
Differentiating Neuroblastoma: A Systematic Review of the Retinoic Acid, Its Derivatives, and Synergistic Interactions



Abstract
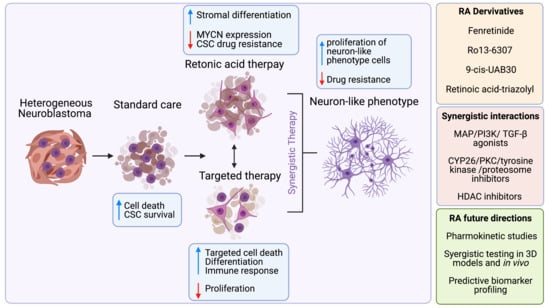
1. Introduction
2. Materials and Methods
2.1. Data Sources and Search Strategy
2.2. Study Selection
2.3. Inclusion and Exclusion Criteria
2.4. Data Extraction
3. Results
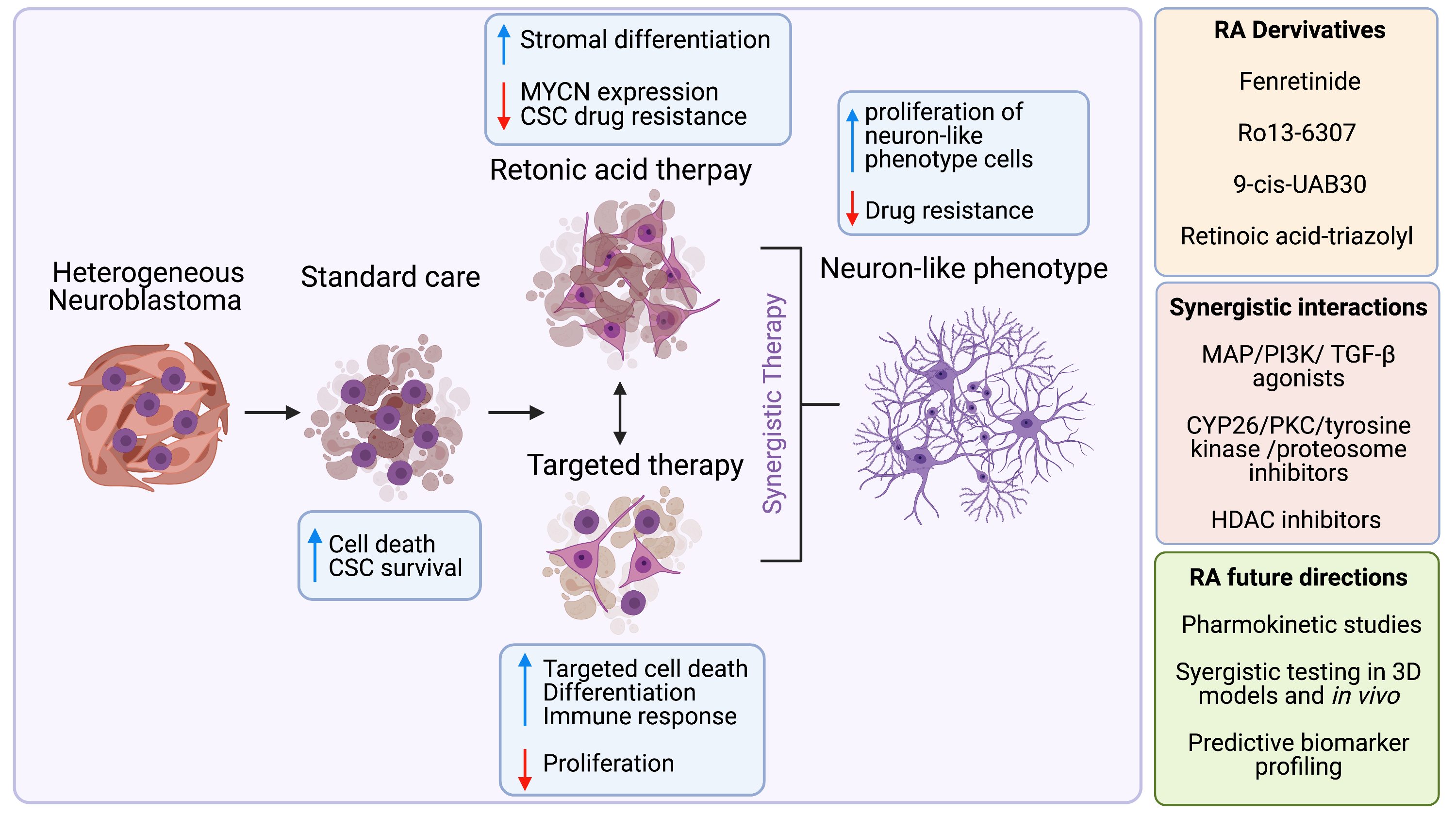
3.1. Search Results
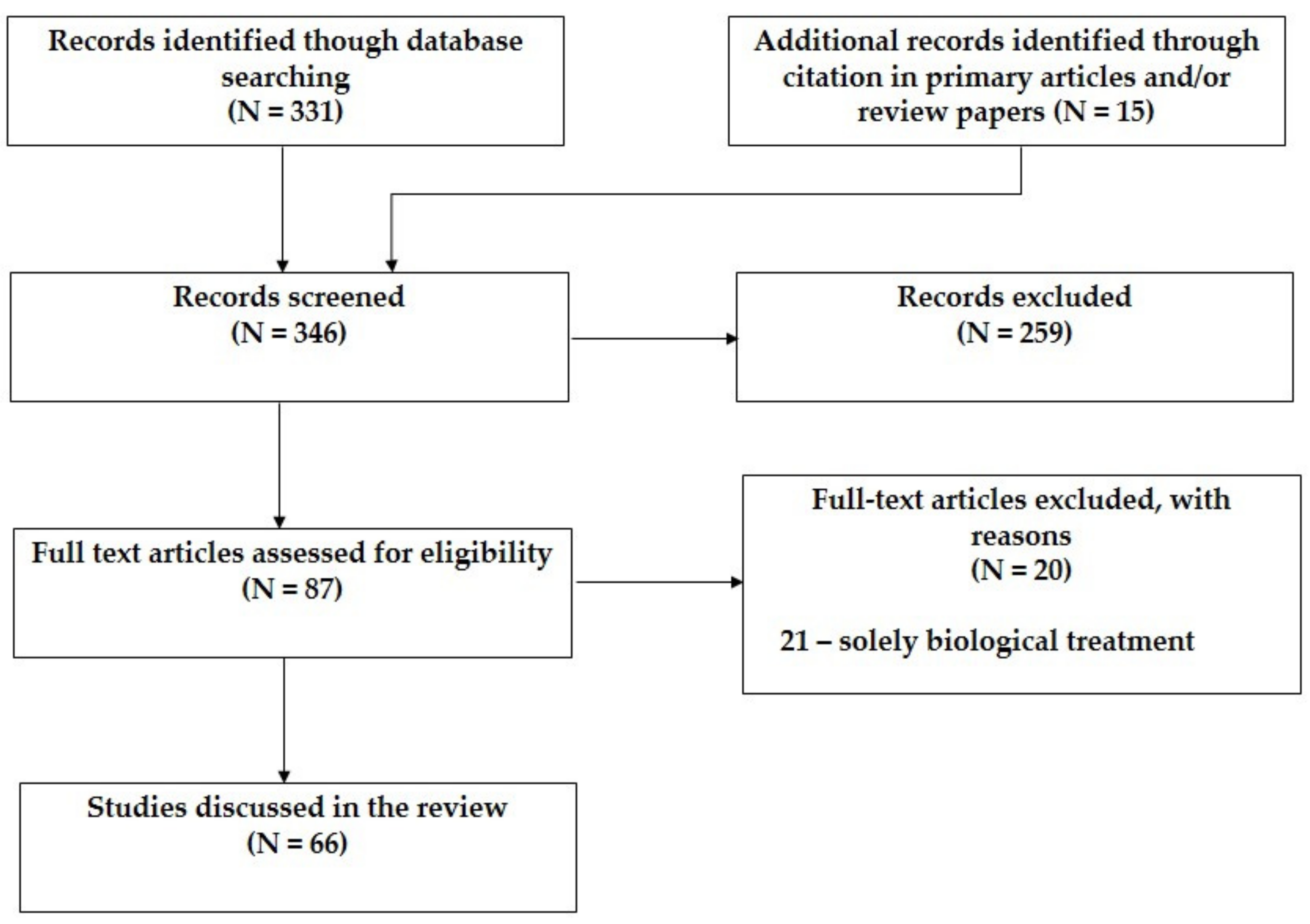
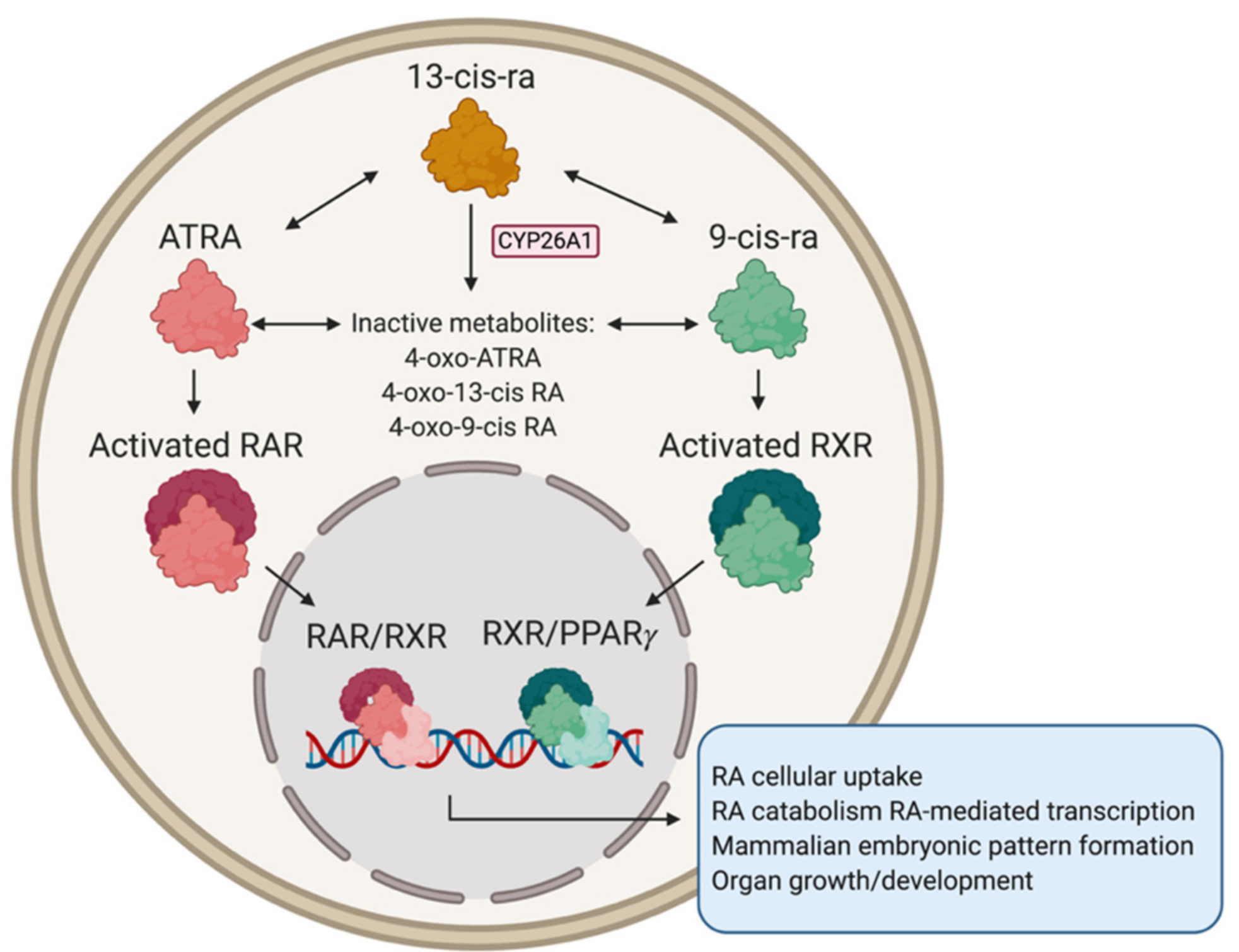
3.2. Retinoic Acid for Neuroblastoma Treatment
3.3. Retinoic Acid Derivatives for Neuroblastoma Treatment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of the Compound | Chemical Structure | Mechanism of Action | Reference |
|---|---|---|---|
| Fenretinide | N-(4-Hydroxyphenyl)retinamide | Low affinity to RAR/RXR, ceramide accumulation | [16,27,28] |
| 9-cis-UAB30 | (2E,4E,6Z,8E)-8-(3,4-dihydro-2H-naphthalen-1-ylidene)-3,7-dimethylocta-2,4,6-trienoic acid | RXR agonist | [30] |
| 6-Methyl-UAB30 | (2E,4E,6Z,8E)-8-(3,4-dihydro-2H-naphthalen-1-ylidene)-3,6,7-trimethylocta-2,4,6-trienoic acid | RXR agonist | [31] |
| LG153 | 4-[1-(3-Chloro-5,5,8,8-tetramethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)-vinyl]-benzoic acid | RXR agonist | [20] |
| LG69, or Bexaroten | 4-[1-(3,5,5,8,8-pentamethyl-6,7-dihydronaphthalen-2-yl)ethenyl]benzoic acid | RXR agonist | [19] |
| CD437, or AHPN | Naphthoic acid | RAR agonist | [28] |
| Ro 13-6307 | Ethyltetrahydrotetramethyl-2-naphthalenyl-3-methyloctatrienoic acid | RXR agonist | [22,24] |
| RA-Triazolyl derivatives | RA-Triazolyl Derivatives | Not specified | [32] |
| Geranylgeranoic acid (GGA) | acyclic retinoid, natural diterpenoid | RXR agonist | [33] |
| All-trans-retinoyl β-glucuronide | All-trans-retinoyl β-glucuronide | Not specified | [34] |
| TTNPB (Ro 13-7410) | (E)-4-[2-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthylenyl)-1 -propenyl] benzoic acid | RAR agonist | [37] |
| N-(4-hydroxyphenyl)amido (4HPTTNPB) | Hydroxyphenyl modification of TTNB | Low affinity to RAR/RXR | [37] |
| 4-hydroxybenzyl (4HBTTNPB) | Hydroxybenzyl modification of TTNB | Low affinity to RAR/RXR | [37] |
3.4. Inhibitors of CYP26
3.5. Drugs That Are Derivatives of Other Vitamins
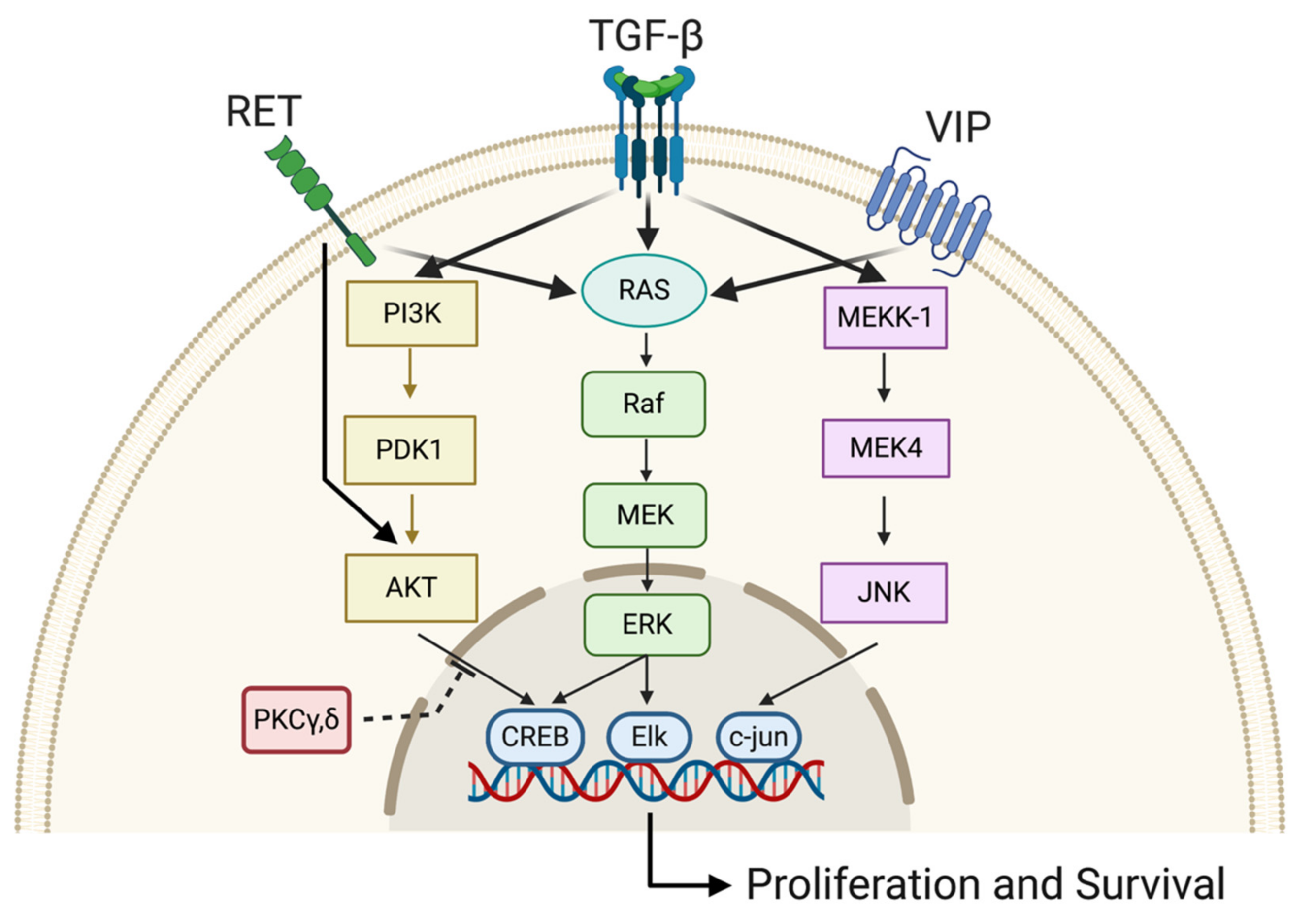
3.6. Drugs Activating the Cellular Cascades That Potentiate RA Efficiency
| Compound | Chemical Structure | Signalling Pathway | Target Protein | Model System | Markers | References |
|---|---|---|---|---|---|---|
| U0126 | Aryl sulfide | MAPK inhibitor | MEK kinase | in vitro | ZNF423, TGM2, pERK, NRF2 | [41,42] |
| PD98059 | Monomethoxyflavone | MAPK inhibitor | MEK | in vitro | PML-NB formation | [43] |
| SB203580 | Imidazole | MAPK inhibitor | Akt&p3 | in vitro | NRF2 | [41] |
| H7 | Hydrochloride anileridine salt | MAPK inhibitor | PKC | in vitro | PML-NB formation | [43] |
| LY294002 | Aryl sulfide | PI3K inhibitor | PI3K | in vitro | NRF2, p-AKT, p21, p27Kip | [41,45] |
| SP600125 | Anthrapyrazole | JNK1/2 inhibitor | JNK1/2 | in vitro | NRF2 | [41] |
| Curcumin | Diferuloylmethane | JNK inhibitor | NFkappaB, AP-1, c-Jun N-term K | in vitro | GAP-43 | [44] |
| CTNF | Peptide | Jak-STAT, MAPK, PI3K activator | CTNF receptor | in vitro | pSTAT3, pAkt, pERK1/2 | [46] |
| STI-571, Imatinib | Benzamide 2-morpholinochromenone | Tyrosine kinase pathways inhibitor | abl, c-kit, PDGF-R | in vitro | - | [53] |
| Sodium orthovanadate | Vanadium oxoanion | Tyrosine phosphatase inhibitor | Tyrosine phosphatase | in vitro | Trk, βIII-tubulin pERK, pAKT, MYCN p21 | [48] |
| ZD6474, Vandetanib | 4-anilinoquinazoline | Growth factor pathways inhibitor | VWGFR2, EGFR, RET | in vitro | pRET | [47] |
| VIP | Peptide | VIP signalling pathway | VIP receptors | in vitro | MYCN | [52] |
| D4 aptamer | 93-base 2′-fluoro-RNA-based aptamer | RET | RET receptor inhibitor | in vitro | VGF,GAP-43, tissue transglutaminase | [49] |
| GDNF | Protein | RET | RET receptor agonist | in vitro | - | [49] |
| Kartogenin | 2-([1,1′-Biphenyl]-4-ylcarbamoyl)benzoic A | TGF-β signalling activator | Filamin A | in vitro | - | [51] |
| Caffeic acid | Hydroxycinnamic A derivative | Processing of arachidonic acid | 5-LOX | in vitro | NF-200, NeuN | [50] |
| Celecoxib | NSAID with diaryl-substituted pyrazole ring | Processing of arachidonic acid | COX2 | in vitro | NF-200, NeuN, CRABP-1 | [50] |
| MG132 | Synthetic peptide aldehydes | Protein degradation inhibitor | Proteasome | in vitro | phospho-histone H, PCNA, NF-κB, Sox2, Oct4, Nestin | [54] |
3.7. Epigenetics and RA Signalling
3.7.1. HDAC Inhibitors
3.7.2. Methylation Inhibitors
3.8. Other Drugs That Exhibit Synergy with RA
3.9. NB Research Models for RA Studies
| Cell line | Treatment/Drug (s) | Dosage | Phenotype | ATRA Sensitive (Y/N) | MYCN Status | Reference |
|---|---|---|---|---|---|---|
| SH-SY5Y | 9-cis RA | 10 μM | Neurite form. | Y | Non-amp | [20] |
| LG153 | 1 μM | ↑ neurit. growth | [20] | |||
| 9-cis-UAB30 | 10 µM | ↑ neurit. growth | [30] | |||
| R116010 | 1 μm | - | [38] | |||
| Minocycline | 100 μm | - | [39] | |||
| Ro 13-6307 | 1 μM | Neurite form | [24] | |||
| GGA | 10 μM | Neurite form | [33] | |||
| Fenretinide | 3 μM | - | [28,29] | |||
| Calcitriol | 10 nM | - | [40] | |||
| 24,25(OH)2D3 | 10 nM | - | [40] | |||
| KH 1060 | 10 nM | - | [40] | |||
