
Rare Variants Associated with Arrhythmogenic Cardiomyopathy: Reclassification Five Years Later

 , , , , , , , ,
, , , , , , , ,  ,
,  ,
,  add
Show full author list
add
Show full author list
Abstract
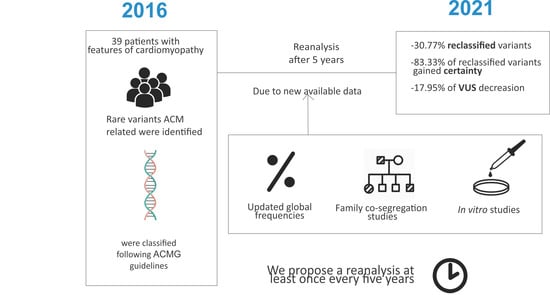
1. Introduction
2. Materials and Methods
2.1. Cohort
2.2. Genetic Analysis
2.3. Data
2.4. Classification
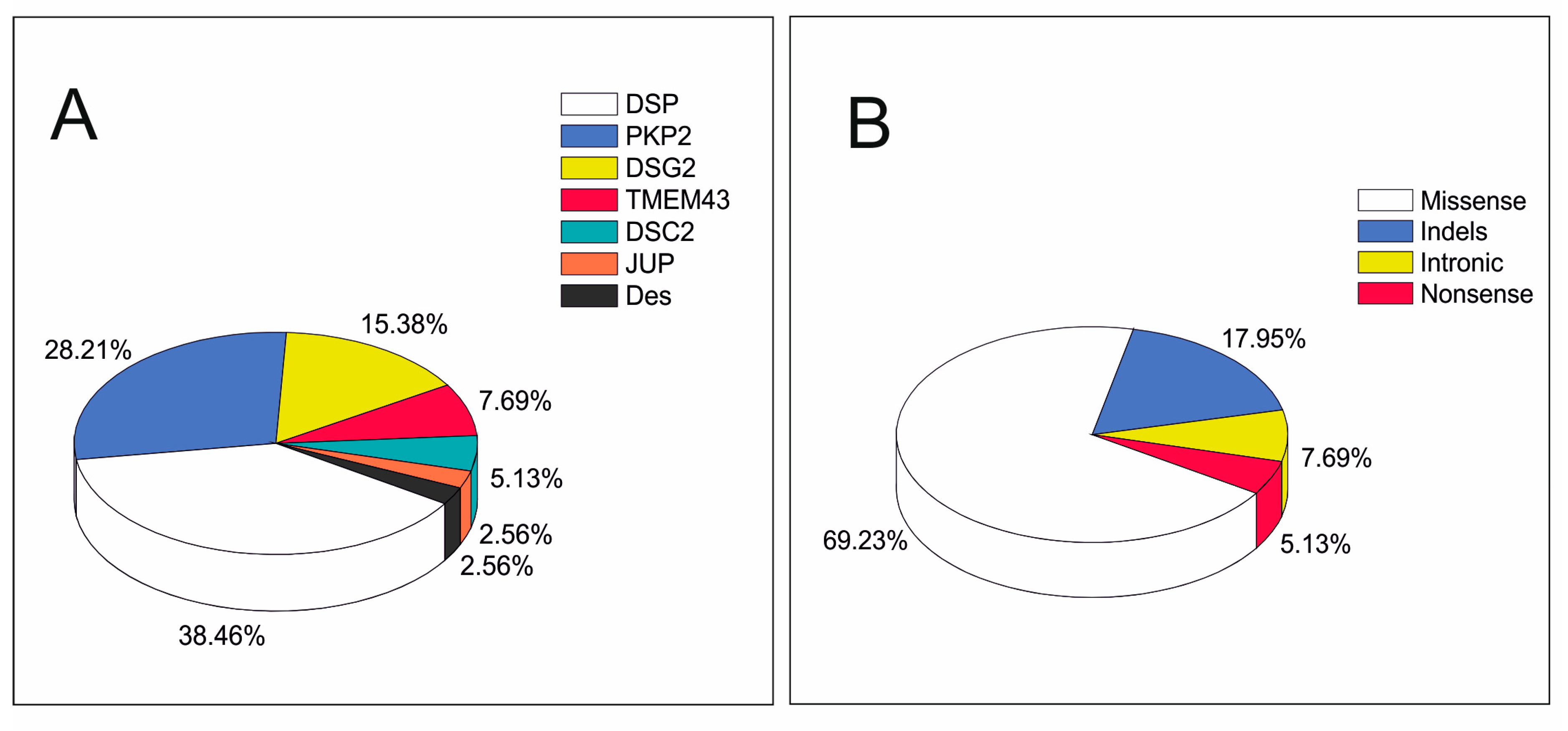
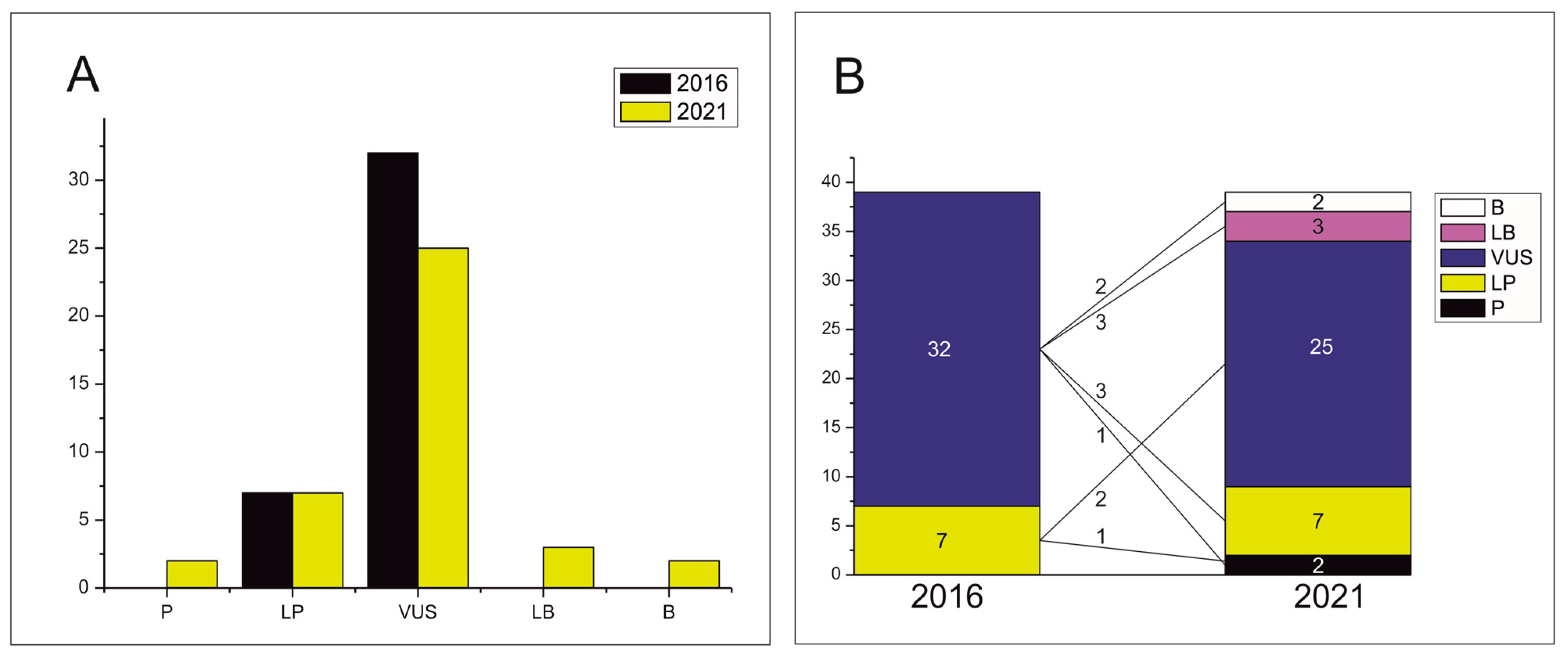
3. Results
4. Discussion
5. Conclusions
6. Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Karmouch, J.; Protonotarios, A.; Syrris, P. Genetic basis of arrhythmogenic cardiomyopathy. Curr. Opin. Cardiol. 2018, 33, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009, 373, 1289–1300. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 784–802. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Xu, J.; Li, G.; Zhou, D.; Li, S.; Zhuang, B.; Chen, X.; Duan, X.; Li, L.; Fan, X.; et al. Arrhythmogenic Left Ventricular Cardiomyopathy: A Clinical and CMR Study. Sci. Rep. 2020, 10, 533. [Google Scholar] [CrossRef]
- Corrado, D.; Link, M.S.; Calkins, H. Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2017, 376, 1489–1490. [Google Scholar] [CrossRef]
- McKenna, W.J.; Thiene, G.; Nava, A.; Fontaliran, F.; Blomstrom-Lundqvist, C.; Fontaine, G.; Camerini, F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br. Heart J. 1994, 71, 215–218. [Google Scholar] [PubMed]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the Task Force Criteria. Eur. Heart J. 2010, 31, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Muller, R.D.; McDonald, T.; Pope, K.; Cragun, D. Evaluation of Clinical Practices Related to Variants of Uncertain Significance Results in Inherited Cardiac Arrhythmia and Inherited Cardiomyopathy Genes. Circ. Genom. Precis. Med. 2020, 13, e002789. [Google Scholar] [CrossRef] [PubMed]
- Bhonsale, A.; Groeneweg, J.A.; James, C.A.; Dooijes, D.; Tichnell, C.; Jongbloed, J.D.; Murray, B.; te Riele, A.S.; van den Berg, M.P.; Bikker, H.; et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur. Heart J. 2015, 36, 847–855. [Google Scholar] [CrossRef]
