
ETS-Domain Transcription Factor Elk-1 Regulates Stemness Genes in Brain Tumors and CD133+ BrainTumor-Initiating Cells



 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and BTIC Isolation from Cell Lines and Primary Tumors
2.2. Dissociation and Culture of Primary GBM Tissue
2.3. Transient Transfection of Cells
2.4. Soft Agar Assay
2.5. Limiting Dilution Analysis (LDA)
2.6. RNA Isolation, cDNA Synthesis, Reverse Transcription Polymerase Chain Reaction (RT-PCR), and Real-Time PCR
2.7. Microarray and Data Analysis
2.8. Promoter Clonings and Site-Directed Mutagenesis
2.9. Luciferase Reporter Assay
2.10. Chromatin Immunoprecipitation (ChIP) Assay
3. Results
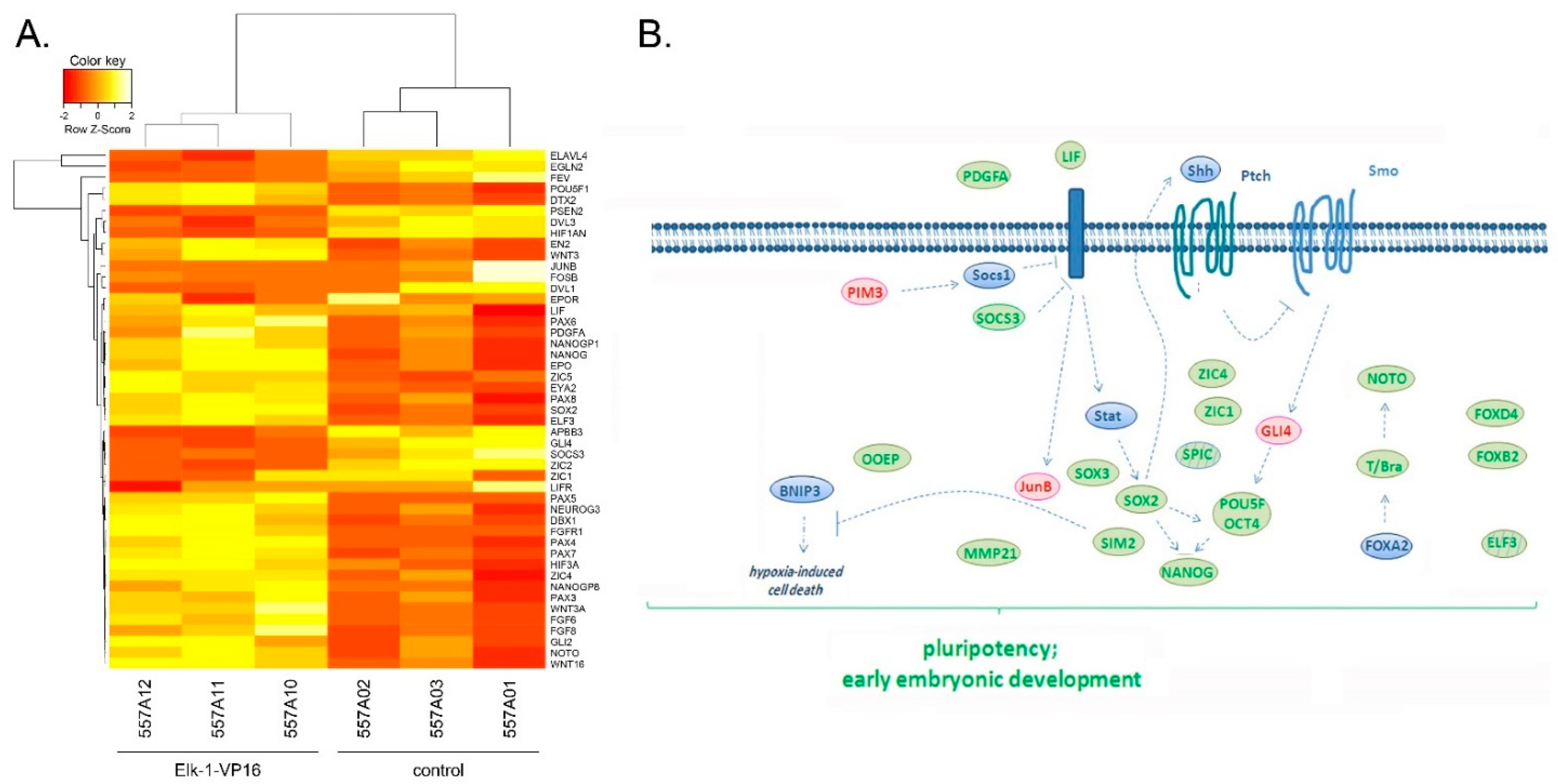
3.1. Microarray Analyses Reveal Novel Targets in Elk-1-VP16 Overexpressing SH-SY5Y Cells
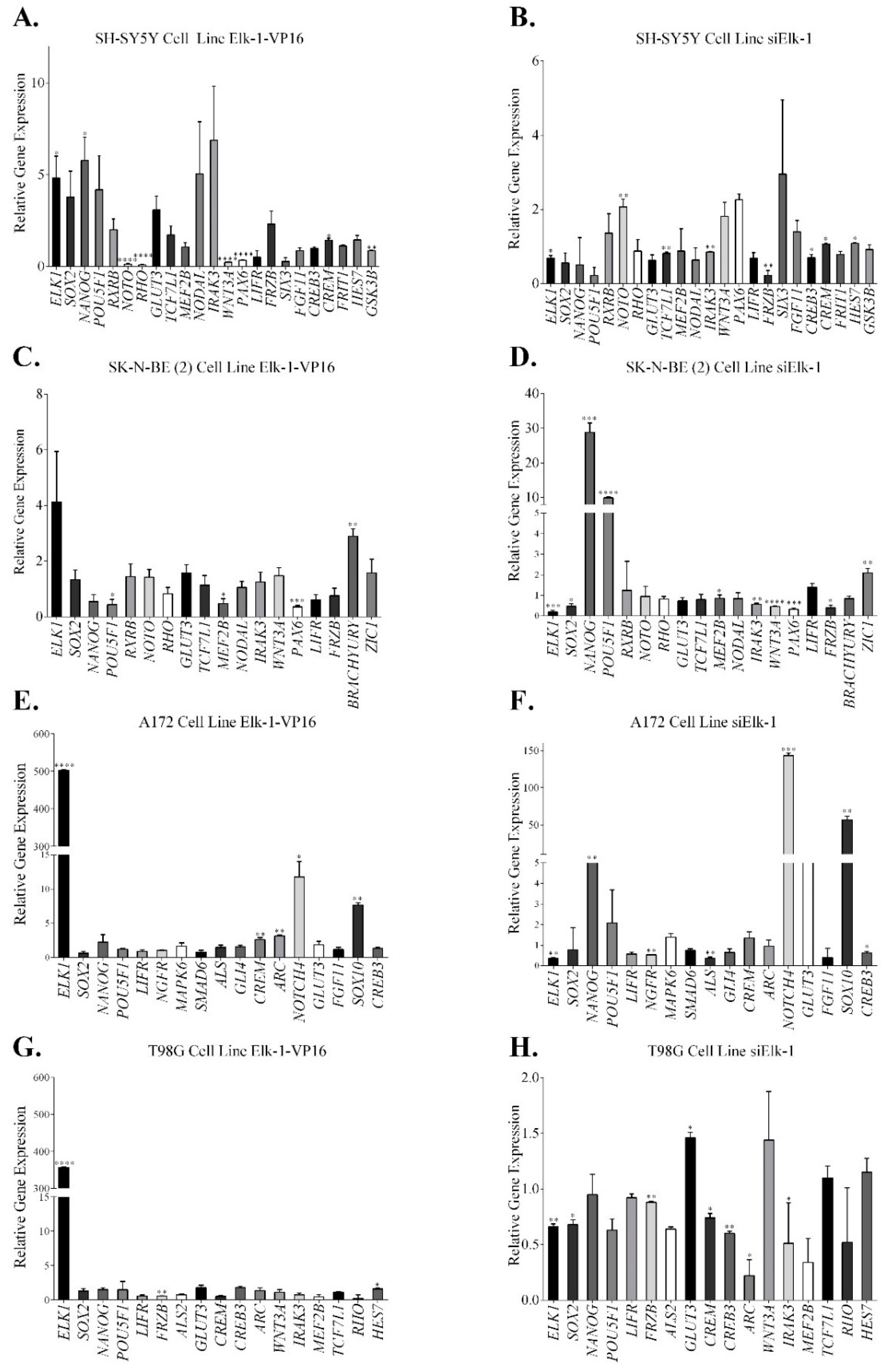
3.2. Regulation of Nervous System Development Related Genes by Elk-1
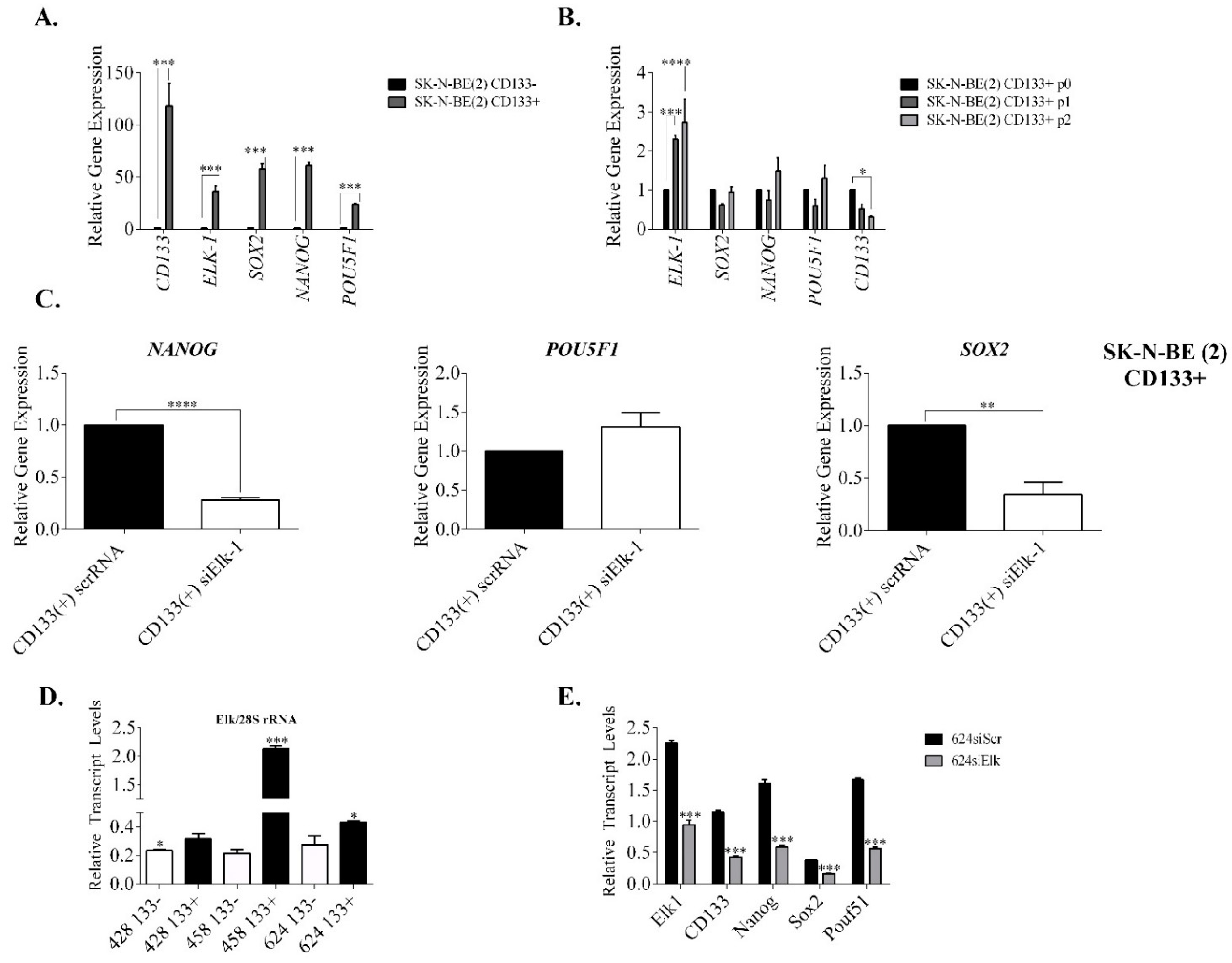
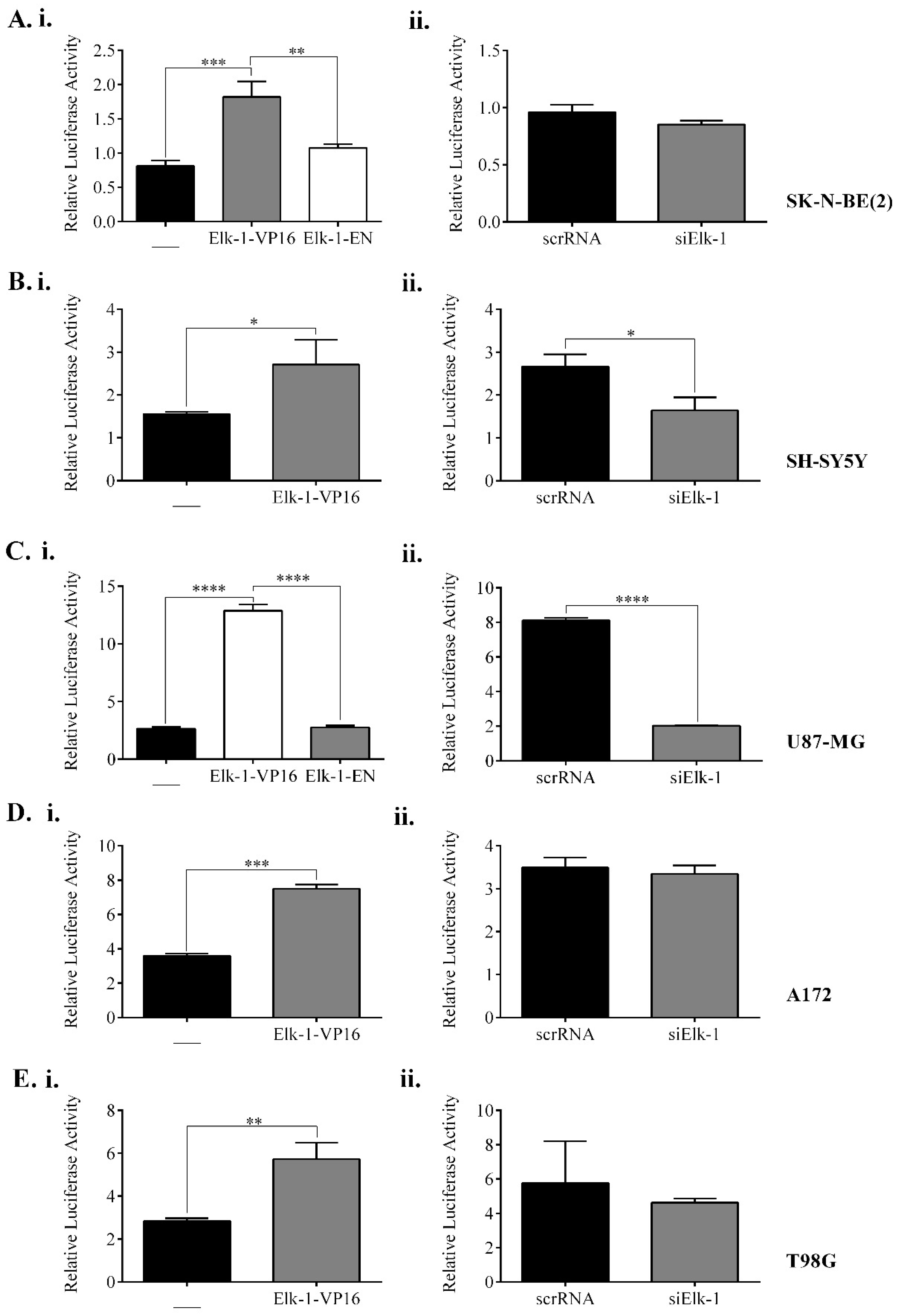
3.3. Regulation of SOX2, NANOG, and POU5F1 by Elk-1 in CD133+ Cells
3.4. Effect of Elk-1 Expression on Colony Formation of SK-N-BE (2) Cells on Soft Agar
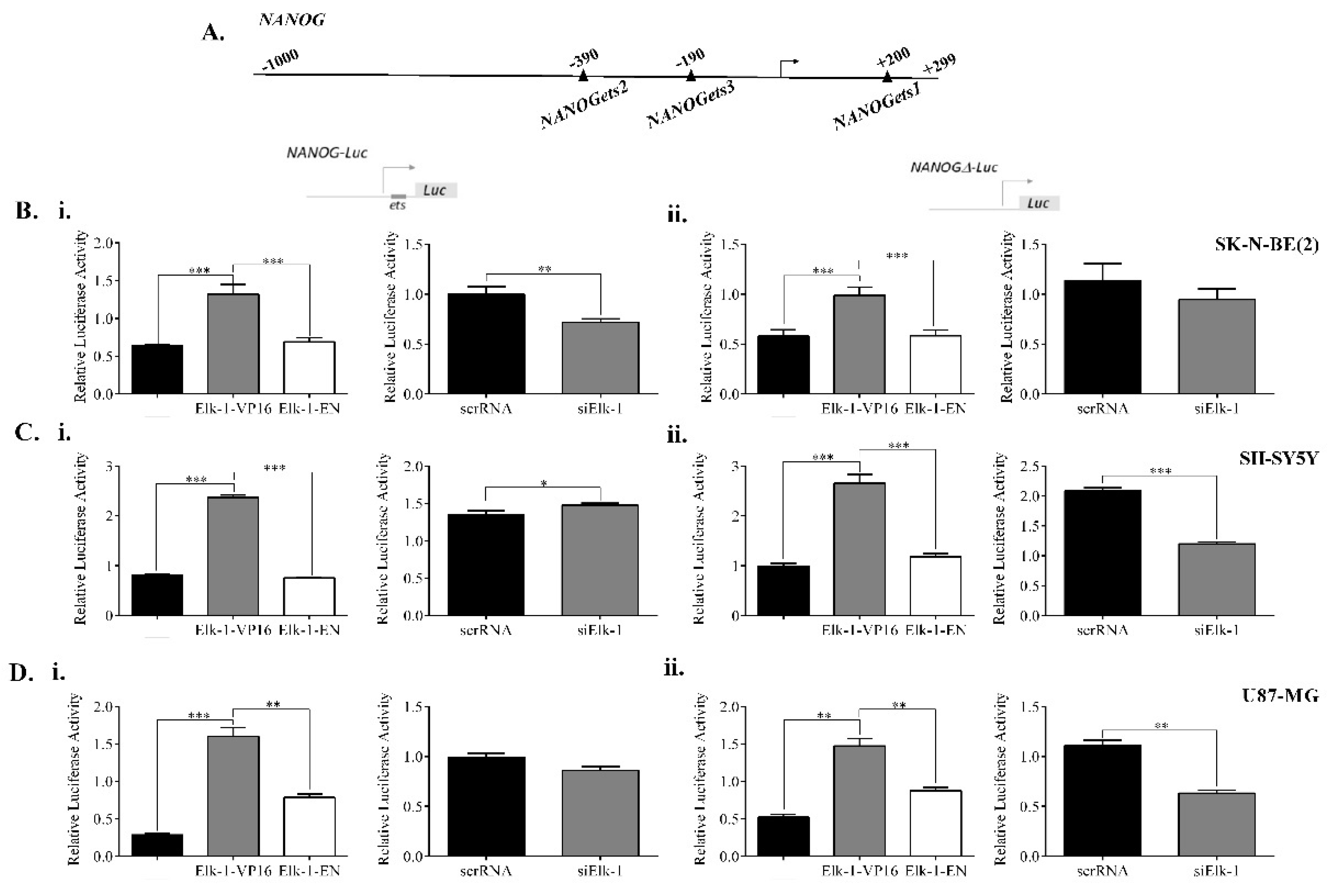
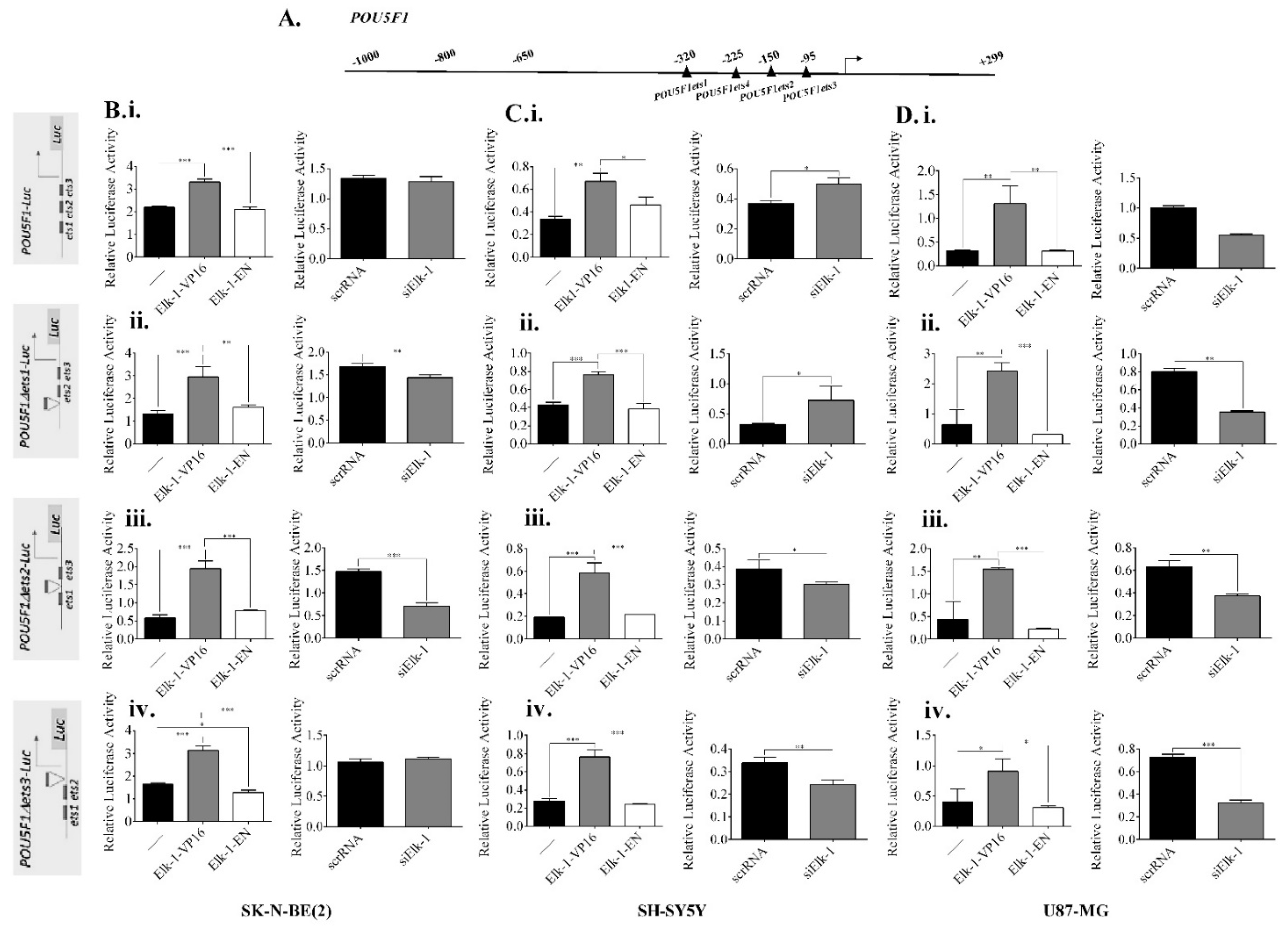
3.5. Regulation of NANOG, POU5F1, and SOX2 Promoters by Elk-1
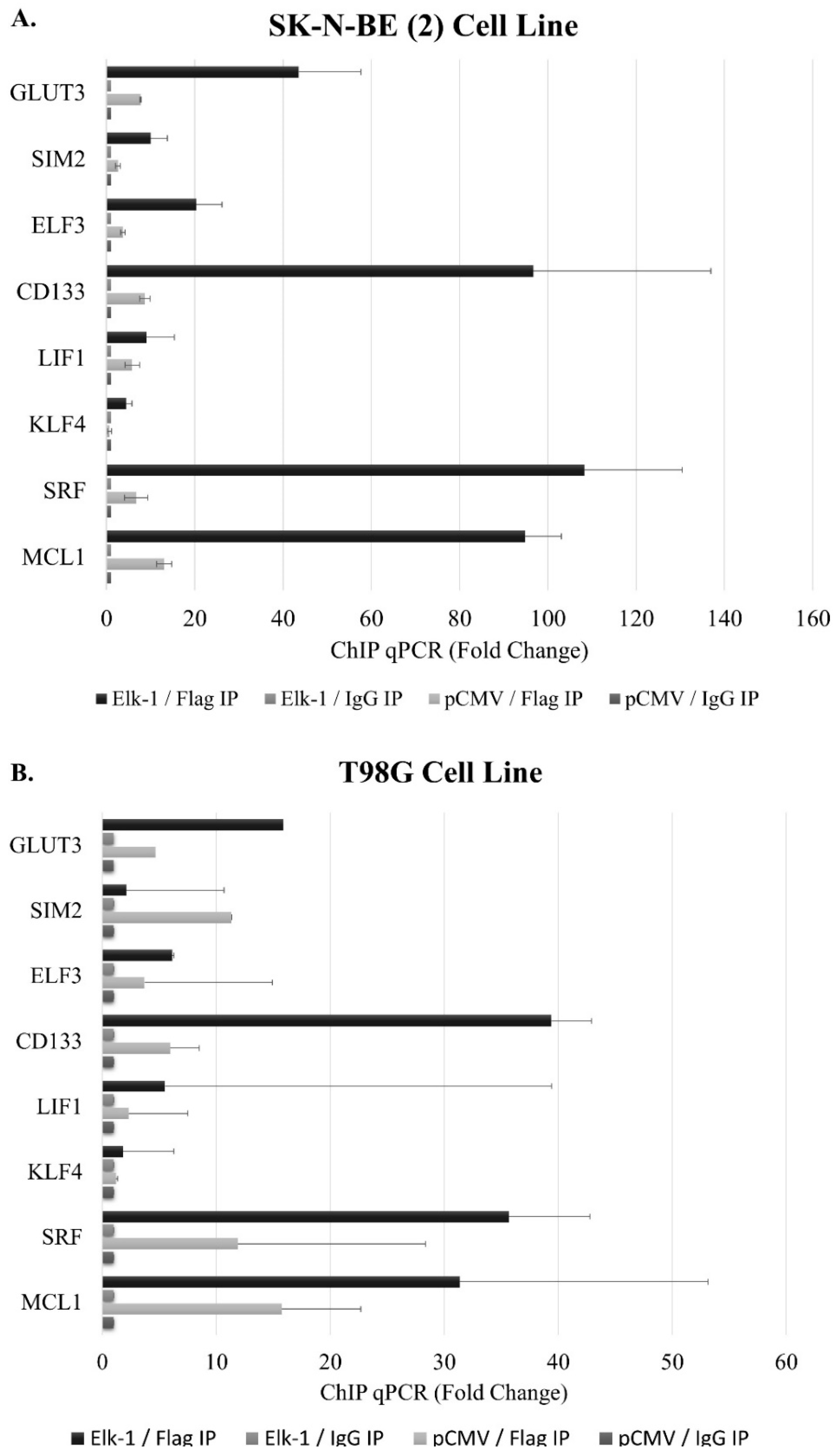
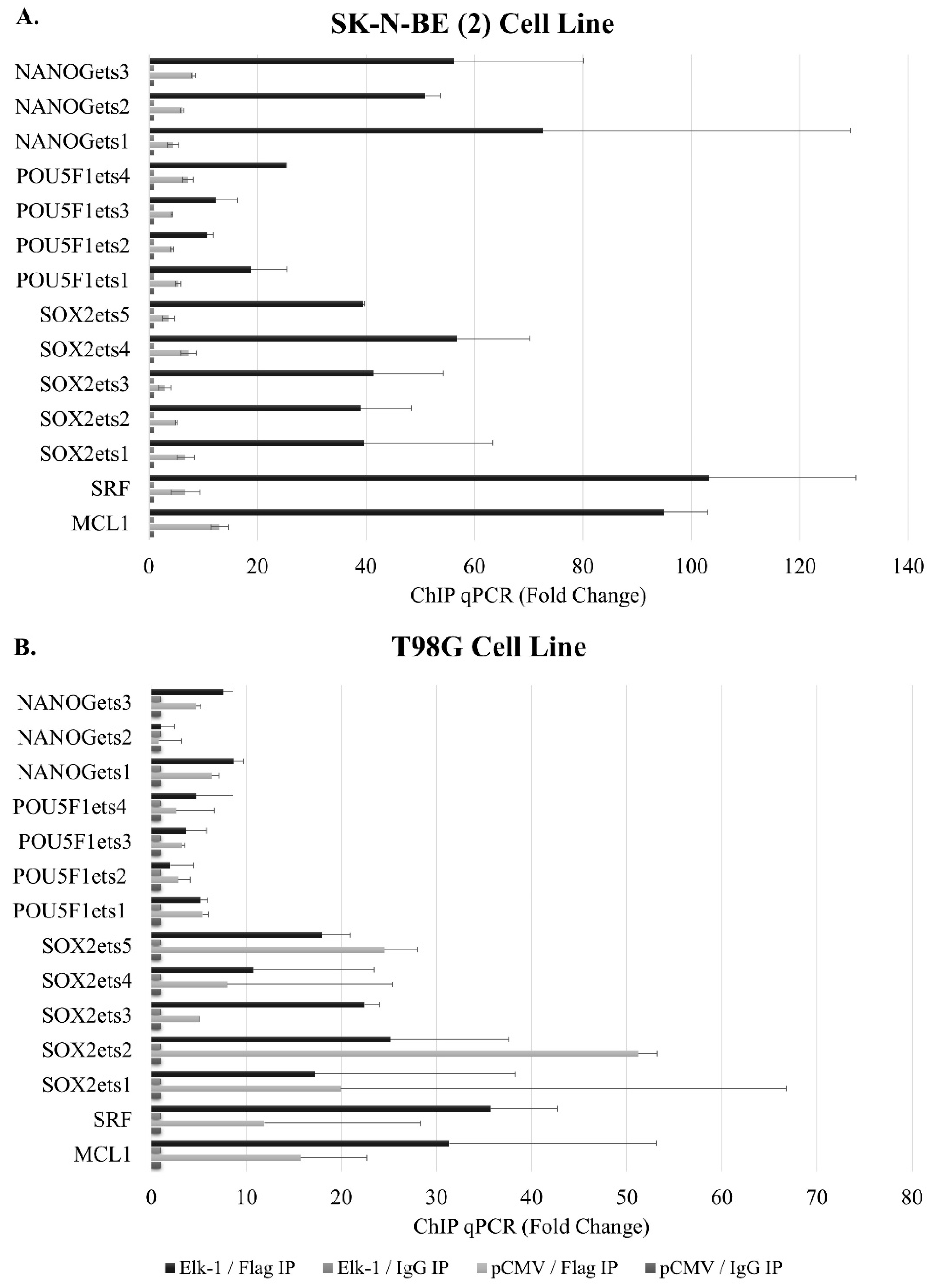
3.6. Binding of Elk-1 to Predicted ets Motifs on SOX2, NANOG, and POU5F1 Promoters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davis, S.P.; Vanhoutte, C.; Pages, J. Caboche and S. Laroche. The MAPK/ERK cascade targets both Elk-1 and cAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. J. Neurosci. 2000, 20, 4563–4572. [Google Scholar] [CrossRef]
- Demir, O.; Ari, O.; Kurnaz, I.A. Elk-1 interacts with dynein upon serum stimulation but independent of serine 383 phosphorylation. Cell. Mol. Neurobiol. 2012, 32, 185–189. [Google Scholar] [CrossRef]
- Demir, O.; Korulu, S.; Yildiz, A.; Karabay, A.; Kurnaz, I.A. Elk-1 interacts with neuronal microtubules and relocalizes to the nucleus upon phosphorylation. Mol. Cell. Neurosci. 2009, 40, 111–119. [Google Scholar] [CrossRef]
- Kaminska, B.; Kaczmarek, L.; Zangenehpour, S.; Chaudhuri, A. Rapid phosphorylation of Elk-1 transcription factor and activation of MAP kinase signal transduction pathways in response to visual stimulation. Mol. Cell. Neurosci. 1999, 13, 405–414. [Google Scholar] [CrossRef]
- Sananbenesi, F.; Fischer, A.; Schrick, C.; Spiess, J.; Radulovic, J. Phosphorylation of hippocampal Erk-1/2, Elk-1, and p90-Rsk-1 during contextual fear conditioning: Interactions between Erk-1/2 and Elk-1. Mol. Cell. Neurosci. 2002, 21, 463–476. [Google Scholar] [CrossRef]
