization of in vitro glucuronidation clearance of a range of drugs in human kidney microsomes: Comparison with liver and intestinal glucuronidation and impact of albumin. Drug Metab. Dispos. 2012, 40, 825–835. [Google Scholar] [CrossRef]
- Breyer-Pfaff, U.; Mey, U.; Green, M.D.; Tephly, T.R. Comparative N-glucuronidation kinetics of ketotifen and amitriptyline by expressed human UDP-glucuronosyltransferases and liver microsomes. Drug Metab. Dispos. 2000, 28, 869–872. [Google Scholar] [PubMed]
- Rowland, A.; Elliot, D.J.; Williams, J.A.; Mackenzie, P.I.; Dickinson, R.G.; Miners, J.O. In vitro characterization of lamotrigine N2-glucuronidation and the lamotrigine-valproic acid interaction. Drug Metab. Dispos. 2006, 34, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Hyland, R.; Osborne, T.; Payne, A.; Kempshall, S.; Logan, Y.R.; Ezzeddine, K.; Jones, B. In vitro and in vivo glucuronidation of midazolam in humans. Br. J. Clin. Pharmacol. 2009, 67, 445–454. [Google Scholar] [CrossRef]
- Haslemo, T.; Loryan, I.; Ueda, N.; Mannheimer, B.; Bertilsson, L.; Ingelman-Sundberg, M.; Molden, E.; Eliasson, E. UGT1A4*3 encodes significantly increased glucuronidation of olanzapine in patients on maintenance treatment and in recombinant systems. Clin. Pharmacol. Ther. 2012, 92, 221–227. [Google Scholar] [CrossRef]
- Uchaipichat, V.; Mackenzie, P.I.; Elliot, D.J.; Miners, J.O. Selectivity of substrate (trifluoperazine) and inhibitor (amitriptyline, androsterone, canrenoic acid, hecogenin, phenylbutazone, quinidine, quinine, and sulfinpyrazone) "probes" for human udp-glucuronosyltransferases. Drug Metab. Dispos. 2006, 34, 449–456. [Google Scholar] [CrossRef]
- Benoit-Biancamano, M.O.; Connelly, J.; Villeneuve, L.; Caron, P.; Guillemette, C. Deferiprone glucuronidation by human tissues and recombinant UDP glucuronosyltransferase 1A6: An in vitro investigation of genetic and splice variants. Drug Metab. Dispos. 2009, 37, 322–329. [Google Scholar] [CrossRef]
- Kostrubsky, S.E.; Sinclair, J.F.; Strom, S.C.; Wood, S.; Urda, E.; Stolz, D.B.; Wen, Y.H.; Kulkarni, S.; Mutlib, A. Phenobarbital and phenytoin increased acetaminophen hepatotoxicity due to inhibition of UDP-glucuronosyltransferases in cultured human hepatocytes. Toxicol. Sci. 2005, 87, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Matsumoto, Y.; Yamazaki, H. Effects of propofol analogs on glucuronidation of propofol, an anesthetic drug, by human liver microsomes. Drug Metab. Lett. 2007, 1, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Lautala, P.; Ethell, B.T.; Taskinen, J.; Burchell, B. The specificity of glucuronidation of entacapone and tolcapone by recombinant human UDP-glucuronosyltransferases. Drug Metab. Dispos. 2000, 28, 1385–1389. [Google Scholar]
- Kuehl, G.E.; Lampe, J.W.; Potter, J.D.; Bigler, J. Glucuronidation of nonsteroidal anti-inflammatory drugs: Identifying the enzymes responsible in human liver microsomes. Drug Metab. Dispos. 2005, 33, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Bernard, O.; Guillemette, C. The main role of UGT1A9 in the hepatic metabolism of mycophenolic acid and the effects of naturally occurring variants. Drug Metab. Dispos. 2004, 32, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Court, M.H.; Duan, S.X.; Guillemette, C.; Journault, K.; Krishnaswamy, S.; Von Moltke, L.L.; Greenblatt, D.J. Stereoselective conjugation of oxazepam by human UDP-glucuronosyltransferases (UGTs): S-oxazepam is glucuronidated by UGT2B15, while R-oxazepam is glucuronidated by UGT2B7 and UGT1A9. Drug Metab. Dispos. 2002, 30, 1257–1265. [Google Scholar] [CrossRef]