| EB 1089 | 10 nM | - | [40] | |||
| Trolox | 200 μM | - | [41] | |||
| n-acetyl-cysteine | 5 mM | - | [41] | |||
| BSO | 0.5 mM | - | [41] | |||
| UO126 | 10 μM | No diff. | [41] | |||
| SB203580 | - | [41] | ||||
| SP600125 | - | [41] | ||||
| LY294002 | 20 μM | No diff. | [41] | |||
| PD98059 | 25 μM | No diff. | [43] | |||
| H7 | 25 μM | ↑ diff. | [43] | |||
| Curcumin | 10 μM | ↑ neurit. growth | [44] | |||
| CNTF | 5/50/500 nM | ↑ neurit. growth | [46] | |||
| ZD6474 | 5 μM | ↑ neurit. growth | [47] | |||
| Sodium orthovanadate | 5 μM | ↑ diff. and neurit. growth | [48] | |||
| Caffeic acid | 13/52 μM | Differentiation | [50] | |||
| Celecoxib | 10/50 μM | Differentiation | [50] | |||
| STI-571 | 1/20 μM | - | [53] | |||
| MG132 | 500 nM | Apoptosis and diff | [54] | |||
| Trichostatin A | 200/500 nM | ↓ neurit. length | [56] | |||
| Sodium butyrate | 2/5 mM | ↓ neurit. length | [56] | |||
| Suberoylanilide hydroxamic acid | 1/2 μM | ↓ neurit. length | [56] | |||
| 5-Aza | 1 μM | - | [57] | |||
| IFN-γ | 100 U/mL | neurit. length | [67] | |||
| shNF1 SH-SY5Y | U0126 | 1/2.5/5 μM | - | N | Non-amp | [42] |
| SK-N-SH | LY294002 | No diff. | Y | Non-amp | [45] | |
| Herbimycin A | 236 nM | Diff. | [15] | |||
| SK-N-AS | 9-cis-UAB30 | 10 µM | ↑ neurit. growth | N | Non-amp | [30] |
| 6-Methyl-UAB30 | 25 μM | ↑ neurit. growth | [31] | |||
| Sodium orthovanadate | 5 μM | ↑ diff. and neurit. growth | [48] | |||
| GSK-J4 | 1 μM | - | [62] | |||
| 5-Aza | 2 μM | Neurite form. | [63] | |||
| SH-EP | 9-cis-UAB30 | 10 µM | ↑ neurit. growth | N | Non-amp | [30] |
| 6-Methyl-UAB30 | 25 μM | ↑ neurit. growth | [31] | |||
| R116010 | 1 μm | - | [38] | |||
| STI-571 | 1/20 μM | - | [53] | |||
| SK-N-BE(2) | 4-oxo-13-cis RA | 10 μM | Neurite form. | Y | Amp | [23] |
| Ro 13-6307 | 1 μM | Neurite form. | [24] | |||
| Fenretinide | 3 μM | - | [28,29] | |||
| 9-cis-UAB30 | 10 µM | ↑ neurit. growth | [30] | |||
| 6-Methyl-UAB30 | 25 μM | ↑ neurit. growth | [31] | |||
| LY294002 | No diff. | [45] | ||||
| ZD6474 | 5 μM | ↓ neurit. growth | [47] | |||
| D4 aptamer | ↓ neurit. growth | [49] | ||||
| GDNF | 100 ng/mL | Neurites form. | [49] | |||
| Caffeic acid | 13/52 μM | Diff. | [50] | |||
| Celecoxib | 10/50 μM | Some diff. | [50] | |||
| MG132 | 500 nM | Apoptosis and diff | [54] | |||
| Sodium butyrate | 1 mM | - | [57] | |||
| 5-Aza | 1 μM | - | [57] | |||
| BRD8430 | 0.6 μM | ↑ diff. | [58] | |||
| compound 60 | 0.3 μM | ↑ diff. | [58] | |||
| PCI-48012 | 30 μM | - | [59] | |||
| TH34 | 5 μM | ↑ neurit. form. | [60] | |||
| GSK-J4 | 1 μM | - | [62] | |||
| DHEA | 100 μM | ↑ neurit. form. | [68] | |||
| Kelly | Sodium orthovanadate | 5 μM | ↑ diff. and neurit. length | Y | Amp | [48] |
| STI-571 | 1/20 μM | - | [53] | |||
| BRD8430 | 0.6 μM | ↑ diff. | [58] | |||
| compound 60 | 0.3 μM | ↑ diff. | [58] | |||
| Herbimycin A | 236 nM | Diff. | [15] | |||
| Vincristine | 0.01 mM | Apoptosis | [69] | |||
| LAN1 | 5-Aza | 2 μM | Neurite form. | Y | Amp | [63] |
| IFN-γ | 100 U/mL | Diff. | [15] | |||
| LAN2 | IFN-γ | 100 U/mL | No diff. | Y | Amp | [15] |
| LAN5 | 9-cis RA, ATRA | 20 μM | Neurite form. | Y | Amp | [18] |
| All-trans-retinoyl β-glucuronide | 10 μM | Neurite form. | [34] | |||
| Fenretinide | 3 μM | - | [27,28] | |||
| PD98059 | 25 μM | No diff. | [43] | |||
| H7 | 25 μM | ↑ diff. | [43] | |||
| Sodium orthovanadate | 5 μM | ↑ diff. and neurite length | [48] | |||
| STI-571 | 1/20 μM | - | [53] | |||
| IFN-γ | 100 U/mL | ↑ diff. | [65,66,67] | |||
| LAN6 | R116010 | 1 μm | - | Y | Non-Amp | [38] |
| IMR32 | R116010 | 1 μm | - | Y | Amp | [38] |
| Fenretinide | 3 μM | - | [29] | |||
| 9-cis-UAB30 | 10 μM | ↑ neurit. growth | [30] | |||
| Kartogenin | 1/5/10 μM | Apoptotic shape | [51] | |||
| Vasoactive intestinal peptide | 1 μm | Diff. | [52] | |||
| PCI-48012 | 30 μM | ↑ diff. | [59] | |||
| IFN-γ | 100 U/mL | ↑ diff. | [65,66] | |||
| NGP | R116010 | 1 μm | - | Y | Amp | [38] |
| Fenretinide | 3 μM | - | [29] | |||
| SMS-LHN | 4-oxo-13-cRA | 10 μM | Neurite form. | Y | Non-AMP | [23] |
| Fenretinide | 3 μM | - | [27] | |||
| SMS-KCN | 4-oxo-13-cRA | 10 μM | Neurite form. | Y | Amp | [23] |
| SMS-KCNR | 4-oxo-13-cRA | 10 μM | Neurite form. | Y | Amp | [23] |
| Fenretinide | 3 μM | - | [27] | |||
| CHLA-20 | 4-oxo-13-cRA | 10 μM | Neurite form. | N | Non-Amp | [23] |
| CHLA-79 | 4-oxo-13-cRA | 10 μM | Neurite form. | Y | Non-Amp | [23] |
| WAC(2) | 9-cis-UAB30 | 10 µM | ↑ neurit. growth | Y | Amp | [30] |
| 6-Methyl-UAB30 | 25 μM | ↑ neurit. growth | [31] | |||
| CHLA-90 | GSK-J4 | 1 μM | - | Y | Non-Amp | [62] |
| CHP-212 | 5-Aza | 2 μM | Neurite form. | Y | Amp | [63] |
| GIMEN | R116010 | 1 μm | - | Y | Non-Amp | [38] |
| Neuro2a a | Retinoic Acid-Triazolyl Derivatives | 10/20 μm | Neurite form. and apoptosis | Y | * | [32] |
| MCF-7 b | 4-HBT(TNPB) | 10 nM | - | Y | NA | [37] |
3.10. RA Drug Delivery Systems
4. Clinical Trials on RA
| Trial Number | Study Title | Treatment | N | Trial Phase | Outcomes | Status | Results Published |
|---|---|---|---|---|---|---|---|
| CCG-3891 | Conventional Dose Chemoradiotherapy vs. Ablative Chemoradiotherapy With Autologous BMT for High Risk Neuroblastoma | 13-cis RA | 539 | III | Event-free survival rate, overall survival rate | Complete | [81,82] |
