
Identification of Somatic Structural Variants in Solid Tumors by Optical Genome Mapping

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Tumor Samples
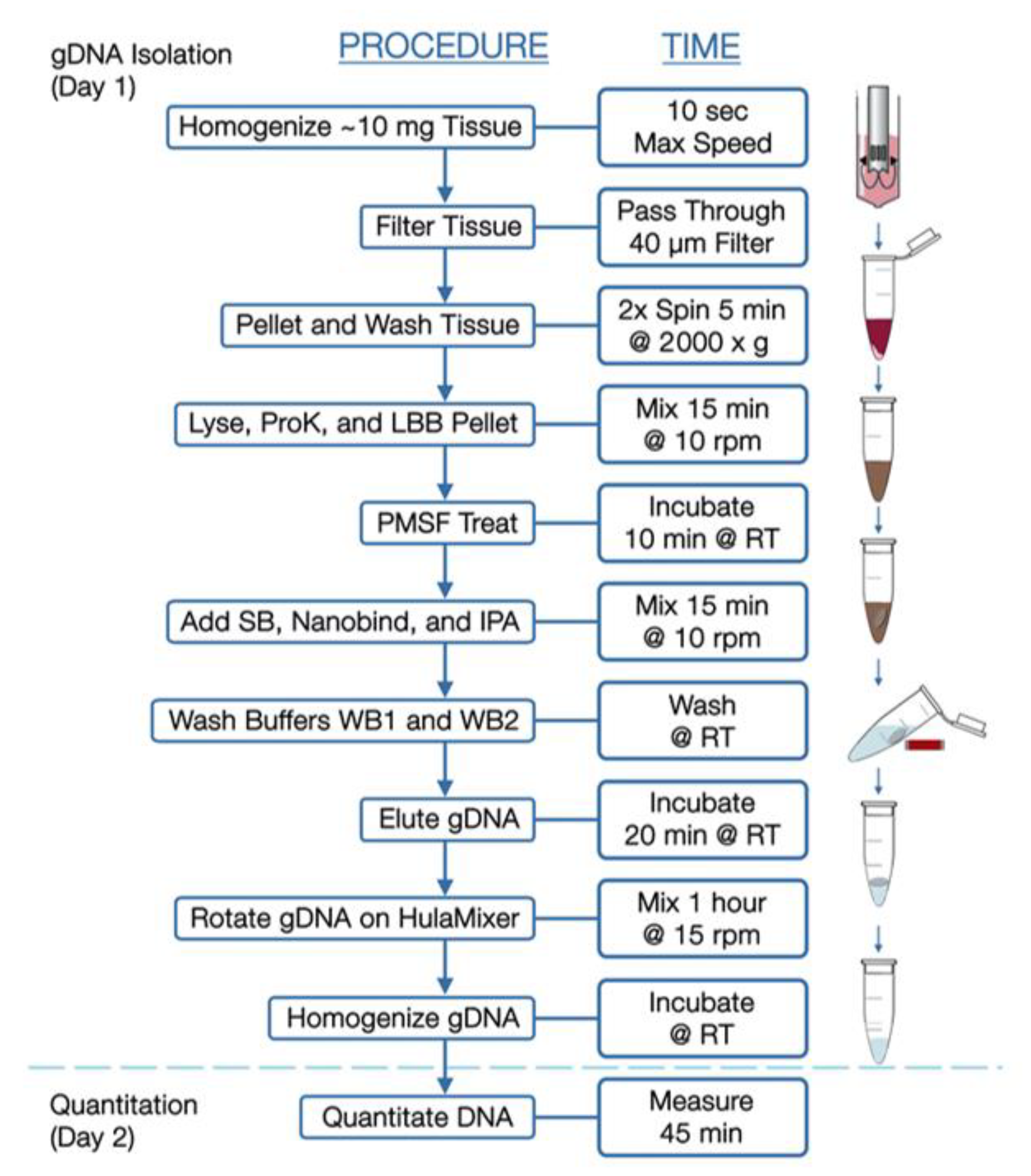
2.2. Bionano Optical Genome Mapping
2.3. Bionano Access and Solve Pipeline
2.4. Data Comparison
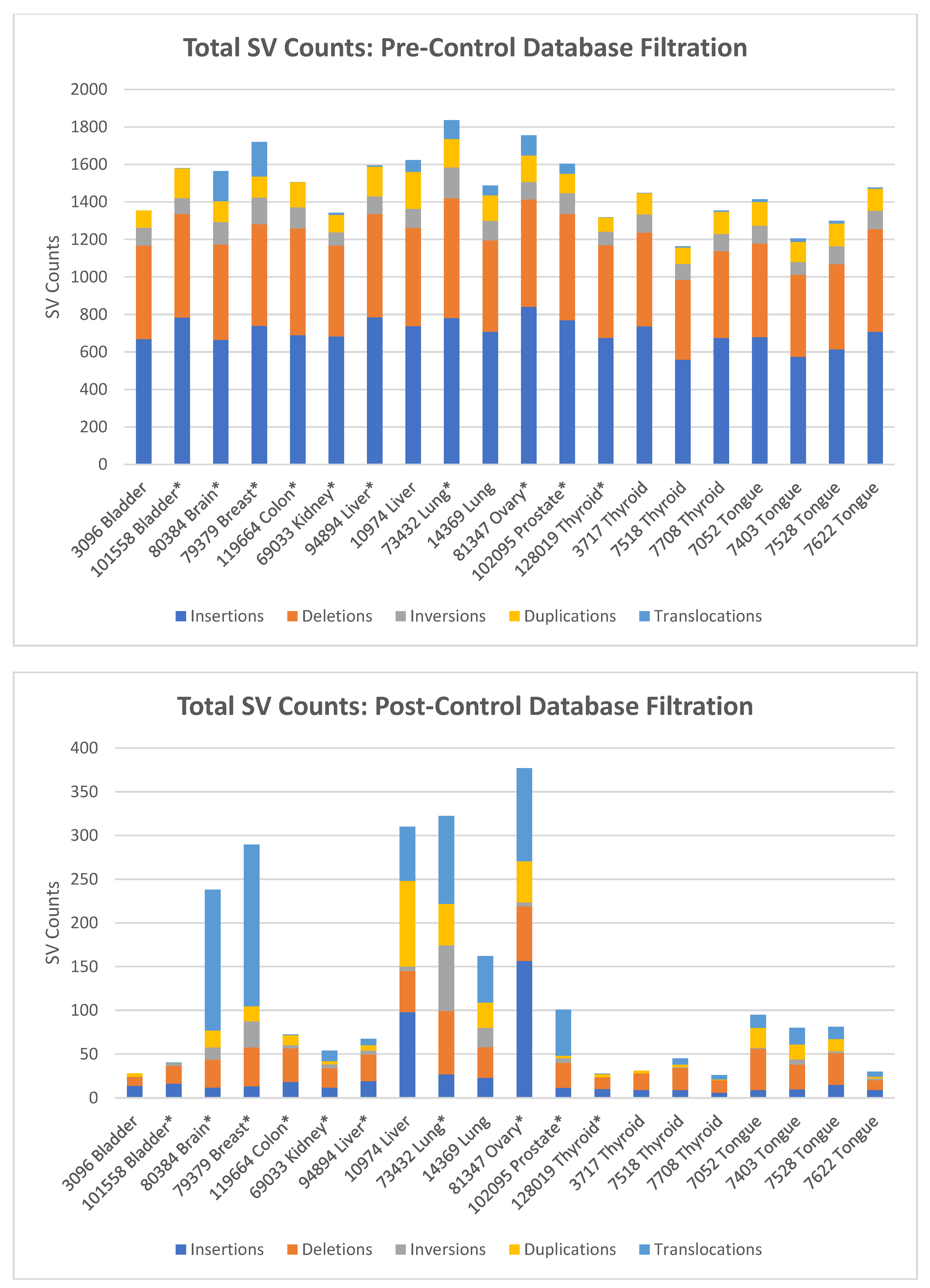
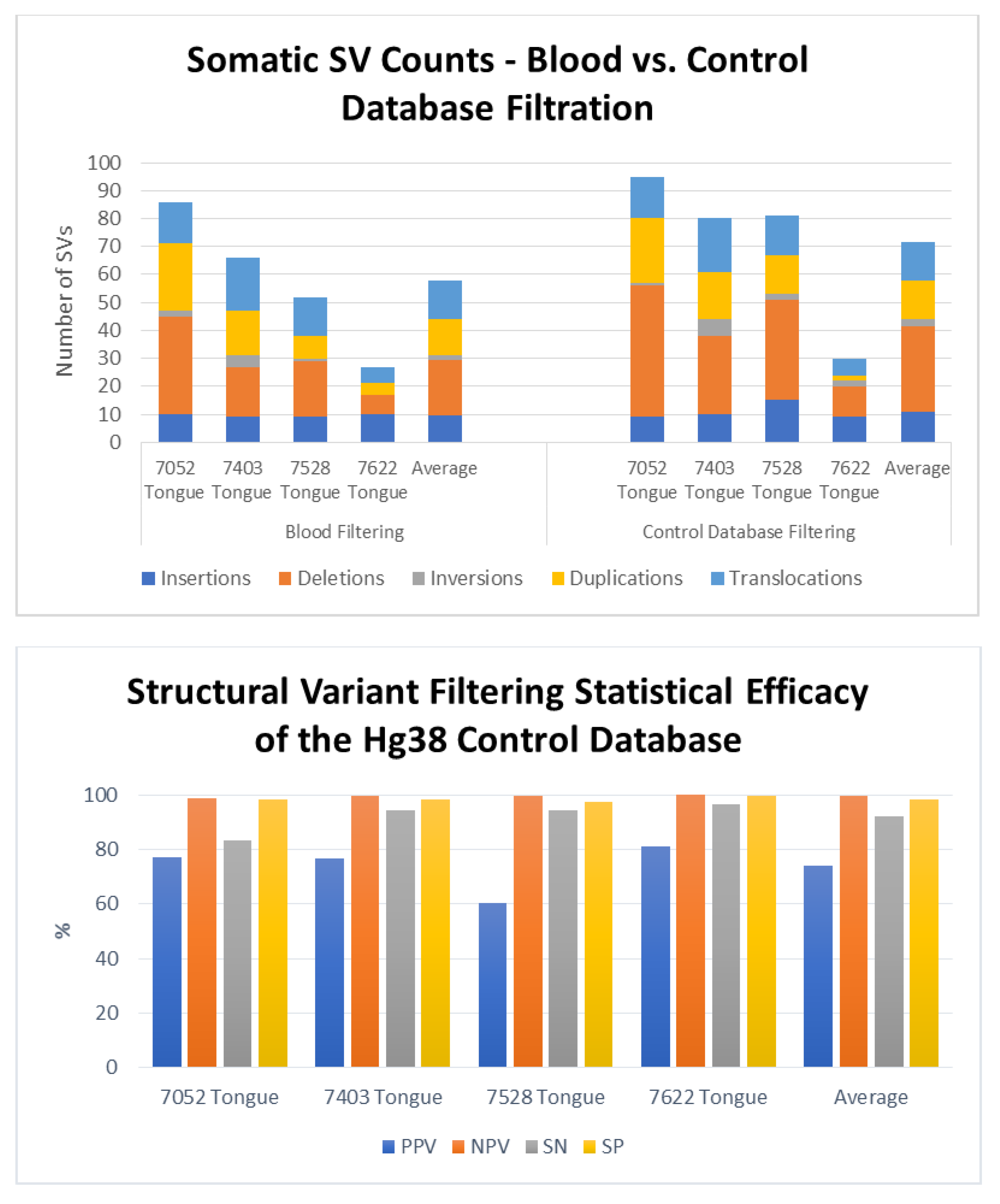
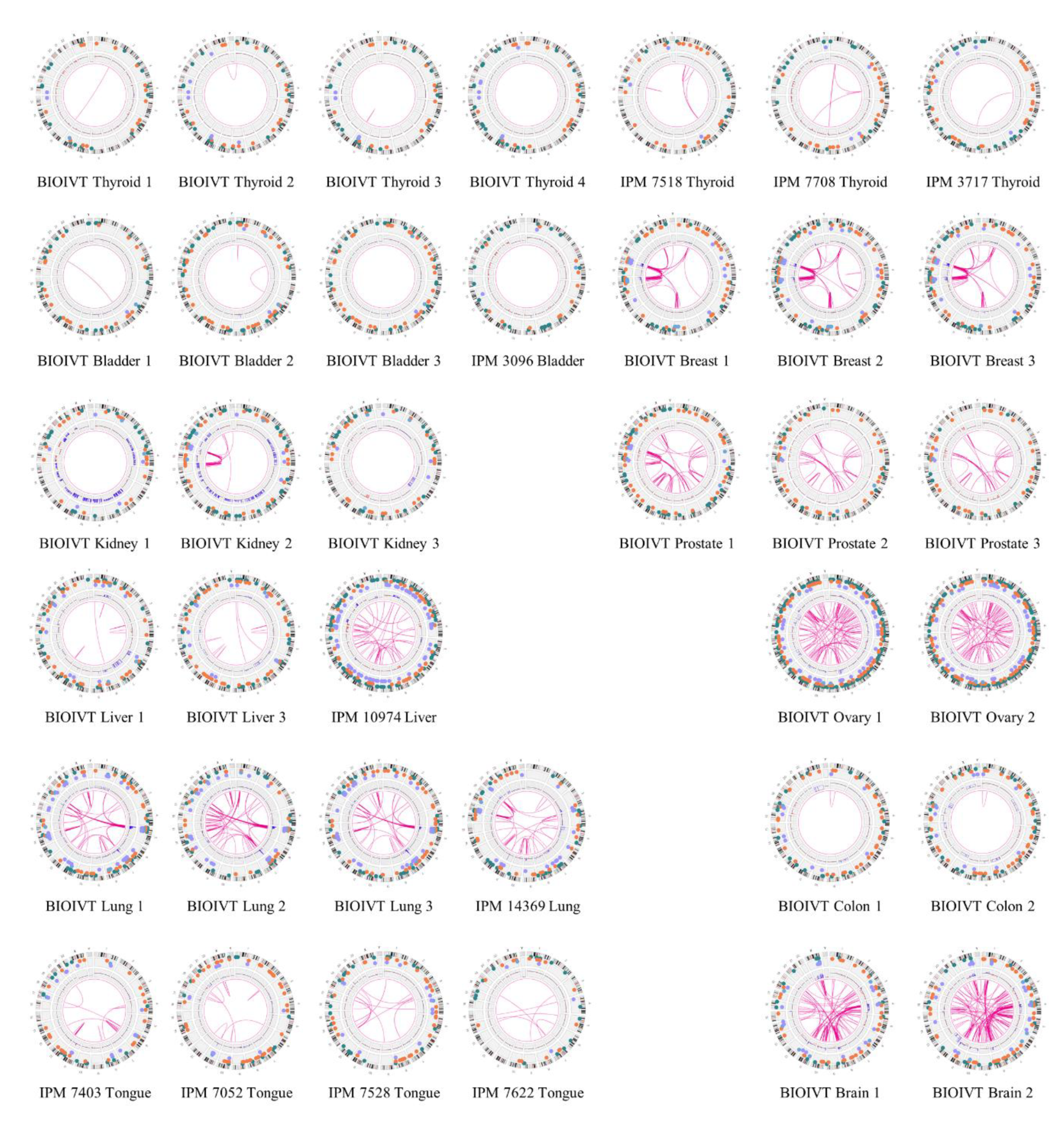
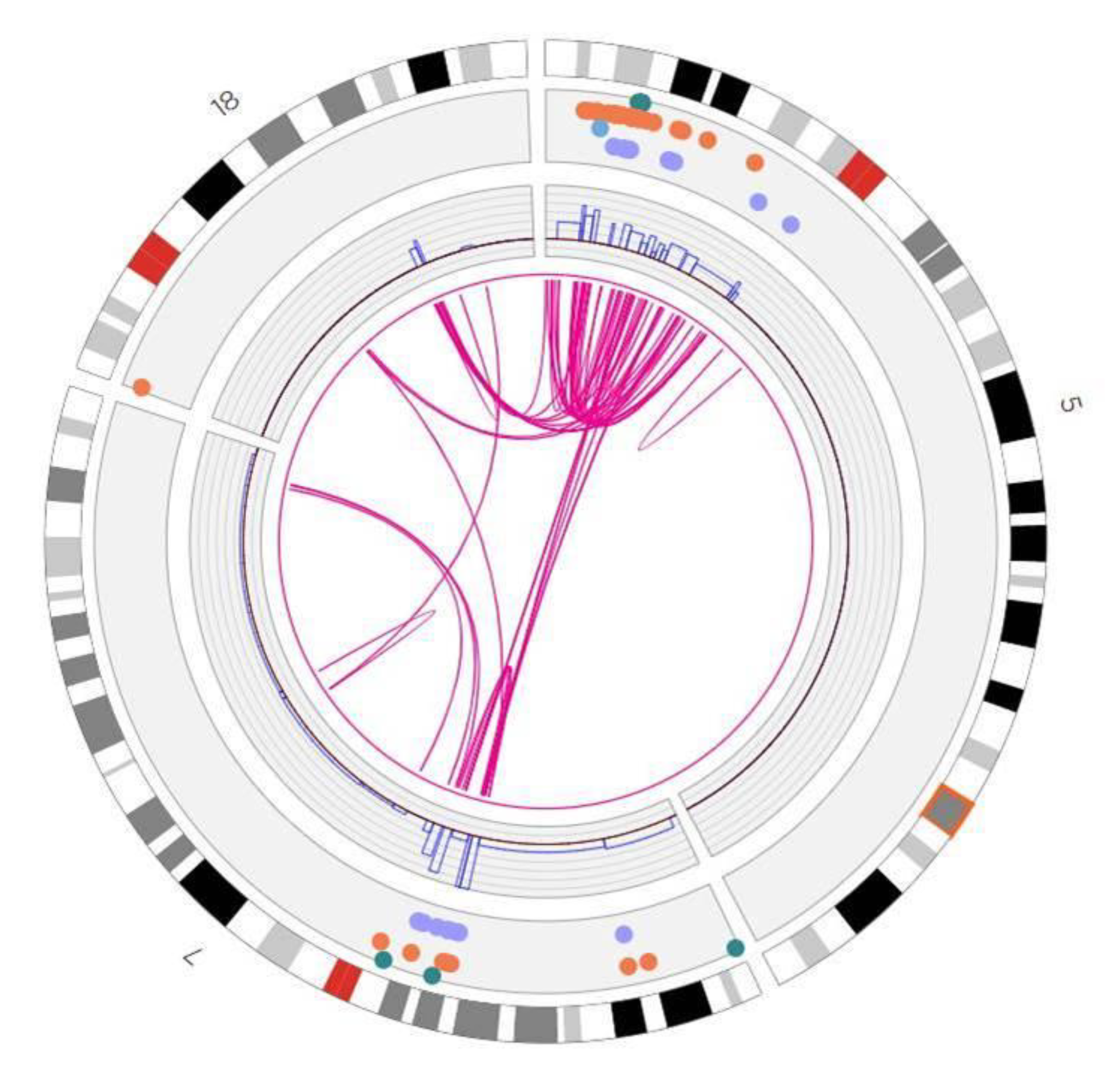
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Sawyers, C.; Hochhaus, A.; Guilhot, F.; Schiffer, C.; Gambacorti-Passerini, C.; Niederwieser, D.; Resta, D.; Capdeville, R.; Zoellner, U.; et al. Hematologic and Cytogenetic Responses to Imatinib Mesylate in Chronic Myelogenous Leukemia. N. Engl. J. Med. 2002, 346, 645–652. [Google Scholar] [CrossRef]
- Kwak, E.L.; Bang, Y.-J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Janne, P.A.; Costa, D.B.; et al. Anaplastic Lymphoma Kinase Inhibition in Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.-I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Xu, J.; Song, F.; Schleicher, E.; Pool, C.; Bann, D.; Hennessy, M.; Sheldon, K.; Batchelder, E.; Annageldiyev, C.; Sharma, A.; et al. An Integrated Framework for Genome Analysis Reveals Numerous Previously Unrecognizable Structural Variants in Leukemia Patients’ Samples. bioRxiv 2019, 563270. [Google Scholar] [CrossRef]
- Zhu, Y.; Brown, H.N.; Zhang, Y.; Stevens, R.G.; Zheng, T. Period3 structural variation: A circadian biomarker associated with breast cancer in young women. Cancer Epidemiol. Biomark. Prev. 2005, 14, 268–270. [Google Scholar]
- Peng, Y.; Yuan, C.; Tao, X.; Zhao, Y.; Yao, X.; Zhuge, L.; Huang, J.; Zheng, Q.; Zhang, Y.; Hong, H.; et al. Integrated analysis of optical mapping and whole-genome sequencing reveals intratumoral genetic heterogeneity in metastatic lung squamous cell carci-noma. Transl. Lung Cancer Res. 2020, 9, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Sajjadian, S.; Eichler, E.E. Limitations of next-generation genome sequence assembly. Nat. Methods 2010, 8, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Song, L.; Cram, D.S.; Xiong, L.; Wang, K.; Wu, R.; Liu, J.; Deng, K.; Jia, B.; Zhong, M.; et al. Traditional karyotyping vs copy number variation sequencing for detection of chromosomal abnormalities associated with spontaneous miscarriage. Ultrasound Obstet. Gynecol. 2015, 46, 472–477. [Google Scholar] [CrossRef]