- Sgambato, V.; Vanhoutte, P.; Pages, C.; Rogard, M.; Hipskind, R.; Besson, M.J.; Caboche, J. In vivo expression and regulation of Elk-1, a target of the extracellular-regulated kinase signaling pathway, in the adult rat brain. J. Neurosci. 1998, 18, 214–226. [Google Scholar] [CrossRef]
- Yang, S.H.; Shore, P.; Willingham, N.; Lakey, J.H.; Sharrocks, A.D. The mechanism of phosphorylation-inducible activation of the ETS-domain transcription factor Elk-1. EMBO J. 1999, 18, 5666–5674. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Whitmarsh, A.J.; Davis, R.J.; Sharrocks, A.D. Differential targeting of MAP kinases to the ETS-domain transcription factor Elk-1. EMBO J. 1998, 17, 1740–1749. [Google Scholar] [CrossRef]
- Sharrocks, A.D. The ETS-domain transcription factor family. Nat. Rev. Mol. Cell Biol. 2001, 2, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Boros, J.; Donaldson, I.J.; O’donnell, A.; Odrowaz, Z.A.; Zeef, L.; Lupien, M.; Meyer, C.A.; Liu, X.S.; Brown, M.; Sharrocks, A.D. Elucidation of the ELK1 target gene network reveals a role in the coordinate regulation of core components of the gene regulation machinery. Genome Res. 2009, 19, 1963–1973. [Google Scholar] [CrossRef]
- Demir, O.; Aysit, N.; Onder, Z.; Turkel, N.; Ozturk, G.; Sharrocks, A.D.; Kurnaz, I.A. ETS-domain transcription factor Elk-1 mediates neuronal survival: SMN as a potential target. Biochim. Biophys. Acta 2011, 1812, 652–662. [Google Scholar] [CrossRef]
- Lenkiewicz, M.; Li, N.; Singh, S.K. Culture and isolation of brain tumor initiating cells. Curr. Protoc. Stem Cell Biol. 2009, 11, 3.3.1–3.3.10. [Google Scholar] [CrossRef]
- Mahller, Y.Y.; Williams, J.P.; Baird, W.H.; Mitton, B.; Grossheim, J.; Saeki, Y.; Cancelas, J.A.; Ratner, N.; Cripe, T.P. Neuroblastoma cell lines contain pluripotent tumor initiating cells that are susceptible to a targeted oncolytic virus. PLoS ONE 2009, 4, e4235. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Ohnishi, S.; Maehara, O.; Nakagawa, K.; Kameya, A.; Otaki, K.; Fujita, H.; Higashi, R.; Takagi, K.; Asaka, M.; Sakamoto, N.; et al. hypoxia-inducible factors activate CD133 promoter through ETS family transcription factors. PLoS ONE 2013, 8, e66255. [Google Scholar] [CrossRef]
- Prise, I.; Sharrocks, A.D. ELK1 has a dual activating and repressive role in human embryonic stem cells. Wellcome Open Res. 2019, 4, 41. [Google Scholar] [CrossRef]