- Court, M.H.; Krishnaswamy, S.; Hao, Q.; Duan, S.X.; Patten, C.J.; Von Moltke, L.L.; Greenblatt, D.J. Evaluation of 3’-azido-3’-deoxythymidine, morphine, and codeine as probe substrates for UDP-glucuronosyltransferase 2B7 (UGT2B7) in human liver microsomes: Specificity and influence of the UGT2B7*2 polymorphism. Drug Metab. Dispos. 2003, 31, 1125–1133. [Google Scholar] [CrossRef]
- Innocenti, F.; Iyer, L.; Ramirez, J.; Green, M.D.; Ratain, M.J. Epirubicin glucuronidation is catalyzed by human UDP-glucuronosyltransferase 2B7. Drug Metab. Dispos. 2001, 29, 686–692. [Google Scholar]
- Coffman, B.L.; Rios, G.R.; King, C.D.; Tephly, T.R. Human UGT2B7 catalyzes morphine glucuronidation. Drug Metab. Dispos. 1997, 25, 1–4. [Google Scholar]
- Di Marco, A.; D’Antoni, M.; Attaccalite, S.; Carotenuto, P.; Laufer, R. Determination of drug glucuronidation and UDP-glucuronosyltransferase selectivity using a 96-well radiometric assay. Drug Metab. Dispos. 2005, 33, 812–819. [Google Scholar] [CrossRef]
- Barbier, O.; Turgeon, D.; Girard, C.; Green, M.D.; Tephly, T.R.; Hum, D.W.; Belanger, A. 3’-azido-3’-deoxythimidine (AZT) is glucuronidated by human UDP-glucuronosyltransferase 2B7 (UGT2B7). Drug Metab. Dispos. 2000, 28, 497–502. [Google Scholar]
- Uchaipichat, V.; Suthisisang, C.; Miners, J.O. The glucuronidation of R- and S-lorazepam: Human liver microsomal kinetics, UDP-glucuronosyltransferase enzyme selectivity, and inhibition by drugs. Drug Metab. Dispos. 2013, 41, 1273–1284. [Google Scholar] [CrossRef]
- Paulson, S.K.; Hribar, J.D.; Liu, N.W.; Hajdu, E.; Bible, R.H., Jr.; Piergies, A.; Karim, A. Metabolism and excretion of [(14)C]celecoxib in healthy male volunteers. Drug Metab. Dispos. 2000, 28, 308–314. [Google Scholar]
- Zhang, J.Y.; Zhan, J.; Cook, C.S.; Ings, R.M.; Breau, A.P. Involvement of human UGT2B7 and 2B15 in rofecoxib metabolism. Drug Metab. Dispos. 2003, 31, 652–658. [Google Scholar] [CrossRef]
- Lu, Y.; Zhu, J.; Chen, X.; Li, N.; Fu, F.; He, J.; Wang, G.; Zhang, L.; Zheng, Y.; Qiu, Z.; et al. Identification of human UDP-glucuronosyltransferase isoforms responsible for the glucuronidation of glycyrrhetinic acid. Drug Metab. Pharmacokinet. 2009, 24, 523–528. [Google Scholar] [CrossRef]
- He, Y.Q.; Liu, Y.; Zhang, B.F.; Liu, H.X.; Lu, Y.L.; Yang, L.; Xiong, A.Z.; Xu, L.L.; Wang, C.H.; Yang, L.; et al. Identification of the UDP-glucuronosyltransferase isozyme involved in senecionine glucuronidation in human liver microsomes. Drug Metab. Dispos. 2010, 38, 626–634. [Google Scholar] [CrossRef]
- Owens, K.H.; Murphy, P.G.; Medlicott, N.J.; Kennedy, J.; Zacharias, M.; Curran, N.; Sreebhavan, S.; Thompson-Fawcett, M.; Reith, D.M. Population pharmacokinetics of intravenous acetaminophen and its metabolites in major surgical patients. J. Pharmacokinet. Pharmacodyn. 2014, 41, 211–221. [Google Scholar] [CrossRef]
- Miyagi, S.J.; Collier, A.C. Pediatric development of glucuronidation: The ontogeny of hepatic UGT1A4. Drug Metab. Dispos. 2007, 35, 1587–1592. [Google Scholar] [CrossRef]
- Court, M.H. Interindividual variability in hepatic drug glucuronidation: Studies into the role of age, sex, enzyme inducers, and genetic polymorphism using the human liver bank as a model system. Drug Metab. Rev. 2009, 42, 209–224. [Google Scholar] [CrossRef]
- Furlan, V.; Demirdjian, S.; Bourdon, O.; Magdalou, J.; Taburet, A.M. Glucuronidation of drugs by hepatic microsomes derived from healthy and cirrhotic human livers. J. Pharmacol. Exp. Ther. 1999, 289, 1169–1175. [Google Scholar]
- Ye, L.; Yang, X.; Guo, E.; Chen, W.; Lu, L.; Wang, Y.; Peng, X.; Yan, T.; Zhou, F.; Liu, Z. Sorafenib metabolism is significantly altered in the liver tumor tissue of hepatocellular carcinoma patient. PLoS ONE 2014, 9, e96664. [Google Scholar] [CrossRef]