- Trask, B.J. Human cytogenetics: 46 chromosomes, 46 years and counting. Nat. Rev. Genet. 2002, 3, 769–778. [Google Scholar] [CrossRef]
- Bickhart, D.M.; Rosen, B.D.; Koren, S.; Sayre, B.L.; Hastie, A.R.; Chan, S.; Lee, J.; Lam, E.T.; Liachko, I.; Sullivan, S.T.; et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat. Genet. 2017, 49, 643–650. [Google Scholar] [CrossRef]
- Cao, H.; Hastie, A.R.; Cao, D.; Lam, E.T.; Sun, Y.; Huang, H.; Liu, X.; Lin, L.; Andrews, W.; Chan, S.; et al. Rapid detection of structural variation in a human genome using nanochannel-based genome mapping technology. GigaScience 2014, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Wu, H.; Luo, R.; Huang, S.; Sun, Y.; Tong, X.; Xie, Y.; Liu, B.; Yang, H.; Zheng, H.; et al. De novo assembly of a haplo-type-resolved human genome. Nat. Biotechnol. 2015, 33, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Chaisson, M.J.P.; Sanders, A.D.; Zhao, X.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; Rodriguez, O.L.; Guo, L.; Collins, R.L.; et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019, 10, 1784. [Google Scholar] [CrossRef]
- Cockburn, I.A.; Mackinnon, M.J.; O’Donnell, A.; Allen, S.J.; Moulds, J.M.; Baisor, M.; Bockarie, M.; Reeder, J.C.; Rowe, J.A. A human com-plement receptor 1 polymorphism that reduces Plasmodium falciparum rosetting confers protection against severe malaria. Proc. Natl. Acad. Sci. USA 2004, 101, 272–277. [Google Scholar] [CrossRef]
- Conrad, D.F.; The Wellcome Trust Case Control Consortium; Pinto, D.; Redon, R.; Feuk, L.; Gokcumen, O.; Zhang, Y.; Aerts, J.; Andrews, T.D.; Barnes, C.; et al. Origins and functional impact of copy number variation in the human genome. Nature 2010, 464, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M.; Zerr, T.; Kidd, J.M.; Eichler, E.E.; Nickerson, D.A. Systematic assessment of copy number variant detection via ge-nome-wide SNP genotyping. Nat. Genet. 2008, 40, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- De Cid, R.; Riveira-Munoz, E.; Zeeuwen, P.L.; Robarge, J.; Liao, W.; Dannhauser, E.N.; Giardina, E.; Stuart, P.E.; Nair, R.; Helms, C.; et al. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat. Genet. 2009, 41, 211–215. [Google Scholar] [CrossRef]