- Vora, P.; Seyfrid, M.; Venugopal, C.; Qazi, M.A.; Salim, S.; Isserlin, R.; Subapanditha, M.; O’farrell, E.; Mahendram, S.; Singh, M.; et al. Bmi1 regulates human glioblastoma stem cells through activation of differential gene networks in CD133+ brain tumor initiating cells. J. Neurooncol. 2019, 143, 417–428. [Google Scholar] [CrossRef]
- Venugopal, C.; Hallett, R.; Vora, P.; Manoranjan, B.; Mahendram, S.; Qazi, M.A.; Mcfarlane, N.; Subapanditha, M.; Nolte, S.M.; Singh, M.; et al. Pyrvinium Targets CD133 in Human Glioblastoma Brain Tumor-Initiating Cells. Clin. Cancer Res. 2015, 21, 5324–5337. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, C.; Mcfarlane, N.M.; Nolte, S.; Manoranjan, B.; Singh, S.K. Processing of primary brain tumor tissue for stem cell assays and flow sorting. J. Vis. Exp. 2012. [Google Scholar] [CrossRef]
- Kandemir, B.; Dag, U.; Bakir Gungor, B.; Durasi, I.M.; Erdogan, B.; Sahin, E.; Sezerman, U.; Aksan Kurnaz, I. In silico analyses and global transcriptional profiling reveal novel putative targets for Pea3 transcription factor related to its function in neurons. PLoS ONE 2017, 12, e0170585. [Google Scholar] [CrossRef]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef]
- Kamburov, A.; Stelzl, U.; Lehrach, H.; Herwig, R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2013, 41, D793–D800. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; Mckay, S.; et al. The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016, 44, D481–D487. [Google Scholar] [CrossRef]
- Nishimura, A. BioCarta. Biotech. Softw. Internet Rep. 2001, 2, 117–120. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Fishilevich, S.; Zimmerman, S.; Kohn, A.; Iny Stein, T.; Olender, T.; Kolker, E.; Safran, M.; Lancet, D. Genic insights from integrated human proteomics in GeneCards. Database 2016, 2016, baw030. [Google Scholar] [CrossRef] [PubMed]
- Messeguer, X.; Escudero, R.; Farre, D.; Nunez, O.; Martinez, J.; Alba, M.M. PROMO: Detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002, 18, 333–334. [Google Scholar] [CrossRef] [PubMed]
- Goke, J.; Chan, Y.S.; Yan, J.; Vingron, M.; Ng, H.H. Genome-wide kinase-chromatin interactions reveal the regulatory network of ERK signaling in human embryonic stem cells. Mol. Cell 2013, 50, 844–855. [Google Scholar] [CrossRef]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Karuppusamy Rathinam, M.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The soft agar colony formation assay. J. Vis. Exp. 2014, e51998. [Google Scholar] [CrossRef]
- Ding, D.C.; Liu, H.W.; Chang, Y.H.; Chu, T.Y. Expression of CD133 in endometrial cancer cells and its implications. J. Cancer 2017, 8, 2142–2153. [Google Scholar] [CrossRef]
