
Phenotyping Rare CFTR Mutations Reveal Functional Expression Defects Restored by TRIKAFTATM



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Culture and Transfection
2.2. CFTR Channel Function in HEK Cells
2.3. Immunoblotting
3. Statistical Analysis
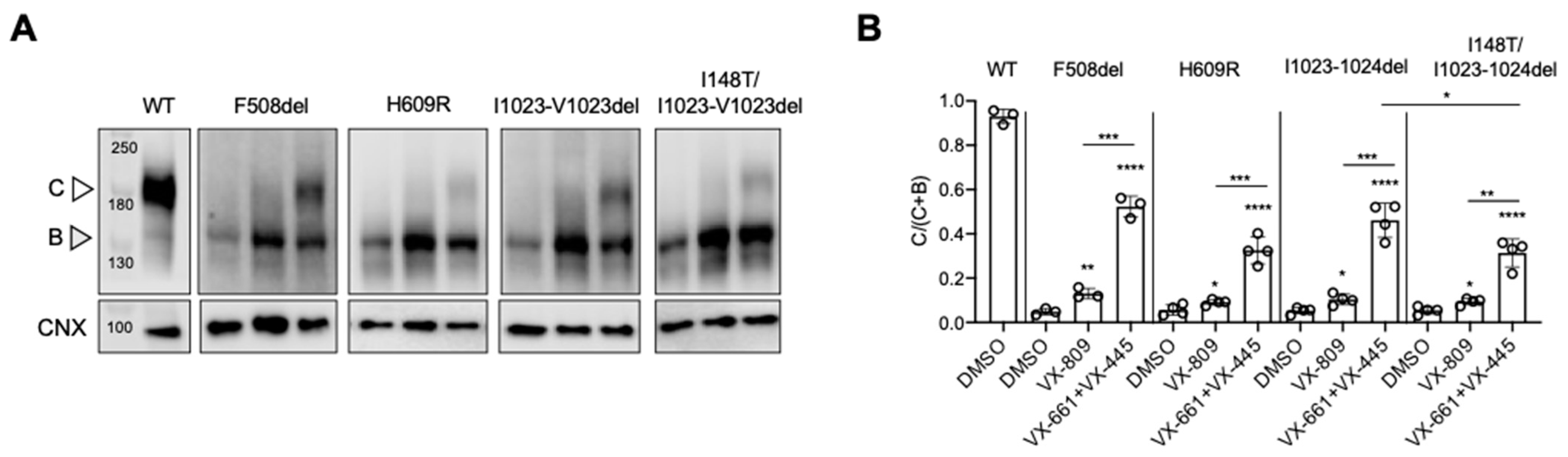
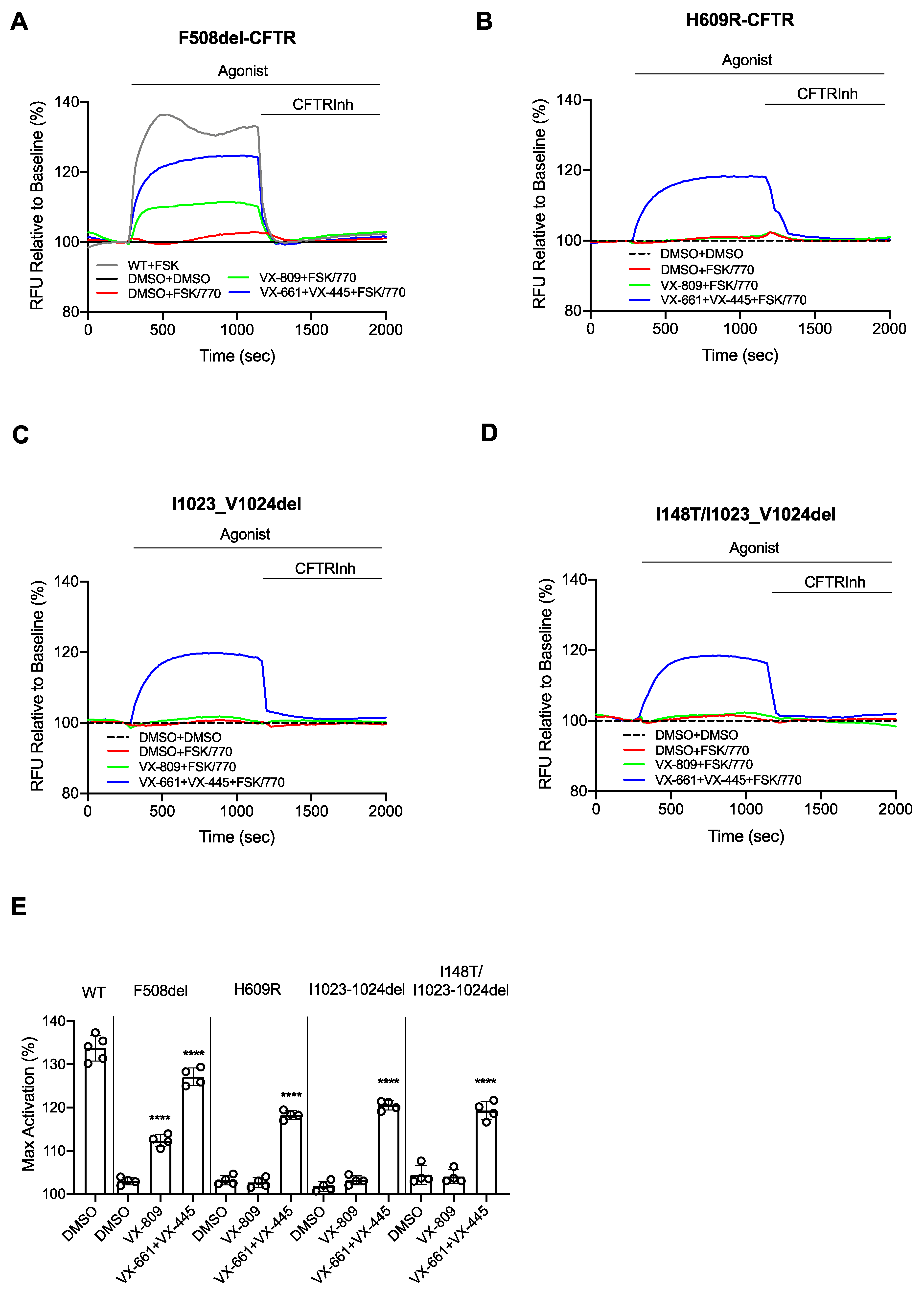
4. Results
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Bear, C.E.; Li, C.H.; Kartner, N.; Bridges, R.J.; Jensen, T.J.; Ramjeesingh, M.; Riordan, J.R. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell 1992, 68, 809–818. [Google Scholar] [CrossRef]
- Rowe, S.M.; Miller, S.; Sorscher, E.J. Cystic fibrosis. N. Engl. J. Med. 2005, 352, 1992–2001. [Google Scholar] [CrossRef]
- CFTR2 Database. Available online: www.CFTR2.org (accessed on 14 April 2021).
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 1783–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, L.; Favia, M.; Di Gioia, S.; Laselva, O.; Bisogno, A.; Casavola, V.; Colombo, C.; Conese, M. The preclinical discovery and development of the combination of ivacaftor + tezacaftor used to treat cystic fibrosis. Expert Opin. Drug Discov. 2020, 15, 873–891. [Google Scholar] [CrossRef]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Expansion of Modulators for People with Certain Rare Mutations. Available online: https://pi.vrtx.com/files/uspi_elexacaftor_tezacaftor_ivacaftor.pdf (accessed on 14 April 2021).
- Monaghan, K.G.; Highsmith, W.E.; Amos, J.; Pratt, V.M.; Roa, B.; Friez, M.; Pike-Buchanan, L.L.; Buyse, I.M.; Redman, J.B.; Strom, C.M.; et al. Genotype-phenotype correlation and frequency of the 3199del6 cystic fibrosis mutation among I148T carriers: Results from a collaborative study. Genet. Med. 2004, 6, 421–425. [Google Scholar] [CrossRef] [Green Version]
- Ruchon, A.F.; Ryan, S.R.; Fetni, R.; Rozen, R.; Scott, P. Frequency and phenotypic consequences of the 3199del6 CFTR mutation in French Canadians. Genet. Med. 2005, 7, 210–211. [Google Scholar] [CrossRef] [Green Version]
- Terlizzi, V.; Castaldo, G.; Salvatore, D.; Lucarelli, M.; Raia, V.; Angioni, A.; Carnovale, V.; Cirilli, N.; Casciaro, R.; Colombo, C.; et al. Genotype-phenotype correlation and functional studies in patients with cystic fibrosis bearing CFTR complex alleles. J. Med. Genet. 2017, 54, 224–235. [Google Scholar] [CrossRef]
- Moya-Quiles, M.R.; Glover, G.; Mondejar-Lopez, P.; Pastor-Vivero, M.D.; Fernandez-Sanchez, A.; Sanchez-Solis, M. CFTR H609R mutation in Ecuadorian patients with cystic fibrosis. J. Cyst. Fibros 2009, 8, 280–281. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.J.; Wang, J.; Zhang, Y.H.; Hsu, E.; Heim, R.A.; Bowman, C.M.; Woo, M.S. Improved detection of CFTR mutations in Southern California Hispanic CF patients. Hum. Mutat. 2001, 18, 296–307. [Google Scholar] [CrossRef]