| ENSG clinical trial | European Neuroblastoma Study Group clinical trial | 13-cis RA | 175 | III | Event-free survival rate | Complete | [83] |
| CCG-0961 | Phase 2 All Trans Retinoic Acid/Interferon alpha 2A | ATRA + IFNα2a | 16 | II | Normalization of urinary catecholamines, significant reduction of tumour size | Complete | [84] |
| NCT00053326 | Fenretinide in Treating Children With Recurrent or Resistant Neuroblastoma | Fenretinide | 59 | II | Progression-free survival, overall survival | Complete | [91] |
| NCT00295919 | N2004-04: Fenretinide LXS in Treating Patients With Recurrent, Refractory, or Persistent Neuroblastoma | Fenretinide/LXS +/− Ketoconazole | 32 | I | Peak plasma concentrations, maximum tolerated dose | Complete | [92] |
| NCT02163356 | Fenretinide Lym-X-Sorb + Ketoconazole + Vincristine for Recurrent or Resistant Neuroblastoma (SPOC2013-001) | Fenretinide/LXS + Ketoconazole + Vincristine | 4 | I | Peak plasma concentrations, maximum tolerated dose | Complete | NP |
| NCT00026312 | Isotretinoin With or Without Dinutuximab, Aldesleukin, and Sargramostim Following Stem Cell Transplant in Treating Patients With Neuroblastoma | Dinutuximab/GM-CSF/IL-2/13-cis RA | 226 | III | Event-free survival rate, overall survival rate | Complete | [87] |
| NCT02743429 | Phase II Study of Monoclonal Antibody ch14.18/CHO Continuous Infusion in Patients With Primary Refractory or Relapsed Neuroblastoma | Dinutuximab/IL-2/13-cis RA | 53 | II | Progression free survival, overall survival | Complete | [89] |
| NCT01183897 | 3F8/GM-CSF Immunotherapy Plus 13-Cis-Retinoic Acid for Primary Refractory Neuroblastoma in Bone Marrow | anti-GD2 3F8 antibody/GM-CSF/13-cis RA | 169 | III | Progression free survival, overall survival | Complete | [90] |
| NCT00533169 | ZD6474 Alone and in Combination With Retinoic Acid in Pediatric Neuroblastoma | ZD6474 + 13-cis RA | 10 | I | Peak plasma concentrations, maximum tolerated dose | Terminated | NP |
| NCT01208454 | Vorinostat and Isotretinoin in Treating Patients With High-Risk Refractory or Recurrent Neuroblastoma | Vorinostat + 13-cis RA | 29 | I | Peak plasma concentrations, maximum tolerated dose | Complete | NP |
| NCT00217412 | Vorinostat With or Without Isotretinoin in Treating Young Patients With Recurrent or Refractory Solid Tumors, Lymphoma, or Leukaemia | Vorinostat + 13-cis RA | 60 | I | Peak plasma concentrations, maximum tolerated dose | Complete | NP |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Davidoff, A.M. Neuroblastoma. Semin. Pediatr. Surg. 2012, 21, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Newman, E.A.; Abdessalam, S.; Aldrink, J.H.; Austin, M.; Heaton, T.E.; Bruny, J.; Ehrlich, P.; Dasgupta, R.; Baertschiger, R.M.; Lautz, T.B.; et al. Update on neuroblastoma. J. Pediatr. Surg. 2019, 54, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Nakagawara, A.; Li, Y.; Izumi, H.; Muramori, K.; Inada, H.; Nishi, M. Neuroblastoma. Jpn. J. Clin. Oncol. 2018, 48, 214–241. [Google Scholar] [CrossRef]
- Vo, K.T.; Matthay, K.K.; Neuhaus, J.; London, W.B.; Hero, B.; Ambros, P.F.; Nakagawara, A.; Miniati, D.; Wheeler, K.; Pearson, A.D.J.; et al. Clinical, biologic, and prognostic differences on the basis of primary tumor site in neuroblastoma: A report from the International Neuroblastoma Risk Group project. J. Clin. Oncol. 2014, 32, 3169–3176. [Google Scholar] [CrossRef]
- Pinto, N.R.; Applebaum, M.A.; Volchenboum, S.L.; Matthay, K.K.; London, W.B.; Ambros, P.F.; Nakagawara, A.; Berthold, F.; Schleiermacher, G.; Park, J.R.; et al. Advances in risk classification and treatment strategies for neuroblastoma. J. Clin. Oncol. 2015, 33, 3008–3017. [Google Scholar] [CrossRef]
- Parikh, N.S.; Howard, S.C.; Chantada, G.; Israels, T.; Khattab, M.; Alcasabas, P.; Lam, C.G.; Faulkner, L.; Park, J.R.; London, W.B.; et al. SIOP-PODC adapted risk stratification and treatment guidelines: Recommendations for neuroblastoma in low- and middle-income settings. Pediatr. Blood Cancer 2015, 62, 1305–1316. [Google Scholar] [CrossRef] [PubMed]
- Gatta, G.; Botta, L.; Rossi, S.; Aareleid, T.; Bielska-Lasota, M.; Clavel, J.; Dimitrova, N.; Jakab, Z.; Kaatsch, P.; Lacour, B.; et al. Childhood cancer survival in Europe 1999-2007: Results of EUROCARE-5-a population-based study. Lancet Oncol. 2014, 15, 35–47. [Google Scholar] [CrossRef]
- Shimada, H.; Chatten, J.; Newton, W.A.; Sachs, N.; Hamoudi, A.B.; Chiba, T.; Marsden, H.B.; Misugi, K. Histopathologic prognostic factors in neuroblastic tumors: Definition of subtypes of ganglioneuroblastoma and an age-linked classification of neuroblastomas. J. Natl. Cancer Inst. 1984, 73, 405–416. [Google Scholar] [CrossRef]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef]
- Masetti, R.; Biagi, C.; Zama, D.; Vendemini, F.; Martoni, A.; Morello, W.; Gasperini, P.; Pession, A. Retinoids in pediatric onco-hematology: The model of acute promyelocytic leukemia and neuroblastoma. Adv. Ther. 2012, 29, 747–762. [Google Scholar] [CrossRef]
- Peinemann, F.; van Dalen, E.C.; Tushabe, D.A.; Berthold, F. Retinoic acid post consolidation therapy for high-risk neuroblastoma patients treated with autologous hematopoietic stem cell transplantation. Cochrane Database Syst. Rev. 2015, 1, CD010685. [Google Scholar] [CrossRef]
- Reynolds, C.P.; Matthay, K.K.; Villablanca, J.G.; Maurer, B.J. Retinoid therapy of high-risk neuroblastoma. Cancer Lett. 2003, 197, 185–192. [Google Scholar] [CrossRef]