- English, A.C.; Salerno, W.J.; Hampton, A.O.; Gonzaga-Jauregui, C.; Ambreth, S.; Ritter, D.I.; Beck, C.R.; Davis, C.F.; Dahdouli, M.; Mahmoud, D.; et al. Assessing structural variation in a personal genome—towards a human reference diploid genome. BMC Genom. 2015, 16, 286. [Google Scholar] [CrossRef] [PubMed]
- Hastie, A.R.; Dong, L.; Smith, A.; Finklestein, J.; Lam, E.T.; Huo, N.; Cao, H.; Kwok, P.Y.; Deal, K.R.; Dvorak, J.; et al. Rapid genome mapping in nanochannel arrays for highly complete and accurate de novo sequence assembly of the complex Aegilops tauschii genome. PLoS ONE 2013, 8, e55864. [Google Scholar] [CrossRef]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.-S.; et al. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef]
- Mak, A.C.; Lai, Y.Y.; Lam, E.T.; Kwok, T.P.; Leung, A.K.; Poon, A.; Mostovoy, Y.; Hastie, A.R.; Stedman, W.; Anantharaman, T.; et al. Genome-Wide Structural Variation Detection by Genome Mapping on Nanochannel Arrays. Genetics 2016, 202, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, M.; Sebra, R.; Pang, A.W.C.; Ummat, A.; Franzen, O.; Rausch, T.; Stütz, A.M.; Stedman, W.; Anantharaman, T.; Hastie, A.; et al. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat. Methods 2015, 12, 780–786. [Google Scholar] [CrossRef]
- Sarsani, V.K.; Raghupathy, N.; Fiddes, I.T.; Armstrong, J.; Thibaud-Nissen, F.; Zinder, O.; Bolisetty, M.; Howe, K.; Hinerfeld, D.; Ruan, X.; et al. The Genome of C57BL/6J “Eve”, the Mother of the Laboratory Mouse Genome Reference Strain. G3 2019, 9, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.-S.; Rhie, A.; Kim, J.; Lee, S.; Sohn, M.-H.; Kim, C.-U.; Hastie, A.; Cao, A.H.H.; Yun, J.-Y.; Kim, J.; et al. De novo assembly and phasing of a Korean human genome. Nature 2016, 538, 243–247. [Google Scholar] [CrossRef]
- Shi, L.; Guo, Y.; Dong, C.; Huddleston, J.; Yang, H.; Han, X.; Fu, A.; Li, Q.; Li, N.; Gong, S.; et al. Long-read sequencing and de novo assembly of a Chinese genome. Nat. Commun. 2016, 7, 12065. [Google Scholar] [CrossRef]