- Choi, J.Y.; Muallem, D.; Kiselyov, K.; Lee, M.G.; Thomas, P.J.; Muallem, S. Aberrant CFTR-dependent HCO3- transport in mutations associated with cystic fibrosis. Nature 2001, 410, 94–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laselva, O.; Marzaro, G.; Vaccarin, C.; Lampronti, I.; Tamanini, A.; Lippi, G.; Gambari, R.; Cabrini, G.; Bear, C.E.; Chilin, A.; et al. Molecular Mechanism of Action of Trimethylangelicin Derivatives as CFTR Modulators. Front. Pharmacol. 2018, 9, 719. [Google Scholar] [CrossRef]
- Laselva, O.; Erwood, S.; Du, K.; Ivakine, Z.; Bear, C.E. Activity of lumacaftor is not conserved in zebrafish Cftr bearing the major cystic fibrosis-causing mutation. FASEB Bioadv. 2019, 1, 661–670. [Google Scholar] [CrossRef]
- Molinski, S.V.; Shahani, V.M.; Subramanian, A.S.; MacKinnon, S.S.; Woollard, G.; Laforet, M.; Laselva, O.; Morayniss, L.D.; Bear, C.E.; Windemuth, A. Comprehensive mapping of cystic fibrosis mutations to CFTR protein identifies mutation clusters and molecular docking predicts corrector binding site. Proteins 2018, 86, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Laselva, O.; Moraes, T.J.; He, G.; Bartlett, C.; Szarics, I.; Ouyang, H.; Gunawardena, T.N.A.; Strug, L.; Bear, C.E.; Gonska, T. The CFTR Mutation c.3453G > C (D1152H) Confers an Anion Selectivity Defect in Primary Airway Tissue that Can Be Rescued by Ivacaftor. J. Pers. Med. 2020, 10, 40. [Google Scholar] [CrossRef]
- Laselva, O.; Stone, T.A.; Bear, C.E.; Deber, C.M. Anti-Infectives Restore ORKAMBI((R)) Rescue of F508del-CFTR Function in Human Bronchial Epithelial Cells Infected with Clinical Strains of P. aeruginosa. Biomolecules 2020, 10, 334. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.S.; Jiang, J.; Ahmadi, S.; Lew, A.; Laselva, O.; Xia, S.; Bartlett, C.; Ip, W.; Wellhauser, L.; Ouyang, H.; et al. ORKAMBI-Mediated Rescue of Mucociliary Clearance in Cystic Fibrosis Primary Respiratory Cultures Is Enhanced by Arginine Uptake, Arginase Inhibition, and Promotion of Nitric Oxide Signaling to the Cystic Fibrosis Transmembrane Conductance Regulator Channel. Mol. Pharmacol. 2019, 96, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Rizwan, M.; Fokina, A.; Kivijärvi, T.; Ogawa, M.; Kufleitner, M.; Laselva, O.; Smith, L.J.; Bear, C.E.; Ogawa, S.; Keller, G.; et al. Photochemically Activated Notch Signaling Hydrogel Preferentially Differentiates Human Derived Hepatoblasts to Cholangiocytes. Adv. Funct. Mater. 2021, 31, 2006116. [Google Scholar] [CrossRef]