- Kageyama, S.; Liu, H.; Nagata, M.; Aoki, F. The role of ETS transcription factors in transcription and development of mouse preimplantation embryos. Biochem. Biophys. Res. Commun. 2006, 344, 675–679. [Google Scholar] [CrossRef]
- Bruck, T.; Yanuka, O.; Benvenisty, N. Human pluripotent stem cells with distinct X inactivation status show molecular and cellular differences controlled by the X-Linked ELK-1 gene. Cell Rep. 2013, 4, 262–270. [Google Scholar] [CrossRef]
- Cesari, F.; Brecht, S.; Vintersten, K.; Vuong, L.G.; Hofmann, M.; Klingel, K.; Schnorr, J.J.; Arsenian, S.; Schild, H.; Herdegen, T.; et al. Mice deficient for the ets transcription factor elk-1 show normal immune responses and mildly impaired neuronal gene activation. Mol. Cell. Biol. 2004, 24, 294–305. [Google Scholar] [CrossRef][Green Version]
- Bunina, D.; Abazova, N.; Diaz, N.; Noh, K.M.; Krijgsveld, J.; Zaugg, J.B. Genomic Rewiring of SOX2 Chromatin Interaction Network during Differentiation of ESCs to Postmitotic Neurons. Cell Syst. 2020, 10, 480–494.e8. [Google Scholar] [CrossRef]
- Zhang, L.; Gong, Y.; Zhao, X.; Zhou, H. Comparative study between hypoxia and hypoxia mimetic agents on osteogenesis of bone marrow mesenchymal stem cells in mouse. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi 2016, 30, 903–908. [Google Scholar]
- Park, S.W.; Do, H.J.; Choi, W.; Song, H.; Chung, H.J.; Kim, J.H. NANOG gene expression is regulated by the ETS transcription factor ETV4 in human embryonic carcinoma NCCIT cells. Biochem. Biophys. Res. Commun. 2017, 487, 532–538. [Google Scholar] [CrossRef]
- Park, S.W.; Do, H.J.; Ha, W.T.; Han, M.H.; Park, K.H.; Song, H.; Kim, N.H.; Kim, J.H. Transcriptional activation of OCT4 by the ETS transcription factor PEA3 in NCCIT human embryonic carcinoma cells. FEBS Lett. 2014, 588, 3129–3136. [Google Scholar] [CrossRef]
- Akagi, T.; Kuure, S.; Uranishi, K.; Koide, H.; Costantini, F.; Yokota, T. ETS-related transcription factors ETV4 and ETV5 are involved in proliferation and induction of differentiation-associated genes in embryonic stem(ES) cells. J. Biol. Chem. 2015, 290, 22460–22473. [Google Scholar] [CrossRef]
- Huh, S.; Song, H.R.; Jeong, G.R.; Jang, H.; Seo, N.H.; Lee, J.H.; Yi, J.Y.; Lee, B.; Choi, H.W.; Do, J.T.; et al. Suppression of the ERK-SRF axis facilitates somatic cell reprogramming. Exp. Mol. Med. 2018, 50, e448. [Google Scholar] [CrossRef]
- Vora, P.; Venugopal, C.; Mcfarlane, N.; Singh, S.K. Culture and Isolation of Brain Tumor Initiating Cells. Curr. Protoc. Stem Cell Biol. 2015, 34, 3.3.1–3.3.13. [Google Scholar] [CrossRef]