- Jarrar, Y.B.; Al-Essa, L.; Kilani, A.; Hasan, M.; Al-Qerem, W. Alterations in the gene expression of drug and arachidonic acid-metabolizing Cyp450 in the livers of controlled and uncontrolled insulin-dependent diabetic mice. Diabetes Metab. Syndr. Obes. 2018, 11, 483–492. [Google Scholar] [CrossRef]
- Dostalek, M.; Court, M.H.; Hazarika, S.; Akhlaghi, F. Diabetes mellitus reduces activity of human UDP-glucuronosyltransferase 2B7 in liver and kidney leading to decreased formation of mycophenolic acid acyl-glucuronide metabolite. Drug Metab. Dispos. 2011, 39, 448–455. [Google Scholar] [CrossRef]
- Gallagher, C.J.; Balliet, R.M.; Sun, D.; Chen, G.; Lazarus, P. Sex differences in UDP-glucuronosyltransferase 2B17 expression and activity. Drug Metab. Dispos. 2010, 38, 2204–2209. [Google Scholar] [CrossRef]
- Picard, N.; Ratanasavanh, D.; Premaud, A.; Le Meur, Y.; Marquet, P. Identification of the UDP-glucuronosyltransferase isoforms involved in mycophenolic acid phase II metabolism. Drug Metab. Dispos. 2005, 33, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Rae, J.M.; Drury, S.; Hayes, D.F.; Stearns, V.; Thibert, J.N.; Haynes, B.P.; Salter, J.; Sestak, I.; Cuzick, J.; Dowsett, M. CYP2D6 and UGT2B7 genotype and risk of recurrence in tamoxifen-treated breast cancer patients. J. Natl. Cancer Inst. 2012, 104, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.K.; Aithal, G.P.; Leathart, J.B.; Swainsbury, R.A.; Dang, T.S.; Day, C.P. Genetic susceptibility to diclofenac-induced hepatotoxicity: Contribution of UGT2B7, CYP2C8, and ABCC2 genotypes. Gastroenterology 2007, 132, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.; Das, S.; Janisch, L.; Wen, M.; Ramirez, J.; Karrison, T.; Fleming, G.F.; Vokes, E.E.; Schilsky, R.L.; Ratain, M.J. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J. 2002, 2, 43–47. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, D.; Kuang, Q.; Liu, G.; Xu, W. Association between UGT1A1*28 polymorphisms and clinical outcomes of irinotecan-based chemotherapies in colorectal cancer: A meta-analysis in Caucasians. PLoS ONE 2013, 8, e58489. [Google Scholar]
- Jarrar, Y.B.; Kim, D.H.; Lee, S.J.; Shin, J.G. Inhibition of 20-hydroxyeicosatetraenoic acid (20-HETE) glucuronidation by non-steroidal anti-inflammatory drugs in human liver microsomes and recombinant UDP-glucuronosyltransferase enzymes. Prostaglandins Leukot. Essent. Fatty Acids 2020, 153, 102055. [Google Scholar] [CrossRef] [PubMed]
- Burger, D.M.; Meenhorst, P.L.; Koks, C.H.; Beijnen, J.H. Pharmacokinetic interaction between rifampin and zidovudine. Antimicrob. Agents Chemother. 1993, 37, 1426–1431. [Google Scholar] [CrossRef]
- Benowitz, N.L. Cigarette smoking and the personalization of irinotecan therapy. J. Clin. Oncol. 2007, 25, 2646–2647. [Google Scholar] [CrossRef]
- Whirl-Carrillo, M.; McDonagh, E.M.; Hebert, J.M.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Altman, R.B.; Klein, T.E. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012, 92, 414–417. [Google Scholar] [CrossRef]
- Choe, P.G.; Park, W.B.; Song, J.S.; Kim, N.H.; Song, K.H.; Park, S.W.; Kim, H.B.; Kim, N.J.; Oh, M.D. Incidence of atazanavir-associated hyperbilirubinemia in Korean HIV patients: 30 months follow-up results in a population with low UDP-glucuronosyltransferase1A1*28 allele frequency. J. Korean Med. Sci. 2010, 25, 1427–1430. [Google Scholar] [CrossRef]
- Onoue, M.; Terada, T.; Kobayashi, M.; Katsura, T.; Matsumoto, S.; Yanagihara, K.; Nishimura, T.; Kanai, M.; Teramukai, S.; Shimizu, A.; et al. UGT1A1*6 polymorphism is most predictive of severe neutropenia induced by irinotecan in Japanese cancer patients. Int. J. Clin. Oncol. 2009, 14, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.B.; Smith, D.M.; Shaak, T.L. Genomic sequencing of uric acid metabolizing and clearing genes in relationship to xanthine oxidase inhibitor dose. Rheumatol. Int. 2017, 37, 445–453. [Google Scholar] [CrossRef]
