Drug Repurposing for Triple-Negative Breast Cancer


 , , ,
, , , 
Abstract
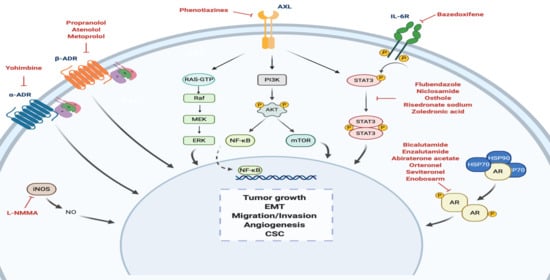
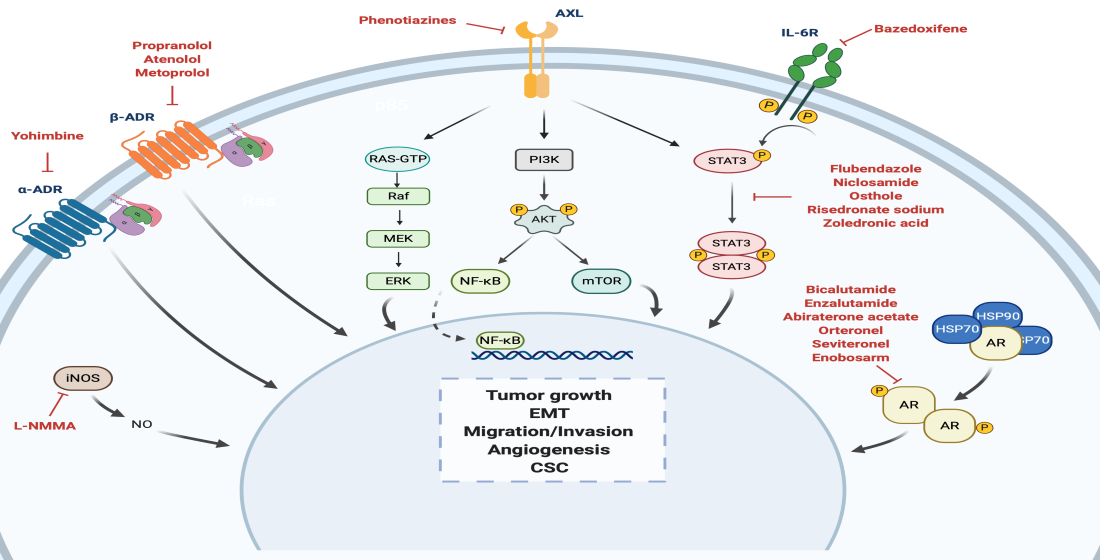
1. Introduction
2. Current Treatments for TNBC
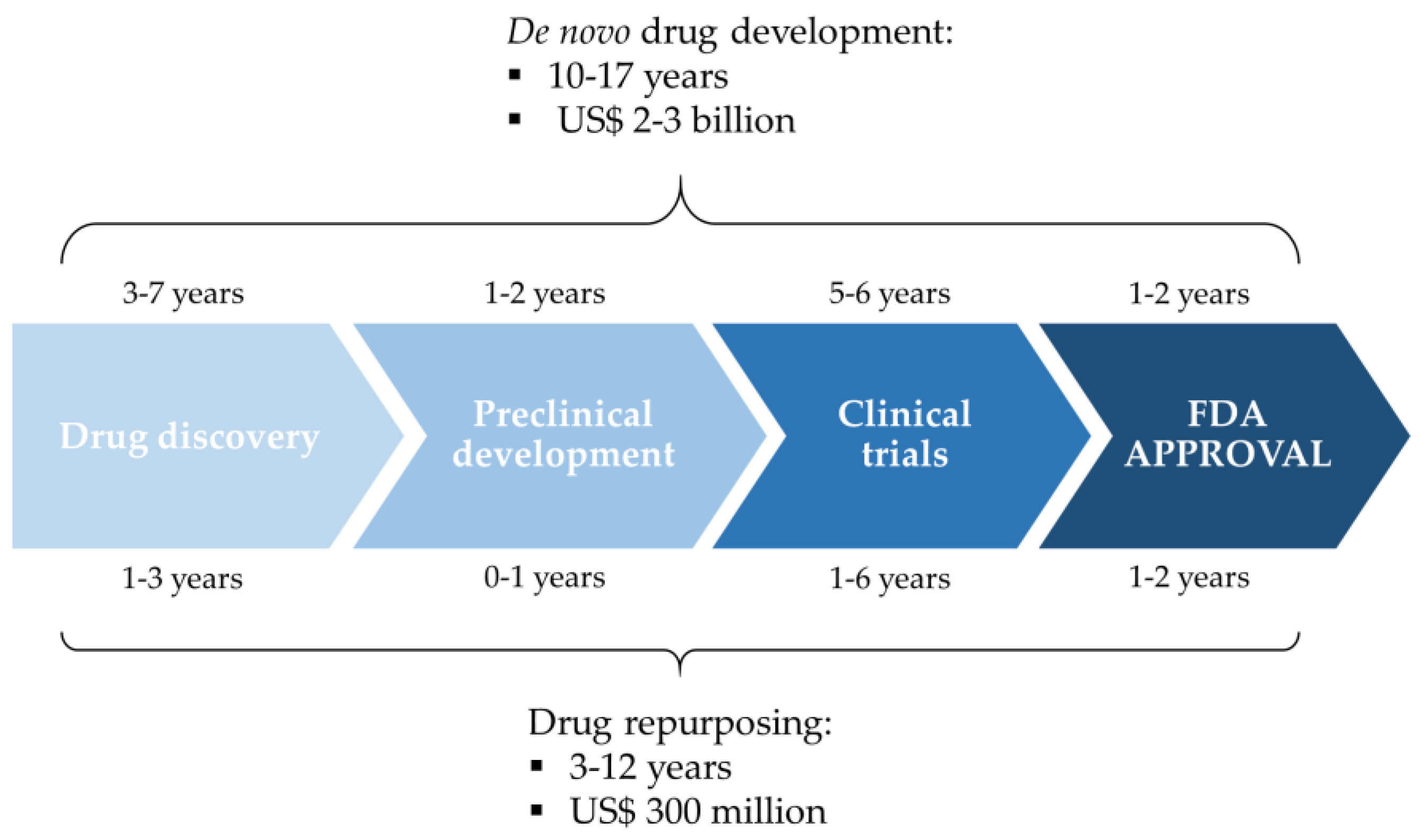
3. Drug Repurposing