- Usher, C.L.; Handsaker, R.E.; Esko, T.; Tuke, M.A.; Weedon, M.N.; Hastie, A.R.; Cao, H.; Moon, J.E.; Kashin, S.; Fuchsberger, C.; et al. Structural forms of the human amylase locus and their relationships to SNPs, haplotypes and obesity. Nat. Genet. 2015, 47, 921–925. [Google Scholar] [CrossRef]
- Zook, J.M.; Hansen, N.F.; Olson, N.D.; Chapman, L.; Mullikin, J.C.; Xiao, C.; Sherry, S.; Koren, S.; Phillippy, A.M.; Boutros, P.C.; et al. A robust benchmark for detection of germline large deletions and insertions. Nat. Biotechnol. 2020, 38, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Neveling, K.; Mantere, T.; Vermeulen, S.; Oorsprong, M.; van Beek, R.; Kater-Baats, E.; Pauper, M.; van der Zande, G.; Smeets, D.; Weghuis, D.O.; et al. Next generation cytogenetics: Comprehensive assessment of 48 leukemia genomes by genome imaging. bioRxiv 2020, preprint. [Google Scholar] [CrossRef]
- Dixon, J.R.; Xu, J.; Dileep, V.; Zhan, Y.; Song, F.; Le, V.T.; Yardimci, G.G.; Chakraborty, A.; Bann, D.V.; Wang, Y.; et al. Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet. 2018, 50, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Lam, E.; Saghbini, M.; Bocklandt, S.; Hastie, A.; Cao, H.; Holmlin, E.; Borodkin, M. Structural Variation Detection and Analysis Using Bionano Optical Mapping. Methods Mol. Biol. 2018, 1833, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Pang, A.; Lee, J.; Anantharaman, T.; Lam, E.; Hastie, A.; Borodkin, M. Comprehensive Detection of Germline and Somatic Structural Mutation in Cancer Genomes by Bionano Genomics Optical Mapping. J. Biomol. Technol. 2019, 30, S9. [Google Scholar]
- Zhang, Y.; Broach, J. Abstract 5125: A novel method for isolating high-quality UHMW DNA from 10 mg of freshly frozen or liquid-preserved animal and human tissue including solid tumors. Cancer Res. 2019, 79, 5125. [Google Scholar]
- Crumbaker, M.; Chan, E.K.F.; Gong, T.; Corcoran, N.; Jaratlerdsiri, W.; Lyons, R.J.; Haynes, A.M.; Kulidjian, A.A.; Kalsbeek, A.M.F.; Petersen, D.C.; et al. The Impact of Whole Genome Data on Therapeutic Decision-Making in Metastatic Prostate Cancer: A Retrospective Analysis. Cancers 2020, 12, 1178. [Google Scholar] [CrossRef]