- Musunuru, K.; Hershberger, R.E.; Day, S.M.; Klinedinst, N.J.; Landstrom, A.P.; Parikh, V.N.; Prakash, S.; Semsarian, C.; Sturm, A.C.; American Heart Association Council on Genomic and Precision Medicine; et al. Genetic Testing for Inherited Cardiovascular Diseases: A Scientific Statement From the American Heart Association. Circ. Genom. Precis. Med. 2020, 13, e000067. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, J.A.; Bhonsale, A.; James, C.A.; te Riele, A.S.; Dooijes, D.; Tichnell, C.; Murray, B.; Wiesfeld, A.C.; Sawant, A.C.; Kassamali, B.; et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ. Cardiovasc. Genet. 2015, 8, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M.; et al. Genetic evaluation of cardiomyopathy: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 899–909. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Sarquella-Brugada, G.; Fernandez-Falgueras, A.; Coll, M.; Iglesias, A.; Ferrer-Costa, C.; Cesar, S.; Arbelo, E.; Garcia-Alvarez, A.; Jorda, P.; et al. Reanalysis and reclassification of rare genetic variants associated with inherited arrhythmogenic syndromes. EBioMedicine 2020, 54, 102732. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: Executive summary. Heart Rhythm 2019, 16, e373–e407. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.S.; Bernhardt, M.; McBride, K.L.; Reshmi, S.C.; Zmuda, E.; Kertesz, N.J.; Garg, V.; Fitzgerald-Butt, S.; Kamp, A.N. Reclassification of Variants of Uncertain Significance in Children with Inherited Arrhythmia Syndromes is Predicted by Clinical Factors. Pediatric Cardiol. 2019, 40, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.; Medeiros-Domingo, A.; Gasperetti, A.; Akdis, D.; Berger, W.; James, C.A.; Ruschitzka, F.; Brunckhorst, C.B.; Duru, F.; Saguner, A.M. Impact of Genetic Variant Reassessment on the Diagnosis of Arrhythmogenic Right Ventricular Cardiomyopathy Based on the 2010 Task Force Criteria. Circulation. Genomic and precision medicine 2021, 14, e003047. [Google Scholar] [CrossRef]
- Harrison, S.M.; Biesecker, L.G.; Rehm, H.L. Overview of Specifications to the ACMG/AMP Variant Interpretation Guidelines. Curr. Protoc. Hum. Genet. 2019, 103, e93. [Google Scholar] [CrossRef] [PubMed]
- Whiffin, N.; Minikel, E.; Walsh, R.; O’Donnell-Luria, A.H.; Karczewski, K.; Ing, A.Y.; Barton, P.J.R.; Funke, B.; Cook, S.A.; MacArthur, D.; et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med. Off. J. Am. Coll. Med. Genet. 2017, 19, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. Off. J. Am. Coll. Med. Genet. 2017, 19, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M. ClinGen Sequence Variant Interpretation Working Group. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef]