- Takekuma, Y.; Takenaka, T.; Kiyokawa, M.; Yamazaki, K.; Okamoto, H.; Kitabatake, A.; Tsutsui, H.; Sugawara, M. Contribution of polymorphisms in UDP-glucuronosyltransferase and CYP2D6 to the individual variation in disposition of carvedilol. J. Pharm. Pharm. Sci. 2006, 9, 101–112. [Google Scholar]
- Lankisch, T.O.; Moebius, U.; Wehmeier, M.; Behrens, G.; Manns, M.P.; Schmidt, R.E.; Strassburg, C.P. Gilbert’s disease and atazanavir: From phenotype to UDP-glucuronosyltransferase haplotype. Hepatology 2006, 44, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Hirvensalo, P.; Tornio, A.; Neuvonen, M.; Tapaninen, T.; Paile-Hyvarinen, M.; Karja, V.; Mannisto, V.T.; Pihlajamaki, J.; Backman, J.T.; Niemi, M. Comprehensive Pharmacogenomic Study Reveals an Important Role of UGT1A3 in Montelukast Pharmacokinetics. Clin. Pharmacol. Ther. 2018, 104, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.K.; Oh, E.S.; Park, K.; Park, M.S.; Chung, J.Y. The UGT1A3*2 polymorphism affects atorvastatin lactonization and lipid-lowering effect in healthy volunteers. Pharmacogenet Genomics 2012, 22, 598–605. [Google Scholar] [CrossRef]
- Allegra, S.; Cusato, J.; De Francia, S.; Longo, F.; Pirro, E.; Massano, D.; Avataneo, V.; De Nicolo, A.; Piga, A.; D’Avolio, A. The effect of vitamin D pathway genes and deferasirox pharmacogenetics on liver iron in thalassaemia major patients. Pharmacogenomics J. 2019, 19, 417–427. [Google Scholar] [CrossRef]
- Levesque, E.; Belanger, A.S.; Harvey, M.; Couture, F.; Jonker, D.; Innocenti, F.; Cecchin, E.; Toffoli, G.; Guillemette, C. Refining the UGT1A haplotype associated with irinotecan-induced hematological toxicity in metastatic colorectal cancer patients treated with 5-fluorouracil/irinotecan-based regimens. J. Pharmacol. Exp. Ther. 2013, 345, 95–101. [Google Scholar] [CrossRef]
- Visscher, H.; Ross, C.J.; Rassekh, S.R.; Sandor, G.S.; Caron, H.N.; van Dalen, E.C.; Kremer, L.C.; van der Pal, H.J.; Rogers, P.C.; Rieder, M.J.; et al. Validation of variants in SLC28A3 and UGT1A6 as genetic markers predictive of anthracycline-induced cardiotoxicity in children. Pediatr. Blood Cancer 2013, 60, 1375–1381. [Google Scholar] [CrossRef]
- Dadheech, S.; Rao, A.V.; Shaheen, U.; Hussien, M.D.; Jain, S.; Jyothy, A.; Munshi, A. Three most common nonsynonymous UGT1A6*2 polymorphisms (Thr181Ala, Arg184Ser and Ser7Ala) and therapeutic response to deferiprone in beta-thalassemia major patients. Gene 2013, 531, 301–305. [Google Scholar] [CrossRef]
- Kim, S.Y.; Hong, Y.S.; Shim, E.K.; Kong, S.Y.; Shin, A.; Baek, J.Y.; Jung, K.H. S-1 plus irinotecan and oxaliplatin for the first-line treatment of patients with metastatic colorectal cancer: A prospective phase II study and pharmacogenetic analysis. Br. J. Cancer 2013, 109, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Woillard, J.B.; Rerolle, J.P.; Picard, N.; Rousseau, A.; Drouet, M.; Munteanu, E.; Essig, M.; Marquet, P.; Le Meur, Y. Risk of diarrhoea in a long-term cohort of renal transplant patients given mycophenolate mofetil: The significant role of the UGT1A8 2 variant allele. Br. J. Clin. Pharmacol. 2010, 69, 675–683. [Google Scholar] [CrossRef]