- Jaratlerdsiri, W.; Chan, E.K.; Petersen, D.C.; Yang, C.; Croucher, P.I.; Bornman, M.R.; Sheth, P.; Hayes, V.M. Next generation mapping reveals novel large genomic rearrangements in prostate cancer. Oncotarget 2017, 8, 23588–23602. [Google Scholar] [CrossRef] [PubMed]
- Whitlock, R.; Hipperson, H.; Mannarelli, M.; Burke, T. A high-throughput protocol for extracting high-purity genomic DNA from plants and animals. Mol. Ecol. Resour. 2008, 8, 736–741. [Google Scholar] [CrossRef]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “per-sonalized” approach to cancer staging. CA Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Cortés-Ciriano, I.; Lee, J.J.K.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.Z.; Pellman, D.S.; et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.F.; Mardis, E.R. The emerging clinical relevance of genomics in cancer medicine. Nat. Rev. Clin. Oncol. 2018, 15, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.-Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study ID | Cancer Type * | Age † | M/F | Ethnicity | Smoking History | Alcohol History | Pathologic TNM ‡ | Cancer Stage |
|---|---|---|---|---|---|---|---|---|
| 7528 | Tongue (SCC) | 25 | M | Caucasian | None | Rare | T3N2bM0 | IVa |
| 7052 | Tongue (SCC) | 35 | M | Caucasian | None | None | T2N3M0 | IVb |
| 7622 | Tongue (SCC) | 60 | F | Caucasian | 50 pack years | 1–2 drinks/week | T3N0M0 | III |
| 7403 | Tongue (SCC) | 65 | M | Caucasian | 45 pack years | Rare | T2N3bM0 | IVb |
| 7518 | Thyroid (AP) | 70 | F | Caucasian | 20 pack years | 2 drinks per day | T4bN1bM1 | IVc |
| 7708 | Thyroid (AP) | 65 | M | Caucasian | None | None | 4aN1bM1 | IVc |
| 3717 | Thyroid (AP) | 80 | M | Hispanic | 25 pack years | Rare | T4aN1aM1 | IV |
| 14369 | Lung (pleomorphic carcinoma) | 60 | M | N/A | 60 pack years | None | T2bN1M0 | IIa |
| 10974 | Liver (metastic adenocarcino-ma of colon) | 65 | F | N/A | Former | None | T3N2aM1 | IVB |
| 3096 | Bladder (urothelial carcinoma) | 55 | M | Caucasian | 60 pack years | None | T2N0M0 | II |
| 73432 | Lung (adeno-squamous carcinoma) | 35 | M | Asian | Former (5 pack years) | Former (1 per day, 10 years) | T2aN1M0 | IIA |
| 94894 | Liver (hepato-cellular carcinoma) | 70 | M | Asian | 7 pack years | 1 per day, 35 years | T1NxM0 | I |