- Quiat, D.; Witkowski, L.; Zouk, H.; Daly, K.P.; Roberts, A.E. Retrospective Analysis of Clinical Genetic Testing in Pediatric Primary Dilated Cardiomyopathy: Testing Outcomes and the Effects of Variant Reclassification. J. Am. Heart Assoc. 2020, 9, e016195. [Google Scholar] [CrossRef] [PubMed]
- Das, K.J.; Ingles, J.; Bagnall, R.D.; Semsarian, C. Determining pathogenicity of genetic variants in hypertrophic cardiomyopathy: Importance of periodic reassessment. Genet. Med. 2014, 16, 286–293. [Google Scholar] [CrossRef]
- Grassi, S.; Campuzano, O.; Coll, M.; Brion, M.; Arena, V.; Iglesias, A.; Carracedo, A.; Brugada, R.; Oliva, A. Genetic variants of uncertain significance: How to match scientific rigour and standard of proof in sudden cardiac death? Legal Med. 2020, 45, 101712. [Google Scholar] [CrossRef] [PubMed]
- Coll, M.; Allegue, C.; Partemi, S.; Mates, J.; Del Olmo, B.; Campuzano, O.; Pascali, V.; Iglesias, A.; Striano, P.; Oliva, A.; et al. Genetic investigation of sudden unexpected death in epilepsy cohort by panel target resequencing. Int. J. Legal Med. 2016, 130, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Partemi, S.; Vidal, M.C.; Striano, P.; Campuzano, O.; Allegue, C.; Pezzella, M.; Elia, M.; Parisi, P.; Belcastro, V.; Casellato, S.; et al. Genetic and forensic implications in epilepsy and cardiac arrhythmias: A case series. Int. J. Legal Med. 2015, 129, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Alcalde, M.; Iglesias, A.; Barahona-Dussault, C.; Sarquella-Brugada, G.; Benito, B.; Arzamendi, D.; Flores, J.; Leung, T.K.; Talajic, M.; et al. Arrhythmogenic right ventricular cardiomyopathy: Severe structural alterations are associated with inflammation. J. Clin. Pathol. 2012, 65, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.B.; Nissen, P.H.; Palmfeldt, J.; Gehmlich, K.; Dalager, S.; Jensen, U.B.; Kim, W.Y.; Heickendorff, L.; Molgaard, H.; Jensen, H.K.; et al. Truncating plakophilin-2 mutations in arrhythmogenic cardiomyopathy are associated with protein haploinsufficiency in both myocardium and epidermis. Circ. Cardiovasc. Genet. 2014, 7, 230–240. [Google Scholar] [CrossRef]
- Qadri, S.; Anttonen, O.; Viikila, J.; Seppala, E.H.; Myllykangas, S.; Alastalo, T.P.; Holmstrom, M.; Helio, T.; Koskenvuo, J.W. Case reports of two pedigrees with recessive arrhythmogenic right ventricular cardiomyopathy associated with homozygous Thr335Ala variant in DSG2. BMC Med. Genet. 2017, 18, 86. [Google Scholar] [CrossRef]