- Tabu, K.; Kimura, T.; Sasai, K.; Wang, L.; Bizen, N.; Nishihara, H.; Taga, T.; Tanaka, S. Analysis of an alternative human CD133 promoter reveals the implication of Ras/ERK pathway in tumor stem-like hallmarks. Mol. Cancer 2010, 9, 39. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Site | Sequence (5′-3′) |
|---|---|---|
| GAPDH | Frw | CAT CTT CCA GGA GCG AGA TCC |
| Rev | AAA TGA GCC CCA GCC TTC TCC | |
| ACTB | Frw | ACG AAA CTA CCT TCA ACT CC |
| Rev | GAT CTT GAT CTT CAT TGT GCT GG | |
| ELK1 | Frw | GCT TCC TAC GCA TAC ATT GAC C |
| Rev | ACT GGA TGG AAA CTG GAA GG | |
| SOX2 | Frw | GGG AAA TGG GAG GGG TGC AAA AGA GG |
| Rev | TTG CGT GAG TGT GGA TGG GAT TGG TG | |
| POU5F1 | Frw | AAG GAT GTG GTC CGA GTG TGG |
| Rev | CCT GAG AAA GGA GAC CCA GCA G | |
| NANOG | Frw | TTC AGA GAC AGA AAT ACC TCA GCC |
| Rev | CCT TCT GCG TCA CAC CAT TGC | |
| WNT3A | Frw | GACAAAGCTACCAGGGAGTC |
| Rev | CTGCTGCAGCCACAGAT | |
| IRAK3 | Frw | ACATACTAGAGTTGGCTGCATATT |
| Rev | TGTCACCTACACACTGCAATC | |
| MEF2B | Frw | CAACCGCCTCTTCCAGTATG |
| Rev | TCAGCGTCTCGAGGATGT | |
| TCF7L1 | Frw | TGAGCGTGAAATCACCAGTC |
| Rev | TGGCCCTCATCTCCTTCATA | |
| RHO | Frw | CATGATGAACAAGCAGTTCCG |
| Rev | AGAGTCCTAGGCAGGTCTTAG | |
| HES7 | Frw | CGGGATCGAGCTGAGAATAG |
| Rev | GTTCCGGAGGTTCTGGTC | |
| NOTO | Frw | GCTGGAAGAGTTGGAGAAAGT |
| Rev | ACTCTCACCTGGTTCTCTGTA | |
| SIX3 | Frw | CAGCAAGAAACGCGAACTG |
| Rev | GTGCTGGAGCCTGTTCTT | |
| CREB3 | Frw | ACCTGCATCTTGGTCCTACTA |
| Rev | GGACAACACTCCATGCTCAG | |
| CREM | Frw | ATCCCAGCATGATGGAAGTATAA |
| Rev | ATTGCTGCTACCTGAGCTAAA | |
| LIFR | Frw | GCTCTGGACAAGTTAAATCCATAC |
| Rev | CCCTTTGAAGGACTGGCT | |
| FRZB | Frw | AAGTTAAGCGCTGGGATATGA |
| Rev | GGGATTTAGTTGCGTGCTTG | |
| GLUT3 | Frw | AGCTCTCTGGGATCAATGCTGTGT |
| Rev | ATGGTGGCATAGATGGGCTCTTGA | |
| RXRB | Frw | GATGTGAAGCCACCAGTCTTAG |
| Rev | GTAGTGTTTGCCTGAGCTTCT | |
| NODAL | Frw | TACATCCAGAGTCTGCTGAAAC |
| Rev | CTAGGAGCACTCTGCCATTATC | |
| PAX6 | Frw | GTGAATGGGCGGAGTTATGA |
| Rev | ATGAGTCCTGTTGAAGTGGTG | |
| GSK3B | Frw | CCGAGGAGAACCCAATGTTT |
| Rev | GCCAGCAGACCATACATCTATAC | |
| FGF11 | Frw | CAAAGGCATCGTCACCAAAC |
| Rev | GATCAGGTTGAAGTGGGTGAA | |
| FRIT1 | Frw | GTGCAGGAAACCGAGTAGAA |
| Rev | GCGCCTTTAGAGTGAGTGAA | |
| GLI4 | Frw | CTCGGAAGGTCCCAGGT |
| Rev | CCCGGTGATGAGAGACTGA | |
| BRACHYURY | Frw | GTAAACTCCACCAGTCCTACTTT |
| Rev | TCTGTCCTTAACAGCTCAACTC | |
| NOTCH4 | Frw | GAGGATATCGATGAGTGCAGAAG |
| Rev | TTCAAAGCCTGGGAGACAC | |