- Rhinn, M.; Dollé, P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Shamseer, L.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A.; Estarli, M.; Barrera, E.S.A.; et al. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Rev. Esp. Nutr. Hum. Diet. 2016, 4, 1. [Google Scholar] [CrossRef]
- Preis, P.N.; Saya, H.; Nadasdi, L.; Hochhaus, G.; Levin, V.; Sadee, W. Neuronal Cell Differentiation of Human Neuroblastoma Cells by Retinoic Acid plus Herbimycin A. Cancer Res. 1988, 48, 6530–6534. [Google Scholar] [PubMed]
- Veal, G.J.; Cole, M.; Errington, J.; Pearson, A.D.J.; Foot, A.B.M.; Whyman, G.; Boddy, A.V. Pharmacokinetics and metabolism of 13-cis-retinoic acid (isotretinoin) in children with high-risk neuroblastoma—a study of the United Kingdom Children’s Cancer Study Group. Br. J. Cancer 2007, 96, 424–431. [Google Scholar] [CrossRef]
- Matthay, K.K.; Villablanca, J.G.; Seeger, R.C.; Stram, D.O.; Harris, R.E.; Ramsay, N.K.; Swift, P.; Shimada, H.; Black, C.T.; Brodeur, G.M.; et al. Treatment of High-Risk Neuroblastoma with Intensive Chemotherapy, Radiotherapy, Autologous Bone Marrow Transplantation, and 13- cis -Retinoic Acid. N. Engl. J. Med. 1999, 341, 1165–1173. [Google Scholar] [CrossRef]
- Han, G.; Chang, B.; Connor, M.J.; Sidell, N. Enhanced potency of 9-cis versus all-trans-retinoic acid to induce the differentiation of human neuroblastoma cells. Differentiation 1995, 59, 61–69. [Google Scholar] [CrossRef]
- Irving, H.; Lovat, P.E.; Hewson, Q.C.; Malcolm, A.J.; Pearson, A.D.J.; Redfern, C.P.F. Retinoid-induced differentiation of neuroblastoma: Comparison between LG69, an RXR-selective analogue and 9-cis retinoic acid. Eur. J. Cancer 1998, 34, 111–117. [Google Scholar] [CrossRef]
- Lovat, P.E.; Irving, H.; Malcolm, A.J.; Pearson, A.D.J.; Redfern, C.P.F. 9-cis retinoic acid—A better retinoid for the modulation of differentiation, proliferation and gene expression in human neuroblastoma. J. Neurooncol. 1998, 31, 85–91. [Google Scholar] [CrossRef]
- Ponthan, F.; Kogner, P.; Bjellerup, P.; Klevenvall, L.; Hassan, M. Bioavailability and dose-dependent anti-tumour effects of 9-cis retinoic acid on human neuroblastoma xenografts in rat. Br. J. Cancer 2001, 85, 2004–2009. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ponthan, F.; Borgström, P.; Hassan, M.; Wassberg, E.; Redfern, C.P.F.; Kogner, P. The vitamin A analogues: 13-cis retinoic acid, 9-cis retinoic acid, and Ro 13-6307 inhibit neuroblastoma tumour growth in vivo. Med. Pediatr. Oncol. 2001. [Google Scholar] [CrossRef]
- Sonawane, P.; Cho, H.E.; Tagde, A.; Verlekar, D.; Yu, A.L.; Reynolds, C.P.; Kang, M.H. Metabolic characteristics of 13-cis-retinoic acid (isotretinoin) and anti-tumour activity of the 13-cis-retinoic acid metabolite 4-oxo-13-cis-retinoic acid in neuroblastoma. Br. J. Pharmacol. 2014, 171, 5330–5344. [Google Scholar] [CrossRef] [PubMed]
- Ponthan, F.; Johnsen, J.I.; Klevenvall, L.; Castro, J.; Kogner, P. The synthetic retinoid Ro 13-6307 induces neuroblastoma differentiation in vitro and inhibits neuroblastoma tumour growth in vivo. Int. J. Cancer 2003, 104, 418–424. [Google Scholar] [CrossRef]
- Lovat, P.E.; Oliverio, S.; Ranalli, M.; Corazzari, M.; Rodolfo, C.; Bernassola, F.; Aughton, K.; Maccarrone, M.; Hewson, Q.D.C.; Pearson, A.D.J.; et al. GADD153 and 12-lipoxygenase mediate fenretinide-induced apoptosis of neuroblastoma. Cancer Res. 2002, 62, 5158–5167. [Google Scholar]
- Osone, S.; Hosoi, H.; Kuwahara, Y.; Matsumoto, Y.; Iehara, T.; Sugimoto, T. Fenretinide induces sustained-activation of JNK/p38 MAPK and apoptosis in a reactive oxygen species-dependent manner in neuroblastoma cells. Int. J. Cancer 2004, 112, 219–224. [Google Scholar] [CrossRef]
- Reynolds, C.P.; Wang, Y.; Melton, L.J.; Einhorn, P.A.; Slamon, D.J.; Maurer, B.J. Retinoic-acid-resistant neuroblastoma cell lines show altered MYC regulation and high sensitivity to fenretinide. Proc. Med. Pediatric Oncol. 2000, 35, 597–602. [Google Scholar] [CrossRef]
- Lovat, P.E.; Ranalli, M.; Bernassola, F.; Tilby, M.; Malcolm, A.J.; Pearson, A.D.J.; Piacentini, M.; Melino, G.; Redfern, C.P.F. Synergistic induction of apoptosis of neuroblastoma by fenretinide or CD437 in combination with chemotherapeutic drugs. Int. J. Cancer 2000, 88, 977–985. [Google Scholar] [CrossRef]
- Veal, G. The impact of retinoic acid treatment on the sensitivity of neuroblastoma cells to fenretinide. Oncol. Rep. 2011, 27, 293–298. [Google Scholar] [CrossRef][Green Version]
- Waters, A.M.; Stewart, J.E.; Atigadda, V.R.; Mroczek-Musulman, E.; Muccio, D.D.; Grubbs, C.J.; Beierle, E.A. Preclinical evaluation of a novel RXR agonist for the treatment of neuroblastoma. Mol. Cancer Ther. 2015, 14, 1559–1569. [Google Scholar] [CrossRef]
- Marayati, R.; Williams, A.P.; Bownes, L.V.; Quinn, C.H.; Stewart, J.E.; Mroczek-Musulman, E.; Atigadda, V.R.; Beierle, E.A. Novel retinoic acid derivative induces differentiation and growth arrest in neuroblastoma. J. Pediatr. Surg. 2020, 55, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Lone, A.M.; Dar, N.J.; Hamid, A.; Shah, W.A.; Ahmad, M.; Bhat, B.A. Promise of Retinoic Acid-Triazolyl Derivatives in Promoting Differentiation of Neuroblastoma Cells. ACS Chem. Neurosci. 2016, 7, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Sakane, C.; Shidoji, Y. Reversible upregulation of tropomyosin-related kinase receptor B by geranylgeranoic acid in human neuroblastoma SH-SY5Y cells. J. Neurooncol. 2011, 104, 705–713. [Google Scholar] [CrossRef]