- Gerull, B.; Kirchner, F.; Chong, J.X.; Tagoe, J.; Chandrasekharan, K.; Strohm, O.; Waggoner, D.; Ober, C.; Duff, H.J. Homozygous founder mutation in desmocollin-2 (DSC2) causes arrhythmogenic cardiomyopathy in the Hutterite population. Circ. Cardiovasc. Genet. 2013, 6, 327–336. [Google Scholar] [CrossRef]
- Lorenzon, A.; Pilichou, K.; Rigato, I.; Vazza, G.; De Bortoli, M.; Calore, M.; Occhi, G.; Carturan, E.; Lazzarini, E.; Cason, M.; et al. Homozygous Desmocollin-2 Mutations and Arrhythmogenic Cardiomyopathy. Am. J. Cardiol. 2015, 116, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Weiss, J.; Debus, J.D.; Stanasiuk, C.; Klauke, B.; Deutsch, M.A.; Fox, H.; Bax, J.; Ebbinghaus, H.; Gartner, A.; et al. A homozygous DSC2 deletion associated with arrhythmogenic cardiomyopathy is caused by uniparental isodisomy. J. Mol. Cell Cardiol. 2020, 141, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Chkourko Gusky, H.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Peters, S. Arrhythmogenic cardiomyopathy and provocable Brugada ECG in a patient caused by missense mutation in plakophilin-2. Int. J. Cardiol. 2014, 173, 317–318. [Google Scholar] [CrossRef]
- Moncayo-Arlandi, J.; Brugada, R. Unmasking the molecular link between arrhythmogenic cardiomyopathy and Brugada syndrome. Nat. Rev. Cardiol. 2017, 14, 744–756. [Google Scholar] [CrossRef]
- Chen, X.; Peng, H.; Zheng, C.; Zhang, H.; Yan, C.; Ma, H.; Dai, X.; Li, X. Two pedigrees with arrhythmogenic right ventricular cardiomyopathy linked with R49H and F531C mutation in DSG2. Hum. Genome Var. 2019, 6, 38. [Google Scholar] [CrossRef]
- Vite, A.; Gandjbakhch, E.; Hery, T.; Fressart, V.; Gary, F.; Simon, F.; Varnous, S.; Hidden Lucet, F.; Charron, P.; Villard, E. Desmoglein-2 mutations in propeptide cleavage-site causes arrhythmogenic right ventricular cardiomyopathy/dysplasia by impairing extracellular 1-dependent desmosomal interactions upon cellular stress. Europace 2020, 22, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Arscott, P.; Caleshu, C.; Kotzer, K.; Kreykes, S.; Kruisselbrink, T.; Orland, K.; Rigelsky, C.; Smith, E.; Spoonamore, K.; Larsen Haidle, J.; et al. A Case for Inclusion of Genetic Counselors in Cardiac Care. Cardiol. Rev. 2016, 24, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Ingles, J.; Semsarian, C. The value of cardiac genetic testing. Trends Cardiovasc. Med. 2014, 24, 217–224. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Proband | Diagnosis 2016 | Gene | Nucleotide | Protein | dbSNP | ExAC (2016) | GnomAD (2021) | 2016 Classification | 2021 Classification |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Possible | DES | c.158T>C | p.(Val53Ala) | NI | NI | NI | VUS | VUS |