- Chin, S.; Ramjeesingh, M.; Hung, M.; Ereno-Oreba, J.; Cui, H.; Laselva, O.; Julien, J.P.; Bear, C.E. Cholesterol Interaction Directly Enhances Intrinsic Activity of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). Cells 2019, 8, 804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erwood, S.; Laselva, O.; Bily, T.M.I.; Brewer, R.A.; Rutherford, A.H.; Bear, C.E.; Ivakine, E.A. Allele-Specific Prevention of Nonsense-Mediated Decay in Cystic Fibrosis Using Homology-Independent Genome Editing. Mol. Ther. Methods Clin. Dev. 2020, 17, 1118–1128. [Google Scholar] [CrossRef]
- Favia, M.; Mancini, M.T.; Bezzerri, V.; Guerra, L.; Laselva, O.; Abbattiscianni, A.C.; Debellis, L.; Reshkin, S.J.; Gambari, R.; Cabrini, G.; et al. Trimethylangelicin promotes the functional rescue of mutant F508del CFTR protein in cystic fibrosis airway cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L48–L61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbattiscianni, A.C.; Favia, M.; Mancini, M.T.; Cardone, R.A.; Guerra, L.; Monterisi, S.; Castellani, S.; Laselva, O.; Di Sole, F.; Conese, M.; et al. Correctors of mutant CFTR enhance subcortical cAMP-PKA signaling through modulating ezrin phosphorylation and cytoskeleton organization. J. Cell Sci. 2016, 129, 1128–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laselva, O.; Bartlett, C.; Gunawardena, T.N.A.; Ouyang, H.; Eckford, P.D.W.; Moraes, T.J.; Bear, C.E.; Gonska, T. Rescue of multiple class II CFTR mutations by elexacaftor+ tezacaftor+ivacaftor mediated in part by the dual activities of Elexacaftor as both corrector and potentiator. Eur. Respir. J. 2020, 57. [Google Scholar] [CrossRef]
- Rohlfs, E.M.; Zhou, Z.; Sugarman, E.A.; Heim, R.A.; Pace, R.G.; Knowles, M.R.; Silverman, L.M.; Allitto, B.A. The I148T CFTR allele occurs on multiple haplotypes: A complex allele is associated with cystic fibrosis. Genet. Med. 2002, 4, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Strom, C.M.; Huang, D.; Buller, A.; Redman, J.; Crossley, B.; Anderson, B.; Entwistle, T.; Sun, W. Cystic fibrosis screening using the College panel: Platform comparison and lessons learned from the first 20,000 samples. Genet. Med. 2002, 4, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goh, J.B.; Ng, S.K. Impact of host cell line choice on glycan profile. Crit. Rev. Biotechnol. 2018, 38, 851–867. [Google Scholar] [CrossRef] [Green Version]
- Buller, A.; Olson, S.; Redman, J.B.; Hantash, F.; Chen, R.; Strom, C.M. Frequency of the cystic fibrosis 3199del6 mutation in individuals heterozygous for I148T. Genet. Med. 2004, 6, 108–109. [Google Scholar] [CrossRef] [Green Version]
- Keyeux, G.; Rodas, C.; Bienvenu, T.; Garavito, P.; Vidaud, D.; Sanchez, D.; Kaplan, J.C.; Aristizabal, G. CFTR mutations in patients from Colombia: Implications for local and regional molecular diagnosis programs. Hum. Mutat. 2003, 22, 259. [Google Scholar] [CrossRef]