- Sidell, N.; Pasquali, M.; Malkapuram, S.; Barua, A.B.; Wanichkul, T.; Wada, R.K. In Vitro and in vivo effects of easily administered, low-toxic retinoid and phenylacetate compounds on human neuroblastoma cells. Br. J. Cancer 2003, 89, 412–419. [Google Scholar] [CrossRef]
- Barua, A.B.; Sidell, N. Retinoyl β-Glucuronide: A Biologically Active Interesting Retinoid. Proc. J. Nutr. 2004, 134, 286S–289S. [Google Scholar] [CrossRef]
- Pignatello, M.A.; Kauffman, F.C.; Levin, A.A. Multiple factors contribute to the toxicity of the aromatic retinoid, TTNPB (Ro 13-7410): Binding affinities and disposition. Toxicol. Appl. Pharmacol. 1997, 142, 319–327. [Google Scholar] [CrossRef]
- Anding, A.L.; Nieves, N.J.; Abzianidze, V.V.; Collins, M.D.; Curley, R.W.; Clagett-Dame, M. 4-hydroxybenzyl modification of the highly teratogenic retinoid, 4-[(1 E)-2-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydro-2-naphthalenyl)-1-propen-1-yl] benzoic acid (TTNPB), yields a compound that induces apoptosis in breast cancer cells and shows reduced tera. Chem. Res. Toxicol. 2011, 24, 1853–1861. [Google Scholar] [CrossRef]
- Armstrong, J.L.; Taylor, G.A.; Thomas, H.D.; Boddy, A.V.; Redfern, C.P.F.; Veal, G.J. Molecular targeting of retinoic acid metabolism in neuroblastoma: The role of the CYP26 inhibitor R116010 in vitro and in vivo. Br. J. Cancer 2007, 96, 1675–1683. [Google Scholar] [CrossRef]
- Regen, F.; Hildebrand, M.; Le Bret, N.; Herzog, I.; Heuser, I.; Hellmann-Regen, J. Inhibition of retinoic acid catabolism by minocycline: Evidence for a novel mode of action? Exp. Dermatol. 2015, 24, 473–476. [Google Scholar] [CrossRef]
- Stio, M.; Celli, A.; Treves, C. Synergistic anti-proliferative effects of vitamin D derivatives and 9-cis retinoic acid in SH-SY5Y human neuroblastoma cells. J. Steroid Biochem. Mol. Biol. 2001. [Google Scholar] [CrossRef]
- de Miranda Ramos, V.; Zanotto-Filho, A.; de Bittencourt Pasquali, M.A.; Klafke, K.; Gasparotto, J.; Dunkley, P.; Gelain, D.P.; Moreira, J.C.F. NRF2 Mediates Neuroblastoma Proliferation and Resistance to Retinoic Acid Cytotoxicity in a Model of In Vitro Neuronal Differentiation. Mol. Neurobiol. 2016, 77, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Hölzel, M.; Huang, S.; Koster, J.; Øra, I.; Lakeman, A.; Caron, H.; Nijkamp, W.; Xie, J.; Callens, T.; Asgharzadeh, S.; et al. NF1 Is a Tumor Suppressor in Neuroblastoma that Determines Retinoic Acid Response and Disease Outcome. Cell 2010, 142, 218–229. [Google Scholar] [CrossRef]
- Delaune, A.; Corbière, C.; Benjelloun, F.D.; Legrand, E.; Vannier, J.P.; Ripoll, C.; Vasse, M. Promyelocytic leukemia-nuclear body formation is an early event leading to retinoic acid-induced differentiation of neuroblastoma cells. J. Neurochem. 2008, 104, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Adlerz, L.; Beckman, M.; Holback, S.; Tehranian, R.; Cortés Toro, V.; Iverfeldt, K. Accumulation of the amyloid precursor-like protein APLP2 and reduction of APLP1 in retinoic acid-differentiated human neuroblastoma cells upon curcumin-induced neurite retraction. Mol. Brain Res. 2003, 119, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Paul, P.; Lee, S.; Qiao, L.; Josifi, E.; Tiao, J.R.; Chung, D.H. PI3K/AKT and ERK regulate retinoic acid-induced neuroblastoma cellular differentiation. Biochem. Biophys. Res. Commun. 2012, 424, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhou, F.; Zhu, X.; Zhang, K.; Huang, B.; Zhu, L.; Zhu, L. Original article Neuroprotective properties of ciliary neurotrophic factor on retinoic acid (RA)-predifferentiated SH-SY5Y neuroblastoma cells. Folia Neuropathol. 2014, 2, 121–127. [Google Scholar] [CrossRef]
- Oppenheimer, O.; Cheung, N.K.; Gerald, W.L. The RET oncogene is a critical component of transcriptional programs associated with retinoic acid-induced differentiation in neuroblastoma. Mol. Cancer Ther. 2007, 6, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Clark, O.; Daga, S.; Stoker, A.W. Tyrosine phosphatase inhibitors combined with retinoic acid can enhance differentiation of neuroblastoma cells and trigger ERK- and AKT-dependent, p53-independent senescence. Cancer Lett. 2013, 328, 44–54. [Google Scholar] [CrossRef]
- Cerchia, L.; D’Alessio, A.; Amabile, G.; Duconge, F.; Pestourie, C.; Tavitian, B.; Libri, D.; De Franciscis, V. An autocrine loop involving ret and glial cell-derived neurotrophic factor mediates retinoic acid-induced neuroblastoma cell differentiation. Mol. Cancer Res. 2006, 4, 481–488. [Google Scholar] [CrossRef]
- Redova, M.; Chlapek, P.; Loja, T.; Zitterbart, K.; Hermanova, M.; Sterba, J.; Veselska, R. Influence of LOX/COX inhibitors on cell differentiation induced by all-trans retinoic acid in neuroblastoma cell lines. Int. J. Mol. Med. 2010, 25, 341. [Google Scholar] [CrossRef]
- Duffy, D.J.; Krstic, A.; Halasz, M.; Schwarzl, T.; Konietzny, A.; Iljin, K.; Higgins, D.G.; Kolch, W. Retinoic acid and TGF-β signalling cooperate to overcome MYCN-induced retinoid resistance. Genome Med. 2017, 9, 1–22. [Google Scholar] [CrossRef]