| 2 | Possible | DSC2 | c.430A>G | p.(Met144Val) | NI | NI | NI | VUS | VUS |
| 3 | Definite | DSC2 | c.2587G>A | p.(Gly863Arg) | rs147109895 | 6/1,3001 (0.04%) | 69/25,1128 (0.02%) | VUS | VUS |
| 4 | Definite | DSG2 | c.146G>A | p.(Arg49His) | rs121913006 | NI | 1/24,9482 (0.0004%) | LP | P |
| 5 | Possible | DSG2 | c.484delG | p.(Asp162Metfs*10) | rs1158782181 | NI | NI | LP | LP |
| 6 | Definite | DSG2 | c.1003A>G | p.(Thr335Ala) | rs191564916 | 5/1,1919 (0.04%) | 130/24,9422 (0.05%) | VUS | LB |
| 7 | Possible | DSG2 | c.1885C>T | p.(Pro629S) | rs200804638 | NI | 6/24,9336 (0.002%) | VUS | VUS |
| 8 | Possible | DSG2 | c.2825C>T | p.(Thr942Ile) | rs771429752 | NI | 1/24,8492 (0.0004%) | VUS | VUS |
| 9 | Possible | DSG2 | c.3266G>A | p.(Gly1089Asp) | rs200264407 | 9/1,2173 (0.07%) | 28/24,9268 (0.01%) | VUS | LB |
| 10 | Possible | DSP | c.130C>T | p.(Arg44Trp) | rs1255744065 | NI | 2/17,7516 (0.001%) | VUS | VUS |
| 11 | Possible | DSP | c.559G>T | p.(Val187Phe) | NI | NI | NI | VUS | VUS |
| 12 | Possible | DSP | c.1063C>T | p.(Gln355*) | rs1561686893 | NI | NI | LP | LP |
| 13 | Definite | DSP | c.1267-2A>G | NI | rs1554106830 | NI | NI | LP | LP |
| 14 | Possible | DSP | c.1297C>T | p.(Arg433Cys) | rs767032884 | NI | 2/25,1302 (0.0007%) | VUS | VUS |
| 15 | Possible | DSP | c.1639delC | p.(Leu547Trpfs*8) | NI | NI | NI | LP | LP |
| 16 | Possible | DSP | c.1696G>A | p.(Ala566Thr) | rs148147581 | 5/1,3001 (0.03%) | 50/25,1036 (0.01%) | LP | VUS |
| 17 | Possible | DSP | c.2515C>T | p.(His839Tyr) | rs1561693806 | NI | 1/25,1454 (0.0003%) | VUS | VUS |
| 18 | Possible | DSP | c.2723G>A | p.(Arg908His) | rs142494121 | 14/1,2992 (0.1%) | 289/25,1322 (0.1%) | VUS | B |
| 19 | Possible | DSP | c.2723G>T | p.(Arg908Leu) | rs142494121 | NI | 4/25,1322 (0.001%) | VUS | VUS |
| 20 | Possible | DSP | c.2867A>G | p.(Asn956Ser) | rs1373071129 | NI | 1/24,8880 (0.0004%) | VUS | VUS |
| 21 | Possible | DSP | c.3398A>G | p.(Asp1133Gly) | NI | NI | NI | VUS | VUS |
| 22 | Possible | DSP | c.3550_3551delCGinsAC | p.(Arg1184Thr) | NI | NI | NI | VUS | VUS |
| 23 | Possible | DSP | c.3643_3644delAAinsTG | p.(Asn1215Cys) | NI | NI | NI | VUS | VUS |