- Lu, Y.; Fang, Y.; Wu, X.; Ma, C.; Wang, Y.; Xu, L. Effects of UGT1A9 genetic polymorphisms on monohydroxylated derivative of oxcarbazepine concentrations and oxcarbazepine monotherapeutic efficacy in Chinese patients with epilepsy. Eur. J. Clin. Pharmacol. 2017, 73, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Lim, H.S.; Park, Y.H.; Lee, S.Y.; Lee, J.S. Integrated pharmacogenetic prediction of irinotecan pharmacokinetics and toxicity in patients with advanced non-small cell lung cancer. Lung Cancer 2009, 63, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Court, M.H.; Freytsis, M.; Wang, X.; Peter, I.; Guillemette, C.; Hazarika, S.; Duan, S.X.; Greenblatt, D.J.; Lee, W.M.; Acute Liver Failure Study Group. The UDP-glucuronosyltransferase (UGT) 1A polymorphism c.2042C>G (rs8330) is associated with increased human liver acetaminophen glucuronidation, increased UGT1A exon 5a/5b splice variant mRNA ratio, and decreased risk of unintentional acetaminophen-induced acute liver failure. J. Pharmacol. Exp. Ther. 2013, 345, 297–307. [Google Scholar]
- Li, J.; Peng, P.; Mei, Q.; Xia, S.; Tian, Y.; Hu, L.; Chen, Y. The impact of UGT2B7 C802T and CYP3A4*1G polymorphisms on pain relief in cancer patients receiving oxycontin. Support Care Cancer 2018, 26, 2763–2767. [Google Scholar] [CrossRef] [PubMed]
- Ning, M.; Tao, Y.; Hu, X.; Guo, L.; Ni, J.; Hu, J.; Shen, H.; Chen, Y. Roles of UGT2B7 C802T gene polymorphism on the efficacy of morphine treatment on cancer pain among the Chinese han population. Niger. J. Clin. Pract. 2019, 22, 1319–1323. [Google Scholar]
- Darbari, D.S.; van Schaik, R.H.; Capparelli, E.V.; Rana, S.; McCarter, R.; van den Anker, J. UGT2B7 promoter variant -840G>A contributes to the variability in hepatic clearance of morphine in patients with sickle cell disease. Am. J. Hematol. 2008, 83, 200–202. [Google Scholar] [CrossRef]
- Tian, J.N.; Ho, I.K.; Tsou, H.H.; Fang, C.P.; Hsiao, C.F.; Chen, C.H.; Tan, H.K.; Lin, L.; Wu, C.S.; Su, L.W.; et al. UGT2B7 genetic polymorphisms are associated with the withdrawal symptoms in methadone maintenance patients. Pharmacogenomics 2012, 13, 879–888. [Google Scholar] [CrossRef]
- Singkham, N.; Towanabut, S.; Lertkachatarn, S.; Punyawudho, B. Influence of the UGT2B7 -161C>T polymorphism on the population pharmacokinetics of lamotrigine in Thai patients. Eur. J. Clin. Pharmacol. 2013, 69, 1285–1291. [Google Scholar] [CrossRef]
- Ma, C.L.; Wu, X.Y.; Jiao, Z.; Hong, Z.; Wu, Z.Y.; Zhong, M.K. SCN1A, ABCC2 and UGT2B7 gene polymorphisms in association with individualized oxcarbazepine therapy. Pharmacogenomics 2015, 16, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Lin, X.Q.; Cai, W.K.; Xu, G.L.; Zhou, M.D.; Yang, M.; He, G.H. Effect of UGT2B7 genotypes on plasma concentration of valproic acid: A meta-analysis. Eur. J. Clin. Pharmacol. 2018, 74, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Woillard, J.B.; Picard, N.; Thierry, A.; Touchard, G.; Marquet, P. Associations between polymorphisms in target, metabolism, or transport proteins of mycophenolate sodium and therapeutic or adverse effects in kidney transplant patients. Pharmacogenet Genomics 2014, 24, 256–262. [Google Scholar] [CrossRef][Green Version]
- Knights, K.M.; Miners, J.O. Renal UDP-glucuronosyltransferases and the glucuronidation of xenobiotics and endogenous mediators. Drug Metab. Rev. 2010, 42, 63–73. [Google Scholar] [CrossRef]
- Bowalgaha, K.; Elliot, D.J.; Mackenzie, P.I.; Knights, K.M.; Miners, J.O. The glucuronidation of Delta4-3-Keto C19- and C21-hydroxysteroids by human liver microsomal and recombinant UDP-glucuronosyltransferases (UGTs): 6alpha- and 21-hydroxyprogesterone are selective substrates for UGT2B7. Drug Metab. Dispos. 2007, 35, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Tsoutsikos, P.; Miners, J.O.; Stapleton, A.; Thomas, A.; Sallustio, B.C.; Knights, K.M. Evidence that unsaturated fatty acids are potent inhibitors of renal UDP-glucuronosyltransferases (UGT): Kinetic studies using human kidney cortical microsomes and recombinant UGT1A9 and UGT2B7. Biochem. Pharmacol. 2004, 67, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Little, J.M.; Williams, L.; Xu, J.; Radominska-Pandya, A. Glucuronidation of the dietary fatty acids, phytanic acid and docosahexaenoic acid, by human UDP-glucuronosyltransferases. Drug Metab. Dispos. 