- Ortiz, S.C.; Aguirre, S.J.; Flores, S.; Maldonado, C.; Mejia, J.; Salinas, L. Spectrum of CFTR gene mutations in Ecuadorian cystic fibrosis patients: The second report of the p.H609R mutation. Mol. Genet. Genom. Med. 2017, 5, 751–757. [Google Scholar] [CrossRef] [Green Version]
- Han, S.T.; Rab, A.; Pellicore, M.J.; Davis, E.F.; McCague, A.F.; Evans, T.A.; Joynt, A.T.; Lu, Z.; Cai, Z.; Raraigh, K.S.; et al. Residual function of cystic fibrosis mutants predicts response to small molecule CFTR modulators. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Pasyk, E.A.; Morin, X.K.; Zeman, P.; Garami, E.; Galley, K.; Huan, L.J.; Wang, Y.; Bear, C.E. A conserved region of the R domain of cystic fibrosis transmembrane conductance regulator is important in processing and function. J. Biol. Chem. 1998, 273, 31759–31764. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Da Paula, A.C.; Bozoky, Z.; Zhang, H.; Bertrand, C.A.; Peters, K.W.; Forman-Kay, J.D.; Frizzell, R.A. Phosphorylation-dependent 14-3-3 protein interactions regulate CFTR biogenesis. Mol. Biol. Cell 2012, 23, 996–1009. [Google Scholar] [CrossRef] [PubMed]
- Laselva, O.; McCormack, J.; Bartlett, C.; Ip, W.; Gunawardena, T.N.A.; Ouyang, H.; Eckford, P.D.W.; Gonska, T.; Moraes, T.J.; Bear, C.E. Preclinical Studies of a Rare CF-Causing Mutation in the Second Nucleotide Binding Domain (c.3700A>G) Show Robust Functional Rescue in Primary Nasal Cultures by Novel CFTR Modulators. J. Pers. Med. 2020, 10, 209. [Google Scholar] [CrossRef] [PubMed]
- Laselva, O.; Bartlett, C.; Popa, A.; Ouyang, H.; Gunawardena, T.N.A.; Gonska, T.; Moraes, T.J.; Bear, C.E. Emerging preclinical modulators developed for F508del-CFTR have the potential to be effective for ORKAMBI resistant processing mutants. J. Cyst. Fibros 2021, 20, 106–119. [Google Scholar] [CrossRef]
- Cao, H.; Ouyang, H.; Laselva, O.; Bartlett, C.; Zhou, Z.P.; Duan, C.; Gunawardena, T.; Avolio, J.; Bear, C.E.; Gonska, T.; et al. A helper-dependent adenoviral vector rescues CFTR to wild-type functional levels in cystic fibrosis epithelial cells harbouring class I mutations. Eur. Respir. J. 2020, 56, 2000205. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5, e139983. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Xu, H.; Dreano, E.; Avramescu, R.G.; Bagdany, M.; Beitel, L.K.; Roldan, A.; Hancock, M.A.; Lay, C.; Li, W.; et al. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat. Med. 2018, 24, 1732–1742. [Google Scholar] [CrossRef]