- Chevrier, L.; Meunier, A.C.; Cochaud, S.; Muller, J.M.; Chadéneau, C. Vasoactive intestinal peptide decreases MYCN expression and synergizes with retinoic acid in a human MYCN-amplified neuroblastoma cell line. Int. J. Oncol. 2008, 33, 1081–1089. [Google Scholar] [CrossRef]
- Rössler, J.; Zambrzycka, I.; Lagodny, J.; Kontny, U.; Niemeyer, C.M. Effect of STI-571 (imatinib mesylate) in combination with retinoic acid and γ-irradiation on viability of neuroblastoma cells. Biochem. Biophys. Res. Commun. 2006, 342, 1405–1412. [Google Scholar] [CrossRef]
- Hämmerle, B.; Yañez, Y.; Palanca, S.; Cañete, A.; Burks, D.J.; Castel, V.; Font de Mora, J. Targeting Neuroblastoma Stem Cells with Retinoic Acid and Proteasome Inhibitor. PLoS ONE 2013, 8, e76761. [Google Scholar] [CrossRef]
- Piskareva, O.; Stallings, R.L.R.L. Neuroblastoma. In Epigenetic Cancer Therapy; Gray, S., Ed.; Academic Press: Cambridge, MA, USA, 2015; ISBN 9780128002063. [Google Scholar]
- De los Santos, M.; Zambrano, A.; Aranda, A. Combined effects of retinoic acid and histone deacetylase inhibitors on human neuroblastoma SH-SY5Y cells. Mol. Cancer Ther. 2007, 6, 1425–1432. [Google Scholar] [CrossRef]
- Almeida, V.R.; Vieira, I.A.; Buendia, M.; Brunetto, A.T.; Gregianin, L.J.; Brunetto, A.L.; Klamt, F.; de Farias, C.B.; Abujamra, A.L.; da Lopez, P.L.C.; et al. Combined Treatments with a Retinoid Receptor Agonist and Epigenetic Modulators in Human Neuroblastoma Cells. Mol. Neurobiol. 2017, 54, 7610–7619. [Google Scholar] [CrossRef] [PubMed]
- Frumm, S.M.; Fan, Z.P.; Ross, K.N.; Duvall, J.R.; Gupta, S.; Verplank, L.; Suh, B.-C.C.; Holson, E.; Wagner, F.F.; Smith, W.B.; et al. Selective HDAC1/HDAC2 Inhibitors Induce Neuroblastoma Differentiation. Chem. Biol. 2013, 20, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Rettig, I.; Koeneke, E.; Trippel, F.; Mueller, W.C.; Burhenne, J.; Kopp-Schneider, A.; Fabian, J.; Schober, A.; Fernekorn, U.; Von Deimling, A.; et al. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis. 2015, 6, e1657. [Google Scholar] [CrossRef] [PubMed]
- Kolbinger, F.R.; Koeneke, E.; Ridinger, J.; Heimburg, T.; Müller, M.; Bayer, T.; Sippl, W.; Jung, M.; Gunkel, N.; Miller, A.K.; et al. The HDAC6/8/10 inhibitor TH34 induces DNA damage-mediated cell death in human high-grade neuroblastoma cell lines. Arch. Toxicol. 2018, 92, 2649–2664. [Google Scholar] [CrossRef] [PubMed]
- Coffey, D.C.; Kutko, M.C.; Glick, R.D.; Butler, L.M.; Heller, G.; Rifkind, R.A.; Marks, P.A.; Richon, V.M.; La Quaglia, M.P. The histone deacetylase inhibitor, CBHA, inhibits growth of human neuroblastoma xenografts in vivo, alone and synergistically with all-trans retinoic acid. Cancer Res. 2001, 61, 3591–3594. [Google Scholar] [PubMed]
- Lochmann, T.L.; Powell, K.M.; Ham, J.; Floros, K.V.; Heisey, D.A.R.; Kurupi, R.I.J.; Calbert, M.L.; Ghotra, M.S.; Greninger, P.; Dozmorov, M.; et al. Targeted inhibition of histone H3K27 demethylation is effective in high-risk neuroblastoma. Sci. Transl. Med. 2018, 10, eaao4680. [Google Scholar] [CrossRef]
- Westerlund, I.; Shi, Y.; Toskas, K.; Fell, S.M.; Li, S.; Surova, O.; Södersten, E.; Kogner, P.; Nyman, U.; Schlisio, S.; et al. Combined epigenetic and differentiation-based treatment inhibits neuroblastoma tumor growth and links HIF2α to tumor suppression. Proc. Natl. Acad. Sci. USA 2017, 114, E6137–E6146. [Google Scholar] [CrossRef]
- Chuang, H.C.; Lin, H.Y.; Liao, P.L.; Huang, C.C.; Lin, L.L.; Hsu, W.M.; Chuang, J.H. Immunomodulator polyinosinic-polycytidylic acid enhances the inhibitory effect of 13-cis-retinoic acid on neuroblastoma through a TLR3-related immunogenic-apoptotic response. Lab. Investig. 2020, 100, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Wada, R.K.; Pai, D.S.M.; Huang, J.; Yamashiro, J.M.; Sidell, N. Interferon-γ and retinoic acid down-regulate N-myc in neuroblastoma through complementary mechanisms of action. Cancer Lett. 1997, 121, 181–188. [Google Scholar] [CrossRef]
- Cetinkaya, C.; Hultquist, A.; Su, Y.; Wu, S.; Bahram, F.; Pahlman, S.; Guzhova, I.; Larsson, L.-G. Combined IFN- and retinoic acid treatment targets the N-Myc/Max/Mad1 network resulting in repression of N-Myc target genes in MYCN-amplified neuroblastoma cells. Mol. Cancer Ther. 2007, 6, 2634–2641. [Google Scholar] [CrossRef]
- Guzhova, I.; Hultquist, A.; Cetinkaya, C.; Nilsson, K.; Phlman, S.; Larsson, L.G. Interferon-γ cooperates with retinoic acid and phorbol ester to induce differentiation and growth inhibition of human neuroblastoma cells. Int. J. Cancer 2001, 94, 97–108. [Google Scholar] [CrossRef]
- Silvagno, F.; Guarnieri, V.; Capizzi, A.; Pescarmona, G.P. Synergistic effect of retinoic acid and dehydroepiandrosterone on differentiation of human neuroblastoma cells. FEBS Lett. 2002, 532, 153–158. [Google Scholar] [CrossRef]
- Aktas, S.; Altun, Z.; Erbayraktar, Z.; Aygun, N.; Olgun, N. Effect of cytotoxic agents and retinoic acid on Myc-N protein expression in neuroblastoma. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 86–89. [Google Scholar] [CrossRef]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef] [PubMed]
- Keshelava, N.; Seeger, R.C.; Groshen, S.; Reynolds, C.P. Drug resistance patterns of human neuroblastoma cell lines derived from patients at different phases of therapy. Cancer Res. 1998, 58, 5396–5405. [Google Scholar]