| 24 | Possible | DSP | c.8498C>G | p.(Ser2833Cys) | rs767961179 | NI | 3/24,6912 (0.001%) | VUS | VUS |
| 25 | Definite | JUP | c.1235C>T | p.(Thr412Met) | rs782551865 | NI | 2/25,1412 (0.0007%) | VUS | VUS |
| 26 | Possible | PKP2 | c.122C>G | p.(Ala41Gly) | rs1220759009 | NI | 1/3,1346 (0.003%) | VUS | VUS |
| 27 | Definite | PKP2 | c.259G>C | p.(Val87Leu) | rs750028032 | NI | 3/25,1410 (0.001%) | VUS | VUS |
| 28 | Possible | PKP2 | c.505A>G | p.(Ser169Gly) | rs139139859 | 21/1,2979 (0.1%) | 294/25,1282 (0.1%) | VUS | B |
| 29 | Possible | PKP2 | c.635G>T | p.(Arg212Leu) | NI | NI | NI | VUS | VUS |
| 30 | Possible | PKP2 | c.1034+2dupT | NI | NI | NI | NI | VUS | LP |
| 31 | Possible | PKP2 | c.1093A>G | p.(Met365Val) | rs143900944 | 2/1,3004 (0.01%) | 67/25,1320 (0.02%) | VUS | LB |
| 32 | Definite | PKP2 | c.1489C>T | p.(Arg497*) | rs151212477 | NI | 2/21,7850 (0.0009%) | VUS | LP |
| 33 | Definite | PKP2 | c.1643delG | p.(Gly548Valfs*15) | rs794729137 | NI | NI | VUS | LP |
| 34 | Definite | PKP2 | c.2104_2111dupTCCTTAGG | p.(Ala705Profs*2) | NI | NI | NI | VUS | LP |
| 35 | Possible | PKP2 | c.2245_2246delGCinsAT | p.(Ala749Ile) | rs1565574704 | NI | NI | VUS | VUS |
| 36 | Possible | PKP2 | c.2633C>T | p.(Ser878Phe) | rs1216433436 | NI | 1/25,1470 (0.0003%) | LP | VUS |
| 37 | Possible | TMEM43 | c.780+3A>G | NI | NI | NI | NI | VUS | VUS |
| 38 | Possible | TMEM43 | c.1026C>G | p.(Asp342Glu) | NI | 1/1,3005 (0.007%) | NI | VUS | VUS |
| 39 | Possible | TMEM43 | c.1145T>C | p.(Leu382Pro) | NI | NI | NI | VUS | VUS |
| Indicators for Gene | Indicators for Variant | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide | Protein | 2021 Class | P Gene 1 (%) 1 | P Variant Rate 2 | Loss of Function 3 | Population Data (MAF) | Allelic Data 4 | Hot Spot 5 | Computational and Predictive Data 6 | Clinically Reported P before 7 | ||
| 1 | Des | c.158T>C | p.(Val53Ala) | VUS | N/A | N/A | - | NI | NI | Yes | 6/13 | No |
| 2 | DSC2 | c.430A>G | p.(Met144Val) | VUS | B.T | B.T | - | NI | NI | Yes | 1/13 | No |
| 3 | DSG2 | c.2587G>A | p.(Gly863Arg) | VUS | B.T | B.T | - | 0.02% | 69/25,1128 | No | 10/13 | No |
| 4 | DSG2 | c.146G>A | p.(Arg49His) | P | B.T | B.T | - | 0.0004% | 1/24,9482 | Yes | 13/13 | >10 and no conflict |
| 5 | DSG2 | c.484delG | p.(Asp162Metfs*10) | LP | N/A | N/A | Yes | NI | NI | N/A | N/A | No |
| 6 | DSG2 | c.1003A>G | p.(Thr335Ala) | LB | B.T | B.T | - | 0.05% | 130/2,4942 | Yes | 4/13 | Unclear |