2002, 30, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Trottier, J.; Perreault, M.; Rudkowska, I.; Levy, C.; Dallaire-Theroux, A.; Verreault, M.; Caron, P.; Staels, B.; Vohl, M.C.; Straka, R.J.; et al. Profiling serum bile acid glucuronides in humans: Gender divergences, genetic determinants, and response to fenofibrate. Clin. Pharmacol. Ther. 2013, 94, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, F.; Radominska, A.; Zimniak, P.; Little, J.M.; Havinga, R.; Vonk, R.J.; Lester, R. Defective biliary secretion of bile acid 3-O-glucuronides in rats with hereditary conjugated hyperbilirubinemia. J. Lipid Res. 1989, 30, 1835–1845. [Google Scholar] [CrossRef]
- Czernik, P.J.; Little, J.M.; Barone, G.W.; Raufman, J.P.; Radominska-Pandya, A. Glucuronidation of estrogens and retinoic acid and expression of UDP-glucuronosyltransferase 2B7 in human intestinal mucosa. Drug Metab. Dispos. 2000, 28, 1210–1216. [Google Scholar]
- Sneitz, N.; Krishnan, K.; Covey, D.F.; Finel, M. Glucuronidation of the steroid enantiomers ent-17beta-estradiol, ent-androsterone and ent-etiocholanolone by the human UDP-glucuronosyltransferases. Mol. Biol. 2012, 127, 282–288. [Google Scholar]
- Shatalova, E.G.; Loginov, V.I.; Braga, E.A.; Kazubskaia, T.P.; Sudomoina, M.A.; Blanchard, R.L.; Favorova, O.O. Association of polymorphisms in SULT1A1 and UGT1A1 Genes with breast cancer risk and phenotypes in Russian women. Mol. Biol. 2006, 40, 263–270. [Google Scholar] [CrossRef]
- Turgeon, D.; Chouinard, S.; Belanger, P.; Picard, S.; Labbe, J.F.; Borgeat, P.; Belanger, A. Glucuronidation of arachidonic and linoleic acid metabolites by human UDP-glucuronosyltransferases. J. Lipid Res. 2003, 44, 1182–1191. [Google Scholar] [CrossRef]
- Little, J.M.; Kurkela, M.; Sonka, J.; Jantti, S.; Ketola, R.; Bratton, S.; Finel, M.; Radominska-Pandya, A. Glucuronidation of oxidized fatty acids and prostaglandins B1 and E2 by human hepatic and recombinant UDP-glucuronosyltransferases. J. Lipid Res. 2004, 45, 1694–1703. [Google Scholar] [CrossRef]
- van Erk, M.J.; Wopereis, S.; Rubingh, C.; van Vliet, T.; Verheij, E.; Cnubben, N.H.; Pedersen, T.L.; Newman, J.W.; Smilde, A.K.; van der Greef, J.; et al. Insight in modulation of inflammation in response to diclofenac intervention: A human intervention study. BMC Med. Genomics 2010, 3, 5. [Google Scholar] [CrossRef]
- Faletti, A.; Bassi, D.; Franchi, A.M.; Gimeno, A.L.; Gimeno, M.A. Effects of morphine on arachidonic acid metabolism, on Ca2(+)-uptake and on cAMP synthesis in uterine strips from spayed rats. Prostaglandins Leukot. Essent Fatty Acids 1990, 41, 151–155. [Google Scholar] [CrossRef]
- Rivera, J.; Ward, N.; Hodgson, J.; Puddey, I.B.; Falck, J.R.; Croft, K.D. Measurement of 20-hydroxyeicosatetraenoic acid in human urine by gas chromatography-mass spectrometry. Clin. Chem. 2004, 50, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Watzer, B.; Reinalter, S.; Seyberth, H.W.; Schweer, H. Determination of free and glucuronide conjugated 20-hydroxyarachidonic acid (20-HETE) in urine by gas chromatography/negative ion chemical ionization mass spectrometry. Prostaglandins Leukot. Essent Fatty Acids. 2000, 62, 175–181. [Google Scholar] [CrossRef]
- Knights, K.M.; Tsoutsikos, P.; Miners, J.O. Novel mechanisms of nonsteroidal anti-inflammatory drug-induced renal toxicity. Expert Opin. Drug Metab. Toxicol. 2005, 1, 399–408. [Google Scholar] [CrossRef]
- Knights, K.M.; Mangoni, A.A.; Miners, J.O. Non-selective nonsteroidal anti-inflammatory drugs and cardiovascular events: Is aldosterone the silent partner in crime? Br. J. Clin. Pharmacol. 2006, 61, 738–740. [Google Scholar] [CrossRef] [PubMed]