| 101558 | Bladder (papillary urothelial carcinoma) | 65 | M | Asian | Former (5 pack years) | 1 per day, 20 years | T2NxM0 | II |
| 69033 | Kidney (renal cell carcinoma) | 60 | F | Asian | None | None | T2bNxM0 | II |
| 79379 | Breast (ductal carcinoma in situ) | 50 | F | Asian | None | None | T3N0M0 | IIB |
| 102095 | Prostate (invasive adeno-carcinoma) | 60 | M | Caucasian | 40 pack years | None | T3bN1M0 | IV |
| 80384 | Brain (anaplastic astrocytoma) | 40 | F | Caucasian | None | None | NA | NA |
| 81347 | Ovarian (serous carcinoma) | 75 | F | Asian | None | None | T1aN0M0 | IA |
| 119664 | Colon Cancer (adenocarcinoma) | 80 | M | Asian | 2 pack years | 1 per day, 40 years | TXNXMX | UNK |
| 128019 | Thyroid (papillary) | 35 | F | Asian | None | None | T3bNxM0 | I |
| Tissue | No. of Duplicates | Input (mg) | DNA (ng/µL) | DNA Yield (µg/mg) | N50 Kbp (>20 Kbp) | N50 Kbp (>150 Kbp) | Labels/100 kbp | Map Rate (%) | Gbp/Scan | Effective Coverage |
|---|---|---|---|---|---|---|---|---|---|---|
| 7528 (tongue) | 1 | 17.5 | 37 | 0.12 | 211 | 317 | 12.3 | 58.8 | 53 | 237× |
| 7052 (tongue) | 1 | 17.1 | 81 | 0.28 | 179 | 287 | 15.2 | 82.4 | 37 | 345× |
| 7622 (tongue) | 1 | 18.7 | 160 | 0.51 | 315 | 361 | 13.4 | 75.8 | 64 | 317× |
| 7403 (tongue) | 1 | 18 | 79 | 0.26 | 148 | 272 | 14.8 | 72.8 | 33 | 304× |
| 7518 (thyroid) | 1 | 8.6 | 28 | 0.20 | 143 | 265 | 14.4 | 76.6 | 26 | 312× |
| 7708 (thyroid) | 1 | 10.6 | 85 | 0.47 | 269 | 356 | 13.1 | 61.2 | 35 | 253× |
| 3717 (thyroid) | 1 | 13.2 | 49 | 0.22 | 250 | 320 | 14.5 | 88.2 | 58 | 371× |
| 14369 (lung) | 1 | 11.4 | 87 | 0.45 | 268 | 323 | 14.0 | 89.6 | 36 | 372× |
| 10974 (liver) | 1 | 6.5 | 82 | 0.74 | 235 | 289 | 15.2 | 87.9 | 49 | 360× |
| 3096 (bladder) | 1 | 9.4 | 59 | 0.37 | 265 | 319 | 13.8 | 78.3 | 39 | 325× |
| 73432 (lung) | 3 | 9.6 | 128 | 0.86 | 248 | 304 | 15.0 | 90.4 | 51 | 339× |
| 94894 (liver) | 2 | 9.0 | 196 | 1.41 | 265 | 306 | 14.9 | 89.3 | 84 | 325× |
| 101558 (bladder) | 3 | 9.7 | 245 | 1.64 | 313 | 357 | 15.2 | 91.8 | 66 | 338× |
| 69033 (kidney) | 3 | 10 | 96 | 0.63 | 201 | 269 | 14.6 | 83.5 | 41 | 296× |
| 79379 (breast) | 3 | 13.3 | 183 | 1.04 | 317 | 395 | 14.2 | 84.1 | 77 | 288× |
| 102095 (prostate) | 3 | 10.3 | 113 | 0.72 | 273 | 361 | 14.8 | 85.1 | 62 | 295× |
| 80384 (brain) | 2 | 10.5 | 168 | 1.06 | 228 | 292 | 14.6 | 90.2 | 42 | 306× |