- Tadeo, I.; Piqueras, M.; Montaner, D.; Villamón, E.; Berbegall, A.P.; Cañete, A.; Navarro, S.; Noguera, R. Quantitative modeling of clinical, cellular, and extracellular matrix variables suggest prognostic indicators in cancer: A model in neuroblastoma. Pediatr. Res. 2014, 75, 302–314. [Google Scholar] [CrossRef]
- Tadeo, I.; Gamero-Sandemetrio, E.; Berbegall, A.P.; Gironella, M.; Ritort, F.; Cañete, A.; Bueno, G.; Navarro, S.; Noguera, R. Lymph microvascularization as a prognostic indicator in neuroblastoma. Oncotarget 2018, 9, 26157–26170. [Google Scholar] [CrossRef][Green Version]
- Nolan, J.C.C.; Frawley, T.; Tighe, J.; Soh, H.; Curtin, C.; Piskareva, O. Preclinical Models for Neuroblastoma: Advances and Challenges. Cancer Lett. 2020, 474, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Kroesen, M.; Nierkens, S.; Ansems, M.; Wassink, M.; Orentas, R.J.; Boon, L.; den Brok, M.H.; Hoogerbrugge, P.M.; Adema, G.J. A transplantable TH-MYCN transgenic tumor model in C57Bl/6 mice for preclinical immunological studies in neuroblastoma. Int. J. Cancer 2014, 134, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Ford, R.; Hawkins, D.; Mayo, B.; Api, A. The in vivo dermal absorption and metabolism of [4-14C]coumarin by rats and by human volunteers under simulated conditions of use in fragrances. Food Chem. Toxicol. 2001, 39, 153–162. [Google Scholar] [CrossRef]
- Giuli, M.V.; Hanieh, P.N.; Giuliani, E.; Rinaldi, F.; Marianecci, C.; Screpanti, I.; Checquolo, S.; Carafa, M. Current Trends in ATRA Delivery for Cancer Therapy. Pharmaceutics 2020, 12, 707. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, N.; Pratap, S.; Akopova, I.; Bowman, P.W.; Lacko, A.G. Pre-Clinical Evaluation of rHDL Encapsulated Retinoids for the Treatment of Neuroblastoma. Front. Pediatr. 2013, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Mohammadniaei, M.; Yoon, J.; Choi, H.K.; Placide, V.; Bharate, B.G.; Lee, T.; Choi, J.-W. Multifunctional Nanobiohybrid Material Composed of Ag@Bi 2 Se 3 /RNA Three-Way Junction/miRNA/Retinoic Acid for Neuroblastoma Differentiation. ACS Appl. Mater. Interfaces 2019, 11, 8779–8788. [Google Scholar] [CrossRef]
- Khan, A.A.; Villablanca, J.G.; Reynolds, C.P.; Avramis, V.I. Pharmacokinetic studies of 13- cis -retinoic acid in pediatric patients with neuroblastoma following bone marrow transplantation. Cancer Chemother. Pharmacol. 1996, 39, 34–41. [Google Scholar] [CrossRef]
- Kurczynski, E.M. Treatment effectiveness of high-risk neuroblastoma is improved with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13- cis -retinoic acid. Evid. Based Oncol. 2000, 1, 20–22. [Google Scholar] [CrossRef]
- Matthay, K.K.; Reynolds, C.P.; Seeger, R.C.; Shimada, H.; Adkins, E.S.; Haas-Kogan, D.; Gerbing, R.B.; London, W.B.; Villablanca, J.G. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: A children’s oncology group study. J. Clin. Oncol. 2009, 27, 1007–1013. [Google Scholar] [CrossRef]
- Kohler, J.A.; Imeson, J.; Ellershaw, C.; Lie, S.O. A randomized trial of 13-Cis retinoic acid in children with advanced neuroblastoma after high-dose therapy. Br. J. Cancer 2000, 83, 1124–1127. [Google Scholar] [CrossRef]
- Adamson, P.C.; Matthay, K.K.; Brien, M.O.; Reaman, G.H.; Sato, J.K.; Balis, F.M. A Phase 2 Trial of All- Trans -Retinoic Acid in Combination with Interferon- a 2A in Children with Recurrent Neuroblastoma or Wilms Tumor: A Pediatric Oncology Branch. NCI Child. Oncol. Group Study 2007, 61, 661–665. [Google Scholar] [CrossRef]
- Park, J.A.; Cheung, N.K.V. Targets and antibody formats for immunotherapy of neuroblastoma. J. Clin. Oncol. 2020, 38, 1836–1848. [Google Scholar] [CrossRef]
- Gilman, A.L.; Ozkaynak, F.M.; Matthay, K.K.; Krailo, M.; Yu, A.L.; Gan, J.; Sternberg, A.; Hank, J.A.; Seeger, R.; Reaman, G.H.; et al. Phase I study of ch 14.18 with granulocyte-macrophage colony-stimulating factor and interleukin-2 in children with neuroblastoma after autologous bone marrow transplantation or stem-cell rescue: A report from the children’s oncology group. J. Clin. Oncol. 2009, 27, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef]
- Siebert, N.; Eger, C.; Seidel, D.; Jüttner, M.; Zumpe, M.; Wegner, D.; Kietz, S.; Ehlert, K.; Veal, G.J.; Siegmund, W.; et al. Pharmacokinetics and pharmacodynamics of ch14.18/CHO in relapsed/refractory high-risk neuroblastoma patients treated by long-term infusion in combination with IL-2. MAbs 2016, 8, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Mueller, I.; Ehlert, K.; Endres, S.; Pill, L.; Siebert, N.; Kietz, S.; Brock, P.; Garaventa, A.; Valteau-Couanet, D.; Janzek, E.; et al. Tolerability, response and outcome of high-risk neuroblastoma patients treated with long-term infusion of anti-GD2 antibody ch14.18/CHO. MAbs 2018, 10, 55–61. [Google Scholar] [CrossRef]
- Cheung, N.K.V.; Cheung, I.Y.; Kushner, B.H.; Ostrovnaya, I.; Chamberlain, E.; Kramer, K.; Modak, S. Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte- macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J. Clin. Oncol. 2012, 30, 3264–3270. [Google Scholar] [CrossRef] [PubMed]
- Villablanca, J.G.; London, W.B.; Naranjo, A.; McGrady, P.; Ames, M.M.; Reid, J.M.; McGovern, R.M.; Buhrow, S.A.; Jackson, H.; Stranzinger, E.; et al. Phase II study of oral capsular 4-hydroxyphenylretinamide (4-HPR/fenretinide) in pediatric patients with refractory or recurrent neuroblastoma: A report from the children’s oncology group. Clin. Cancer Res. 2011, 17, 6858–6866. [Google Scholar] [CrossRef] [PubMed]