| ZIC1 | Frw | GAGCGACAAGCCCTATCTTT |
| Rev | GGATTCGTGGACCTTCATGT | |
| ARC | Frw | TCAGCTCATGACTCACCCA |
| Rev | CTTGAGACCTGTTGTCACTCTC | |
| ALS2 | Frw | GGACTCAAAGAAGAGAAGCTCAA |
| Rev | TGGCAATCTCTCTGGTGTTATG | |
| SOX10 | Frw | CTTCATGGTGTGGGCTCA |
| Rev | CGTTCAGCAGCCTCCAG | |
| SMAD6 | Frw | CCTACCGTGTGCTGCAA |
| Rev | GGAATCGGACAGATCCAGTG | |
| NGFR | Frw | CATAGCCTTCAAGAGGTGGAAC |
| Rev | CACTGTCGCTGTGGAGTTT | |
| MAPK6 | Frw | AGGAGCTTCTCAGCGTAATTC |
| Rev | CCAGGAAATCCAGTGCTTCT | |
| NGFR | Frw | CATAGCCTTCAAGAGGTGGAAC |
| Rev | CACTGTCGCTGTGGAGTTT | |
| CD133 | Frw | GCGTCTTCCTCATGGTTGGAG |
| Rev | CTTGCTCGTGTAAGGTTCACAG |
| Number of Predicted ets Binding Motifs with Different Dissimilarity Scores (DS) in Promo 3.0 | |||
|---|---|---|---|
| DS: 0–1 Percent | DS: 1–5 Percent | DS: 5–10 Percent | |
| SRF | - | 2 | 1 |
| MCL1 | 2 | - | 3 |
| LIF | - | 2 | 1 |
| SOX2 (TRED) | - | 1 | 1 |
| NANOG (TRED) | - | 1 | - |
| POU5F1 (TRED) | - | 1 | 2 |
| SOX2 (EPD) | 1 | 2 | 3 |
| NANOG (EPD) | - | 1 | 1 |
| POU5F1 (EPD) | 1 | 2 | 1 |
| Promoter | Forward Primer (5′-3′) | RE Site | Reverse Primer (5′-3′) | RE Site | Product (bp) |
|---|---|---|---|---|---|
| POU5F1 | AGACggtaccAGGGCTG TTGGCTTTGGACA | KpnI | CTGTagatctAGCCATTTAA GAATTCCAGAGTAGG | BglII | 993 |
| SOX2 | CTGTggtaccGGGGAGTG ATTATGGGAAGAA | KpnI | CTGTagatctCACTAGACTG TCTTCATTCAACCGTAGC | BglII | 993 |
| NANOG | CTGTggtaccTTTCTGCC TAAACTAGCCA | KpnI | CTGTagatctAGGTGAAGA TTCTTTACAGTCG | BglII | 988 |
| Primer | Site | Oligo | Length | Tm (°C) | Ta (°C) |
|---|---|---|---|---|---|
| NANOG-etsΔ | Fwd | TACTAACATGAGTGTGGATC | 20 | 59 | 58 |
| Rev | AGGAGGAAAAAATTTAAGAGG | 21 | 57 | ||
| POU5F1- etsΔ1 | Fwd | CCTTTCCCCCTGTCTCTG | 18 | 64 | 65 |
| Rev | CAGGGAAAGGGACCGAGG | 18 | 68 | ||
| POU5F1- etsΔ2 | Fwd | GAATTGGGAACACAAAGG | 18 | 57 | 57 |
| Rev | TGAATGAAGAACTTAATCCC | 20 | 56 | ||
| POU5F1- etsΔ3 | Fwd | GTGAAGTTCAATGATGCTCTTG | 22 | 61 | 62 |
| Rev | AACCAGTTGCCCCAAACT | 18 | 64 | ||
| SOX2- etsΔ1 | Fwd | TTGAAATCACCCTCCCCC | 18 | 64 | 65 |
| Rev | ATCCCACGGCACTGTATG | 18 | 65 | ||
| SOX2- etsΔ2 | Fwd | GTGTCTTTCCCCAGCCCC | 18 | 69 | 68 |
| Rev | GGCGCTCAAAAGTGCAGG | 18 | 67 |
| Name | Primer | Sequence | PCR Product Size |
|---|---|---|---|
| ChIP_MCL1 | Frw Rev | GCCGCCCTAAAACCGTGATA CGCCTGGCTGAGAAAACTG | 99 |
| ChIP_SRF | Frw Rev | TGACAGCAACGAGTTCGGTA CCCCCATATAAAGAGATACAATGTT | 130 |
| ChIP_SOX2_ETS1 | Frw Rev | TGGGAGGGAGTTTGTGACT AAAGTGCAGGCGATGGG | 97 |
| ChIP_SOX2_ETS2 | Frw Rev | GTGGGATGCCAGGAAGTT GTCGTGCGGCTTTCAAATG | 102 |
| ChIP_SOX2_ETS3 | Frw Rev | AGACAGTCTAGTGGGAGATGTG CGGACCATAAGGCAGACTCTA | 138 |