| 81347 (ovary) | 2 | 10.5 | 168 | 1.05 | 228 | 292 | 14.6 | 90.2 | 42 | 330× |
| 119664 (colon) | 2 | 11.3 | 231 | 1.33 | 263 | 330 | 14.9 | 88.6 | 42 | 274× |
| 128019 (thyroid) | 4 | 10 | 126 | 0.77 | 213 | 294 | 14.5 | 87.6 | 64 | 294× |
| Average | 1.9 | 11.8 | 120. | 0.71 | 241. | 315. | 14.4 | 82.6 | 50.0 | 314× |
| Total | % | Insertion | % | Deletion | % | Inversion | % | Duplication | % | Translocation | % | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Brain * | 134 | 70 | 5 | 63 | 21 | 78 | 9 | 69 | 13 | 72 | 86 | 69 |
| Colon * | 63 | 93 | 15 | 83 | 36 | 97 | 2 | 100 | 9 | 90 | 1 | 100 |
| Liver * | 45 | 70 | 14 | 74 | 21 | 81 | 1 | 17 | 4 | 67 | 5 | 71 |
| Ovary ‡ | 338 | 86 | 136 | 82 | 59 | 87 | 4 | 80 | 40 | 85 | 99 | 91 |
| Bladder ‡ | 30 | 88 | 11 | 79 | 18 | 100 | 1 | 100 | 0 | 100 | 0 | 0 |
| Breast ‡ | 221 | 92 | 9 | 82 | 33 | 85 | 23 | 88 | 14 | 88 | 142 | 95 |
| Kidney ‡ | 19 | 76 | 6 | 67 | 11 | 85 | 2 | 100 | 0 | 0 | 0 | 100 |
| Lung ‡ | 221 | 66 | 18 | 69 | 53 | 75 | 59 | 73 | 26 | 50 | 65 | 63 |
| Prostate ‡ | 69 | 48 | 8 | 47 | 22 | 61 | 3 | 38 | 1 | 25 | 35 | 44 |
| Thyroid ‡ | 19 | 86 | 7 | 88 | 10 | 91 | 0 | 100 | 2 | 100 | 0 | 0 |
| Average (all) | 116 | 78 | 23 | 73 | 28 | 84 | 10 | 76 | 11 | 68 | 43 | 63 |
| Duplicate Average | 145 | 80 | 43 | 75 | 34 | 86 | 4 | 66 | 17 | 78 | 48 | 83 |
| Triplicate Average | 97 | 76 | 10 | 72 | 25 | 83 | 15 | 83 | 7 | 60 | 40 | 50 |
| Sample | Oncogene | Tumor Suppressor | Gene Fusion |
|---|---|---|---|
| Prostate | ERBB2 (Dup) | PTEN (Del) | PTEN-LINC01374 |
| GATA2 (T) | NF1 (Del) | DHX30-GATA2 | |
| NUP98 (T) | CASC15-NUP98 | ||
| PRKAR1A-FRMPD4 | |||
| ERG-TMPRSS2 | |||
| FREM1-MYH9 | |||
| Ovarian | NUMA1 (T) | NBEA-ZFHX3 | |
| NF1 (I) | HMGN2P46-BLOC1S6 | ||
| SMARCA4 (I) | LPP-PIEZO1 | ||
| Kidney | PRKAR1A (T) | CDKN2A (Del) | PRKAR1A-FRMPD4 |
| ERBB2 (Dup) | ZFHX3 (Del) | ||
| Colon | FHIT (Del) | ||
| Breast | ERBB4 (Dup) | USP8 (T) | USP8-PRPSAP |
| ERBB2 (Dup) | PRKAR1A (T) | PRKAR1A-FRMPD4 | |
| RAD51B (Del) | LINC01476-BRIP1 | ||
| CDKN2A (Del) | SYK-CFAP77 | ||
| Brain | SETBP1 (T) | LARP4B (T) | CCDC158-LARP4B |
| CSMD3 (T) | CA13-CSMD3 | ||
| LRP1B (Del) | DPYD-SETBP1 | ||