| 7 | DSG2 | c.1885C>T | p.(Pro629S) | VUS | B.T | B.T | - | 0.002% | 6/24,9336 | No | 8/13 | No |
| 8 | DSG2 | c.2825C>T | p.(Thr942Ile) | VUS | B.T | B.T | - | 0.0004% | 1/24,8492 | N/A | 5/13 | No |
| 9 | DSG2 | c.3266G>A | p.(Gly1089Asp) | LB | B.T | B.T | - | 0.01% | 28/24,9268 | No | 0/13 | No |
| 10 | DSP | c.130C>T | p.(Arg44Trp) | VUS | B.T | B.T | - | 0.001% | 2/17,7516 | Yes | 6/13 | No |
| 11 | DSP | c.559G>T | p.(Val187Phe) | VUS | B.T | B.T | - | NI | NI | Yes | 4/13 | No |
| 12 | DSP | c.1063C>T | p.(Gln355*) | LP | N/A | N/A | Yes | NI | NI | N/A | N/A | No |
| 13 | DSP | c.1267-2A>G | NI | LP | N/A | N/A | Yes | NI | NI | N/A | Moderate | No |
| 14 | DSP | c.1297C>T | p.(Arg433Cys) | VUS | B.T | B.T | - | 0.0007% | 2/25,1302 | Yes | 10/13 | No |
| 15 | DSP | c.1639delC | p.(Leu547Trpfs*8) | LP | N/A | N/A | Yes | NI | NI | N/A | N/A | No |
| 16 | DSP | c.1696G>A | p.(Ala566Thr) | VUS | B.T | B.T | - | 0.01% | 50/25,1036 | Yes | 0/13 | No |
| 17 | DSP | c.2515C>T | p.(His839Tyr) | VUS | B.T | B.T | - | 0.0003% | 1/25,1454 | Yes | 4/13 | No |
| 18 | DSP | c.2723G>A | p.(Arg908His) | B | B.T | B.T | - | 0.10% | 289/25,1322 | Yes | 8/13 | No |
| 19 | DSP | c.2723G>T | p.(Arg908Leu) | VUS | B.T | B.T | - | 0.001% | 4/25,1322 | Yes | 7/13 | No |
| 20 | DSP | c.2867A>G | p.(Asn956Ser) | VUS | B.T | B.T | - | 0.0004% | 1/24,8880 | Yes | 3/13 | No |
| 21 | DSP | c.3398A>G | p.(Asp1133Gly) | VUS | B.T | B.T | - | NI | NI | No | 6/13 | No |
| 22 | DSP | c.3550_3551delCGinsAC | p.(Arg1184Thr) | VUS | B.T | B.T | - | NI | NI | No | N/A | No |
| 23 | DSP | c.3643_3644delAAinsTG | p.(Asn1215Cys) | VUS | B.T | B.T | - | NI | NI | No | N/A | No |
| 24 | DSP | c.8498C>G | p.(Ser2833Cys) | VUS | B.T | B.T | - | 0.001% | 3/24,6912 | Yes | 8/13 | No |
| 25 | JUP | c.1235C>T | p.(Thr412Met) | VUS | B.T | B.T | - | 0.0007% | 2/25,1412 | No | 8/13 | No |
| 26 | PKP2 | c.122C>G | p.(Ala41Gly) | VUS | A.T | A.T | - | 0.003% | 1/3,1346 | No | 5/13 | No |
| 27 | PKP2 | c.259G>C | p.(Val87Leu) | VUS | A.T | A.T | - | 0.001% | 3/25,1410 | No | 9/13 | No |
| 28 | PKP2 | c.505A>G | p.(Ser169Gly) | B | A.T | A.T | - | 0.10% | 294/25,1282 | No | 12/13 | No |
| 29 | PKP2 | c.635G>T | p.(Arg212Leu) | VUS | A.T | A.T | - | NI | NI | No | 5/13 | No |
| 30 | PKP2 | c.1034+2dupT | NI | LP | N/A | N/A | Yes | NI | NI | N/A | N/A | No |
| 31 | PKP2 | c.1093A>G | p.(Met365Val) | LB | A.T | A.T | - | 0.02% | 67/25,1320 | No | 2/13 | No |