- Knights, K.M.; Winner, L.K.; Elliot, D.J.; Bowalgaha, K.; Miners, J.O. Aldosterone glucuronidation by human liver and kidney microsomes and recombinant UDP-glucuronosyltransferases: Inhibition by NSAIDs. Br. J. Clin. Pharmacol. 2009, 68, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Crilly, M.A.; Mangoni, A.A. Non-steroidal anti-inflammatory drug (NSAID) related inhibition of aldosterone glucuronidation and arterial dysfunction in patients with rheumatoid arthritis: A cross-sectional clinical study. BMJ Open 2011, 1, e000076. [Google Scholar] [CrossRef]
- Crilly, M.A.; Mangoni, A.A.; Knights, K.M. Aldosterone glucuronidation inhibition as a potential mechanism for arterial dysfunction associated with chronic celecoxib and diclofenac use in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2013, 31, 691–698. [Google Scholar]
- Sten, T.; Finel, M.; Ask, B.; Rane, A.; Ekstrom, L. Non-steroidal anti-inflammatory drugs interact with testosterone glucuronidation. Steroids 2009, 74, 971–977. [Google Scholar] [CrossRef]
- Graziani, G.; Tentori, L.; Portarena, I.; Vergati, M.; Navarra, P. Valproic acid increases the stimulatory effect of estrogens on proliferation of human endometrial adenocarcinoma cells. Endocrinology 2003, 144, 2822–2828. [Google Scholar] [CrossRef]
- Ethell, B.T.; Anderson, G.D.; Burchell, B. The effect of valproic acid on drug and steroid glucuronidation by expressed human UDP-glucuronosyltransferases. Biochem. Pharmacol. 2003, 65, 1441–1449. [Google Scholar] [CrossRef]


{kind=link}
| Factor | Effect on Glucuronidation | References |
|---|---|---|
| Age | Neonates have a low capacity for drug glucuronidation, such as paracetamol, due to low expression of UGT enzymes. The expression and activity of UGTs reach maximum at around 20 months of age. | [58,59,60] |
| Disease | Liver cirrhosis, cancer, and diabetes mellitus decrease glucuronidation capacity. | [61,62,63] |
| Gender | Males have higher glucuronidation activity against (S)-oxazepam than females. | [64,65] |
| Genetic variants | UGT2B7*2 decreases glucuronidation capacity towards mycophenolic acid and fatty acids, such as arachidonic acid metabolites. UGT1A1*28 decreases the metabolism of irinotecan. | [66,67,69,70] |
| Environmental | Smoking induces the UGT1A family, which increases the metabolism of SN-28. | [73] |
| Genetic Variant | Rs Number | Clinical Impact on Drug Responses | References |
|---|---|---|---|
| UGT1A1 (TA)6TAA> (TA)7TAA (UGT1A1 *28) | rs8175347 | Associated with increased hyperbilirubinemia after treatment with the protease inhibitor atazanavir. | [75] |
| UGT1A1 211G > A (UGT1A1*6) | rs4148323 | The UGT1A1*6 allele increases the likelihood of neutropenia among Asian patients treated with the anticancer drug irinotecan. In addition, UGT1A1*6 can affect the metabolism of carvedilol. | [76,78] |
| UGT1A3 −66T > C | rs3806596 | Associated with hyperbilirubinemia in HIV patients treated with atazanavir and ritonavir. | [79] |
| UGT1A3 IVS1 −17564C > T | rs7604115 | The UGT1A3T allele is associated with decreased concentrations of plasma montelukast levels in healthy individuals. | [80] |
| UGT1A3 −751T > C (UGT1A3 *2) | rs1983023 | The UGT1A3*2C allele increases the response to atorvastatin in healthy subjects compared to the wild-type UGT1A3 *1 allele. The UGT1A3*2T allele can increase the response to deferasirox. | [81,82] |
| UGT1A6 A > G (UGT1A6*5) | rs2070959 | Can increase the risk for severe neutropenia among patients on irinotecan treatment. | [83] |
| UGT1A6 19A > G | rs6759892 | May increase the risk of cardiotoxicity of anticancer anthracyclines. In addition, this genetic variant is associated with adverse drug reactions to deferiprone in patients with beta-thalassemia. | [84,85] |