- Maurer, B.J.; Kang, M.H.; Villablanca, J.G.; Janeba, J.; Groshen, S.; Matthay, K.K.; Sondel, P.M.; Maris, J.M.; Jackson, H.A.; Goodarzian, F.; et al. Phase I trial of fenretinide delivered orally in a novel organized lipid complex in patients with relapsed/refractory neuroblastoma: A report from the new approaches to neuroblastoma therapy (NANT) consortium. Pediatr. Blood Cancer 2013, 60, 1801–1808. [Google Scholar] [CrossRef]
- Dong, R.; Yang, R.; Zhan, Y.; Lai, H.D.; Ye, C.J.; Yao, X.Y.; Luo, W.Q.; Cheng, X.M.; Miao, J.J.; Wang, J.F.; et al. Single-Cell Characterization of Malignant Phenotypes and Developmental Trajectories of Adrenal Neuroblastoma. Cancer Cell 2020, 38, 716–733.e6. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Thiele, C.J. Unraveling the Enigmatic Origin of Neuroblastoma. Cancer Cell 2020, 38, 618–620. [Google Scholar] [CrossRef]
- Garner, E.F.; Beierle, E.A. Cancer Stem Cells and Their Interaction with the Tumor Microenvironment in Neuroblastoma. Cancers 2015, 8, 5. [Google Scholar] [CrossRef]
- Borriello, L.; Seeger, R.C.; Asgharzadeh, S.; DeClerck, Y.A. More than the genes, the tumor microenvironment in neuroblastoma. Cancer Lett. 2015, 380, 304–314. [Google Scholar] [CrossRef]
- Peinemann, F.; Tushabe, D.A.; van Dalen, E.C.; Berthold, F. Rapid COJEC versus standard induction therapies for high-risk neuroblastoma. Cochrane Database Syst. Rev. 2015, 5, CD010774. [Google Scholar] [CrossRef] [PubMed]
- Sell, S.; Ilic, Z. Comparison of survivor scores for differentiation therapy of cancer to those for checkpoint inhibition: Half full or half empty. Tumor Biol. 2019, 41. [Google Scholar] [CrossRef] [PubMed]
- Marayati, R.; Bownes, L.V.; Stafman, L.L.; Williams, A.P.; Quinn, C.H.; Atigadda, V.; Aye, J.M.; Stewart, J.E.; Yoon, K.J.; Beierle, E.A. 9-cis-UAB30, a novel rexinoid agonist, decreases tumorigenicity and cancer cell stemness of human neuroblastoma patient-derived xenografts. Transl. Oncol. 2021, 14, 100893. [Google Scholar] [CrossRef] [PubMed]
- Mathur, S.; Sutton, J. Personalized medicine could transform healthcare. Biomed. Reports 2017, 7, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Dobrotkova, V.; Chlapek, P.; Jezova, M.; Adamkova, K.; Mazanek, P.; Sterba, J.; Veselska, R. Prediction of neuroblastoma cell response to treatment with natural or synthetic retinoids using selected protein biomarkers. PLoS ONE 2019, 14, e0218269. [Google Scholar] [CrossRef] [PubMed]
- Dobrotkova, V.; Chlapek, P.; Mazanek, P.; Sterba, J.; Veselska, R. Traffic lights for retinoids in oncology: Molecular markers of retinoid resistance and sensitivity and their use in the management of cancer differentiation therapy. BMC Cancer 2018, 18, 1059. [Google Scholar] [CrossRef] [PubMed]



| Treatment (s) | Mouse Strain | Site of Injection | Cell Lines | Dosage | Efficacy | Reference |
|---|---|---|---|---|---|---|
| 9-cis RA | HsdHan: RNU-rnu rats | Ectopic subcutaneous | SH-SY5Y | 2 × 2.5 mg/day orally | ↓ tumour V | [21] |
| 9-cis RA | Rowett rnu/rnu mice | Ectopic subcutaneous | SH-SY5Y | 5 mg/day orally | ↓ tumour V&W, animal weight loss | [22] |
| Ro 13-6307 | Rowett rnu/rnu mice | Ectopic subcutaneous | SH-SY5Y | 0.3 mg/day orally | ↓ tumour V&W, ↓ animal weight | [22] |
| Ro 13-6307 | HsdHan: RNU-rnu rats | Ectopic subcutaneous | SH-SY5Y | 0.12 mg/day orally | ↓ tumour V&W, ↑ animal weight gain | [24] |
| R116010. RA | CD-1 nu/nu mice | Ectopic subcutaneous | SH-SY5Y | 1.25/2.5 mg/kg 100 mg/kg | ↑ RA concentration | [38] |
| All-trans-retinoyl β-glucuronide | CD-1 nu/nu mice | Ectopic subcutaneous | LAN5 | 25 μmol/kg; 30 μmol/kg | ↓ tumour incidence and growth rate | [34,35] |
| PCI-48012 13-cis RA | NMRI nude mice | Ectopic subcutaneous | BE(2)-C | 40 mg/kg/day 10 mg/kg/day | ↓ tumour growth rate | [59] |
| CBHA ATRA | SCID mice | Ectopic subcutaneous | SMS-KCN-69 | 50/100/200 mg/kg 2.5 mg/kg | ↓ tumour growth rate | [62] |
| 5-Aza RA | Crl:Nu (Ncr)-Foxn1 nu mice | Ectopic subcutaneous | SK-N-AS, LAN-1 | 0.1 mg/kg/day 10 mg/kg/day | ↓ tumour growth rate, ↑ survival | [63] |
| PIC 13-cis RA | NOD/SCID mice | Ectopic subcutaneous | SK-N-DZ | 10 mg/kg 5 mg/kg | ↓ tumour growth rate | [64] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bayeva, N.; Coll, E.; Piskareva, O. Differentiating Neuroblastoma: A Systematic Review of the Retinoic Acid, Its Derivatives, and Synergistic Interactions. J. Pers. Med. 2021, 11, 211. https://doi.org/10.3390/jpm11030211
Bayeva N, Coll E, Piskareva O. Differentiating Neuroblastoma: A Systematic Review of the Retinoic Acid, Its Derivatives, and Synergistic Interactions. Journal of Personalized Medicine. 2021; 11(3):211. https://doi.org/10.3390/jpm11030211
Chicago/Turabian StyleBayeva, Nadiya, Erin Coll, and Olga Piskareva. 2021. "Differentiating Neuroblastoma: A Systematic Review of the Retinoic Acid, Its Derivatives, and Synergistic Interactions" Journal of Personalized Medicine 11, no. 3: 211. https://doi.org/10.3390/jpm11030211
APA StyleBayeva, N., Coll, E., & Piskareva, O. (2021). Differentiating Neuroblastoma: A Systematic Review of the Retinoic Acid, Its Derivatives, and Synergistic Interactions. Journal of Personalized Medicine, 11(3), 211. https://doi.org/10.3390/jpm11030211