| RAD21 (Del) | CNBD1-AC083836.1 | ||
| Bladder | DDX10 (Del) | ||
| Tongue | BRAF (T) | CDKN2A (Del) | EPHB1-BRAF |
| CDK6 (T) | PTPRD (Del) | CDK6--AC091551.1 | |
| CCND2 (T) | RAD51B (T) | PCLO-RAD51B | |
| CCND1 (Dup) | LRP1B (Del) | ||
| CDKN1B (Del) | |||
| Thyroid | YWHAE (T) | ABR-YWHAE | |
| PTPRD (Del) | CDK12-CSF3 | ||
| RAD18-SRGAP3 | |||
| SHROOM3-AFF1 | |||
| Liver | VTI1A (T) | RMI2 (T) | VTI1A-NHLRC2 |
| MAP3K13 (T) | NCOR (T) | C3orf70-MAP3K13 | |
| MACC1 (T) | CBLC (T) | AC005062.1-MACC1 | |
| NSD3 (T) | MSH2 (T) | NSD3-AC087623.2 | |
| RASGEF1B-VTI1A | |||
| RMI2-TOX3 | |||
| NCOR1-LRRC75A | |||
| MSH2-CYP3A43 | |||
| Lung | CTNND2 (Del,T) | PTPRD (Del) | CTNND2-TRIO |
| IKBKB (T) | RAD51B (T) | DUSP10-CTNND2 | |
| FUS (T) | IKBKB-FAM91A1 | ||
| LRP1B (T) | FUS-CNOT1 | ||
| PDE6D-RAD51B | |||
| PRKCH-HIF1A | |||
| GAS7-LYRM9 | |||
| EHBP1-LRP1B |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goldrich, D.Y.; LaBarge, B.; Chartrand, S.; Zhang, L.; Sadowski, H.B.; Zhang, Y.; Pham, K.; Way, H.; Lai, C.-Y.J.; Pang, A.W.C.; et al. Identification of Somatic Structural Variants in Solid Tumors by Optical Genome Mapping. J. Pers. Med. 2021, 11, 142. https://doi.org/10.3390/jpm11020142
Goldrich DY, LaBarge B, Chartrand S, Zhang L, Sadowski HB, Zhang Y, Pham K, Way H, Lai C-YJ, Pang AWC, et al. Identification of Somatic Structural Variants in Solid Tumors by Optical Genome Mapping. Journal of Personalized Medicine. 2021; 11(2):142. https://doi.org/10.3390/jpm11020142
Chicago/Turabian StyleGoldrich, David Y., Brandon LaBarge, Scott Chartrand, Lijun Zhang, Henry B. Sadowski, Yang Zhang, Khoa Pham, Hannah Way, Chi-Yu Jill Lai, Andy Wing Chun Pang, and et al. 2021. "Identification of Somatic Structural Variants in Solid Tumors by Optical Genome Mapping" Journal of Personalized Medicine 11, no. 2: 142. https://doi.org/10.3390/jpm11020142
APA StyleGoldrich, D. Y., LaBarge, B., Chartrand, S., Zhang, L., Sadowski, H. B., Zhang, Y., Pham, K., Way, H., Lai, C.-Y. J., Pang, A. W. C., Clifford, B., Hastie, A. R., Oldakowski, M., Goldenberg, D., & Broach, J. R. (2021). Identification of Somatic Structural Variants in Solid Tumors by Optical Genome Mapping. Journal of Personalized Medicine, 11(2), 142. https://doi.org/10.3390/jpm11020142