| ChIP_SOX2_ETS4 | Frw Rev | CTTATGGTCCGAGCAGGATTT TCCCGACTAGAAGTTAGGAGAC | 103 |
| ChIP_SOX2_ETS5 | Frw Rev | CGCACCTTAGCTGCTTCC GTCACACCACACGCCTTT | 143 |
| ChIP_NANOG_ETS1 | Frw Rev | CTGGAGGTCCTATTTCTCTAACATC ATGCTTCAAAGCAAGGCAAG | 155 |
| ChIP_NANOG_ETS2 | Frw Rev | GCAGAGGAGAATGAGTCAAAGA CCCAAACCCAACATTCAAGAAA | 131 |
| ChIP_NANOG_ETS3 | Frw Rev | CTTAGTCCAGCCTGTTCCAAA AGTGAAAGACCAAAGGGAAGG | 136 |
| ChIP_POU5F1_ETS1 | Frw Rev | CTTCACTGCACTGTACTCCTC CACCTCAGTTTGAATGCATGG | 101 |
| ChIP_POU5F1_ETS2 | Frw Rev | GGAGTTTGTGCCAGGGTT CCCTCCAACCAGTTGCC | 105 |
| ChIP_POU5F1_ETS3 | Frw Rev | GTTGGAGGGAAGGTGAAGTT TACTGTGTCCCAAGCTTCTTTAT | 93 |
| ChIP_POU5F1_ETS4 | Frw Rev | CATCATCTCTGCTGGGATTACC CTGACTCTAGTTGACGTGTTGG | 143 |
| ChIP_KLF4 | Frw Rev | CATAGCAACGATGGAAGGGA TCTCTCTGGTCGGGAAACT | 149 |
| ChIP_LIF1 | Frw Rev | CTTCCATTCATAATTTCCTATGATGCAC CCTGATTTATATACTGGAGCCTGTG | 111 |
| ChIP_SIM2 | Frw Rev | TCTTGGACCTGCAACACC TCTCGGGAAGGTCACCA | 138 |
| ChIP_ELF3 | Frw Rev | CAGAGGGTGCGGGATGA GCCTTTAGACTGGGCTCCT | 135 |
| ChIP_GLUT3 | Frw Rev | CCACCCTTTGCGGAGATTAT TCCTTCTCAGCAGCAAGTTT | 115 |
| ChIP_CD133 | Frw Rev | TCTACAGGAAATGGATGCTGTC GGATCTGCCTCAGTCACTTAAA | 131 |
| Elk-1-VP16 Overexpression | siElk-1 Silencing | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene ID | SH-SY5Y | SK-N-BE (2) | T98G | A172 | Microarray Data | SH-SY5Y | SK-N-BE (2) | T98G | A172 |
| ALS2 | N/A* | N/A | −0.5 | 0.60 | −6.06 | N/A | N/A | −0.64 | −1.44 |
| ARC | N/A | N/A | 0.44 | 1.67 | −6.87 | N/A | N/A | −2.17 | −0.06 |
| BRACHYURY | N/A | 0.32 | N/A | N/A | 1.54 | N/A | −0.25 | N/A | N/A |
| CREB3 | −0.01 | N/A | 0.83 | 0.46 | −1.92 | 0.09 | N/A | −0.74 | −0.67 |
| CREM | N/A | N/A | −0.91 | 1.40 | N/A | −0.51 | N/A | −0.43 | 0.44 |
| ELK-1 | 1.50 | 2.04 | 8.48 | 8.98 | 13.11 | −0.54 | −2.36 | −0.60 | −1.49 |
| FGF11 | −0.23 | N/A | N/A | 0.28 | −2.40 | 0.48 | N/A | N/A | −1.31 |
| FRIT1 | 0.17 | N/A | N/A | N/A | 1.82 | −0.34 | N/A | N/A | N/A |
| FRZB | 1.21 | −0.72 | −0.86 | N/A | 1.91 | −2.20 | −1.34 | −0.18 | N/A |
| GLI4 | N/A | N/A | N/A | 0.695 | −3.81 | N/A | N/A | N/A | 0.61 |
| GLUT3 | 1.62 | 0.07 | 0.83 | 0.92 | 2.43 | −0.67 | −0.46 | 0.54 | 2.64 |
| GSK3B | −0.22 | N/A | N/A | N/A | −1.61 | −0.12 | N/A | N/A | N/A |
| HES7 | N/A | N/A | 0.68 | N/A | −1.77 | 0.12 | N/A | 0.20 | N/A |
| IRAK3 | 2.78 | 0.51 | −0.46 | N/A | 1.70 | −0.23 | −0.81 | −0.97 | N/A |
| LIFR | −0.91 | −1.52 | −0.84 | −0.08 | −2.01 | −0.53 | 0.48 | −0.12 | −0.82 |
| MAPK6 | N/A | N/A | N/A | 0.76 | 1.64 | N/A | N/A | N/A | N/A |