- Pranke, I.M.; Hatton, A.; Simonin, J.; Jais, J.P.; Le Pimpec-Barthes, F.; Carsin, A.; Bonnette, P.; Fayon, M.; Stremler-Le Bel, N.; Grenet, D.; et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep. 2017, 7, 7375. [Google Scholar] [CrossRef]
- Awatade, N.T.; Uliyakina, I.; Farinha, C.M.; Clarke, L.A.; Mendes, K.; Sole, A.; Pastor, J.; Ramos, M.M.; Amaral, M.D. Measurements of Functional Responses in Human Primary Lung Cells as a Basis for Personalized Therapy for Cystic Fibrosis. EBioMedicine 2015, 2, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Mutyam, V.; Sharma, J.; Li, Y.; Peng, N.; Chen, J.; Tang, L.P.; Falk Libby, E.; Singh, A.K.; Conrath, K.; Rowe, S.M. Novel Correctors and Potentiators Enhance Functional Rescue of CFTR Nonsense Mutation Translational Readthrough. Am. J. Respir. Cell Mol. Biol. 2021. [Google Scholar] [CrossRef]
- Berkers, G.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Dekkers, J.F.; de Winter-de Groot, K.M.; Arets, H.G.M.; Marck-van der Wilt, R.E.P.; Dijkema, J.S.; Vanderschuren, M.M.; et al. Rectal Organoids Enable Personalized Treatment of Cystic Fibrosis. Cell Rep. 2019, 26, 1701–1708.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekkers, J.F.; Wiegerinck, C.L.; de Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; de Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.; Bijvelds, M.J.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, A.S.; Furstova, E.; Vonk, A.M.; Ferrante, M.; Verfaillie, C.; Dupont, L.; Boon, M.; Proesmans, M.; Beekman, J.M.; Sarouk, I.; et al. Correction of CFTR function in intestinal organoids to guide treatment of cystic fibrosis. Eur. Respir. J. 2021, 57, 1902426. [Google Scholar] [CrossRef]
- Cuevas-Ocana, S.; Laselva, O.; Avolio, J.; Nenna, R. The era of CFTR modulators: Improvements made and remaining challenges. Breathe 2020, 16, 200016. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laselva, O.; Ardelean, M.C.; Bear, C.E. Phenotyping Rare CFTR Mutations Reveal Functional Expression Defects Restored by TRIKAFTATM. J. Pers. Med. 2021, 11, 301. https://doi.org/10.3390/jpm11040301
Laselva O, Ardelean MC, Bear CE. Phenotyping Rare CFTR Mutations Reveal Functional Expression Defects Restored by TRIKAFTATM. Journal of Personalized Medicine. 2021; 11(4):301. https://doi.org/10.3390/jpm11040301
Chicago/Turabian StyleLaselva, Onofrio, Maria C. Ardelean, and Christine E. Bear. 2021. "Phenotyping Rare CFTR Mutations Reveal Functional Expression Defects Restored by TRIKAFTATM" Journal of Personalized Medicine 11, no. 4: 301. https://doi.org/10.3390/jpm11040301
APA StyleLaselva, O., Ardelean, M. C., & Bear, C. E. (2021). Phenotyping Rare CFTR Mutations Reveal Functional Expression Defects Restored by TRIKAFTATM. Journal of Personalized Medicine, 11(4), 301. https://doi.org/10.3390/jpm11040301