| 32 | PKP2 | c.1489C>T | p.(Arg497*) | LP | A.T | A.T | Yes | 0.0009% | 2/21,7850 | N/A | N/A | 1 |
| 33 | PKP2 | c.1643delG | p.(Gly548Valfs*15) | LP | N/A | N/A | Yes | NI | NI | N/A | Very Strong | >17 and no conflict |
| 34 | PKP2 | c.2104_2111dupTCCTTAGG | p.(Ala705Profs*2) | LP | N/A | N/A | Yes | NI | NI | N/A | N/A | No |
| 35 | PKP2 | c.2245_2246delGCinsAT | p.(Ala749Ile) | VUS | A.T | A.T | - | NI | NI | No | N/A | No |
| 36 | PKP2 | c.2633C>T | p.(Ser878Phe) | VUS | A.T | A.T | - | 0.00030% | 1/25,1470 | No | 11/13 | No |
| 37 | TMEM43 | c.780+3A>G | NI | VUS | N/A | N/A | No | NI | NI | No | N/A | No |
| 38 | TMEM43 | c.1026C>G | p.(Asp342Glu) | VUS | B.T | B.T | - | NI | NI | No | 2/13 | No |
| 39 | TMEM43 | c.1145T>C | p.(Leu382Pro) | VUS | B.T | B.T | - | NI | NI | No | 4/13 | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallverdú-Prats, M.; Alcalde, M.; Sarquella-Brugada, G.; Cesar, S.; Arbelo, E.; Fernandez-Falgueras, A.; Coll, M.; Pérez-Serra, A.; Puigmulé, M.; Iglesias, A.; et al. Rare Variants Associated with Arrhythmogenic Cardiomyopathy: Reclassification Five Years Later. J. Pers. Med. 2021, 11, 162. https://doi.org/10.3390/jpm11030162
Vallverdú-Prats M, Alcalde M, Sarquella-Brugada G, Cesar S, Arbelo E, Fernandez-Falgueras A, Coll M, Pérez-Serra A, Puigmulé M, Iglesias A, et al. Rare Variants Associated with Arrhythmogenic Cardiomyopathy: Reclassification Five Years Later. Journal of Personalized Medicine. 2021; 11(3):162. https://doi.org/10.3390/jpm11030162
Chicago/Turabian StyleVallverdú-Prats, Marta, Mireia Alcalde, Georgia Sarquella-Brugada, Sergi Cesar, Elena Arbelo, Anna Fernandez-Falgueras, Mónica Coll, Alexandra Pérez-Serra, Marta Puigmulé, Anna Iglesias, and et al. 2021. "Rare Variants Associated with Arrhythmogenic Cardiomyopathy: Reclassification Five Years Later" Journal of Personalized Medicine 11, no. 3: 162. https://doi.org/10.3390/jpm11030162
APA StyleVallverdú-Prats, M., Alcalde, M., Sarquella-Brugada, G., Cesar, S., Arbelo, E., Fernandez-Falgueras, A., Coll, M., Pérez-Serra, A., Puigmulé, M., Iglesias, A., Fiol, V., Ferrer-Costa, C., del Olmo, B., Picó, F., Lopez, L., Jordà, P., García-Álvarez, A., Tirón de Llano, C., Toro, R., ... Campuzano, O. (2021). Rare Variants Associated with Arrhythmogenic Cardiomyopathy: Reclassification Five Years Later. Journal of Personalized Medicine, 11(3), 162. https://doi.org/10.3390/jpm11030162