| MEF2B | 0.09 | −0.41 | −1.19 | N/A | −2.74 | −0.19 | −0.33 | −1.57 | N/A |
| NANOG | 1.96 | −0.91 | 0.58 | 1.18 | 2.54 | −0.99 | 4.85 | −0.07 | 3.12 |
| NODAL | 2.33 | 0.66 | N/A | N/A | 1.64 | −0.66 | −0.26 | N/A | N/A |
| NOTCH4 | N/A | N/A | N/A | 3.56 | 3.39 | N/A | N/A | N/A | 7.16 |
| NOTO | −2.94 | 0.18 | −0.34 | N/A | 2.15 | 1.05 | −0.08 | −0.76 | N/A |
| PAX6 | −1.54 | −0.28 | N/A | N/A | 2.61 | 1.18 | −1.62 | N/A | N/A |
| POU5F1 | 1.62 | −1.20 | 0.56 | 0.32 | 3.68 | −2.19 | 3.31 | −0.67 | 1.06 |
| RHO | −3.56 | −1.07 | −2.26 | N/A | 2.27 | −0.20 | −0.30 | −0.93 | N/A |
| RXRB | 1.01 | 0.66 | 0.83 | 0.46 | 5.95 | 0.45 | 0.31 | N/A | N/A |
| SIX3 | −1.94 | N/A | 0.61 | N/A | −5.72 | 1.56 | N/A | 0.41 | N/A |
| SMAD6 | N/A | N/A | N/A | −0.29 | −3.78 | N/A | N/A | N/A | −0.42 |
| SOX2 | 1.54 | 0.41 | 0.37 | −0.59 | 2.75 | −0.84 | −1.08 | −0.56 | −0.36 |
| SOX10 | N/A | N/A | N/A | 2.94 | 2.41 | N/A | N/A | N/A | 5.83 |
| TCF7L1 | 0.79 | 0.56 | 0.15 | N/A | 2.34 | −0.29 | −0.25 | 0.14 | N/A |
| WNT3A | −2.11 | 0.53 | 0.11 | N/A | 2.25 | 0.86 | −1.11 | 0.52 | N/A |
| ZIC1 | N/A | 1.53 | N/A | N/A | 2.26 | N/A | 1.07 | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sogut, M.S.; Venugopal, C.; Kandemir, B.; Dag, U.; Mahendram, S.; Singh, S.; Gulfidan, G.; Arga, K.Y.; Yilmaz, B.; Kurnaz, I.A. ETS-Domain Transcription Factor Elk-1 Regulates Stemness Genes in Brain Tumors and CD133+ BrainTumor-Initiating Cells. J. Pers. Med. 2021, 11, 125. https://doi.org/10.3390/jpm11020125
Sogut MS, Venugopal C, Kandemir B, Dag U, Mahendram S, Singh S, Gulfidan G, Arga KY, Yilmaz B, Kurnaz IA. ETS-Domain Transcription Factor Elk-1 Regulates Stemness Genes in Brain Tumors and CD133+ BrainTumor-Initiating Cells. Journal of Personalized Medicine. 2021; 11(2):125. https://doi.org/10.3390/jpm11020125
Chicago/Turabian StyleSogut, Melis Savasan, Chitra Venugopal, Basak Kandemir, Ugur Dag, Sujeivan Mahendram, Sheila Singh, Gizem Gulfidan, Kazim Yalcin Arga, Bayram Yilmaz, and Isil Aksan Kurnaz. 2021. "ETS-Domain Transcription Factor Elk-1 Regulates Stemness Genes in Brain Tumors and CD133+ BrainTumor-Initiating Cells" Journal of Personalized Medicine 11, no. 2: 125. https://doi.org/10.3390/jpm11020125
APA StyleSogut, M. S., Venugopal, C., Kandemir, B., Dag, U., Mahendram, S., Singh, S., Gulfidan, G., Arga, K. Y., Yilmaz, B., & Kurnaz, I. A. (2021). ETS-Domain Transcription Factor Elk-1 Regulates Stemness Genes in Brain Tumors and CD133+ BrainTumor-Initiating Cells. Journal of Personalized Medicine, 11(2), 125. https://doi.org/10.3390/jpm11020125