| UGT1A7622T > C (UGT1A7*3) | rs11692021 | UGT1A7*3 may increase the risk of vomiting when treated with a combination of anticancer drugs S-1, irinotecan, and oxaliplatin. | [86] |
| UGT1A8 518C > G (UGT1A8*2) | rs1042597 | This genetic variant can increase the risk of diarrhea among patients with kidney transplants on immune suppressant treatment. | [87] |
| UGT1A8 I399C > T | rs2741049 | Can lower the response to oxcarbazepine among epileptic patients. | [88] |
| UGT1A9-118T10/T9 | rs3832043 | The UGT1A9T9 variant decreased the elimination rate of the active metabolite of irinotecan SN-38 in non-small cell lung cancer patients. | [89] |
| Genetic Variant | Rs Number | Clinical Impacts on Drug Responses | References |
|---|---|---|---|
| UGT2B7802C > T (UGT2B7*2) | rs7439366 | Can decrease the response to oxycodone and the dosage of codeine. Additionally, can decrease oxcarbazepine metabolism. | [91,96] |
| UGT2B7 −840C > T | rs7668282 | The UGT2B7 rs7668282 TT genotype is associated with decreased morphine glucuronidation capacity. | [93] |
| UGT2B7 −900G > A | rs7438135 | Patients with the UGT2B7 rs7438135 G-allele have a reduced severity of opiate withdrawal symptoms than those with the wild-type A-allele. Additionally, the UGT2B7 rs7438135 G-allele was associated with mycophenolate mofetil-induced anemia in kidney transplant patients. | [94,98] |
| UGT2B7 −1759A > T | rs6600880 | Patients with the UGT2B7 rs6600880 A-allele may have a reduced severity of opiate withdrawal symptoms than those with the wild-type A-allele. | [94] |
| UGT2B7 −1112C > T | rs11940316 | Patients with the UGT2B7 rs11940316 T-allele may have a reduced severity of opiate withdrawal symptoms than those with the wild-type C-allele. | [94] |
| UGT2B7 −161C > T | rs28365063 | The UGT2B7 rs28365063 T-allele is associated with increased clearance of the antiepileptic drug lamotrigine. | [95] |
| UGT 2B7 211G > T (UGT 2B7*3) | rs12233719 | The UGT 2B7*3 G-allele is associated with increased valproic acid concentrations in the plasma. | [97] |
| Drugs | Potential Toxicity | Mechanisms | References |
|---|---|---|---|
| NSAIDs | Elevation of blood aldosterone levels that increase water reabsorption. | Inhibition of aldosterone glucuronidation by inhibiting UGT2B7 and 15. | [116,117,118] |
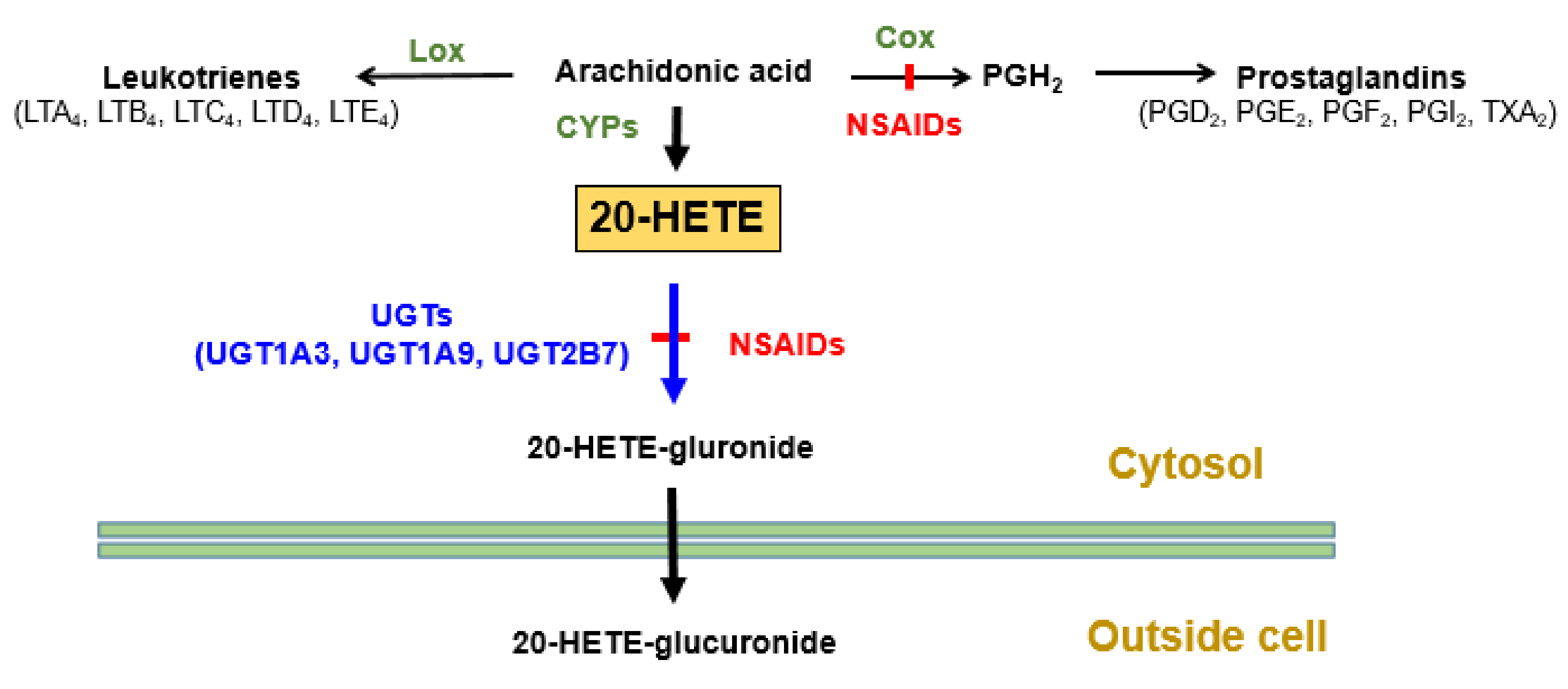
| NSAIDs | Elevation of blood cardiotoxic 20-HETE levels. | Inhibition of 20-HETE glucuronidation by inhibiting UGT2B7, 1A3, and 1A9 isoforms. | [71] |
| Valproic acid | Imbalance of blood steroidal hormones. | Inhibition of UGT2B15. | [121] |
| Diclofenac | Elevation of testosterone levels. | Inhibition of testosterone glucuronidation. | [119] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jarrar, Y.; Lee, S.-J. The Functionality of UDP-Glucuronosyltransferase Genetic Variants and their Association with Drug Responses and Human Diseases. J. Pers. Med. 2021, 11, 554. https://doi.org/10.3390/jpm11060554
Jarrar Y, Lee S-J. The Functionality of UDP-Glucuronosyltransferase Genetic Variants and their Association with Drug Responses and Human Diseases. Journal of Personalized Medicine. 2021; 11(6):554. https://doi.org/10.3390/jpm11060554
Chicago/Turabian StyleJarrar, Yazun, and Su-Jun Lee. 2021. "The Functionality of UDP-Glucuronosyltransferase Genetic Variants and their Association with Drug Responses and Human Diseases" Journal of Personalized Medicine 11, no. 6: 554. https://doi.org/10.3390/jpm11060554
APA StyleJarrar, Y., & Lee, S.-J. (2021). The Functionality of UDP-Glucuronosyltransferase Genetic Variants and their Association with Drug Responses and Human Diseases. Journal of Personalized Medicine, 11(6), 554. https://doi.org/10.3390/jpm11060554