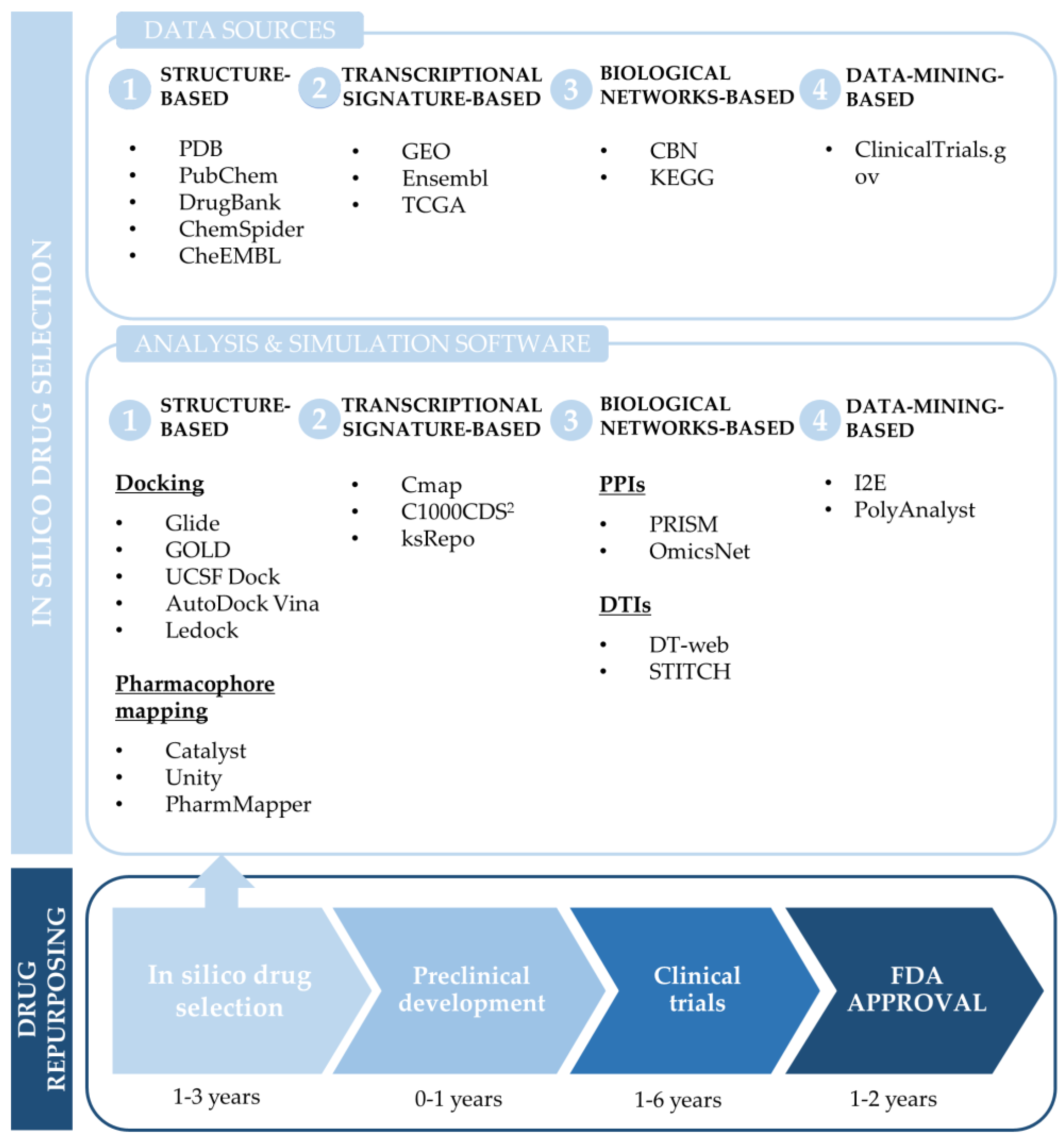
3.1. Common Computational Approaches for Drug Repurposing
3.1.1. Structure-Based Drug Repurposing
3.1.2. Transcriptional Signature-Based Drug Repurposing
3.1.3. Network-Based Drug Repurposing
3.1.4. Data-Mining-Based Drug Repurposing
4. Drug Repurposing for TNBC
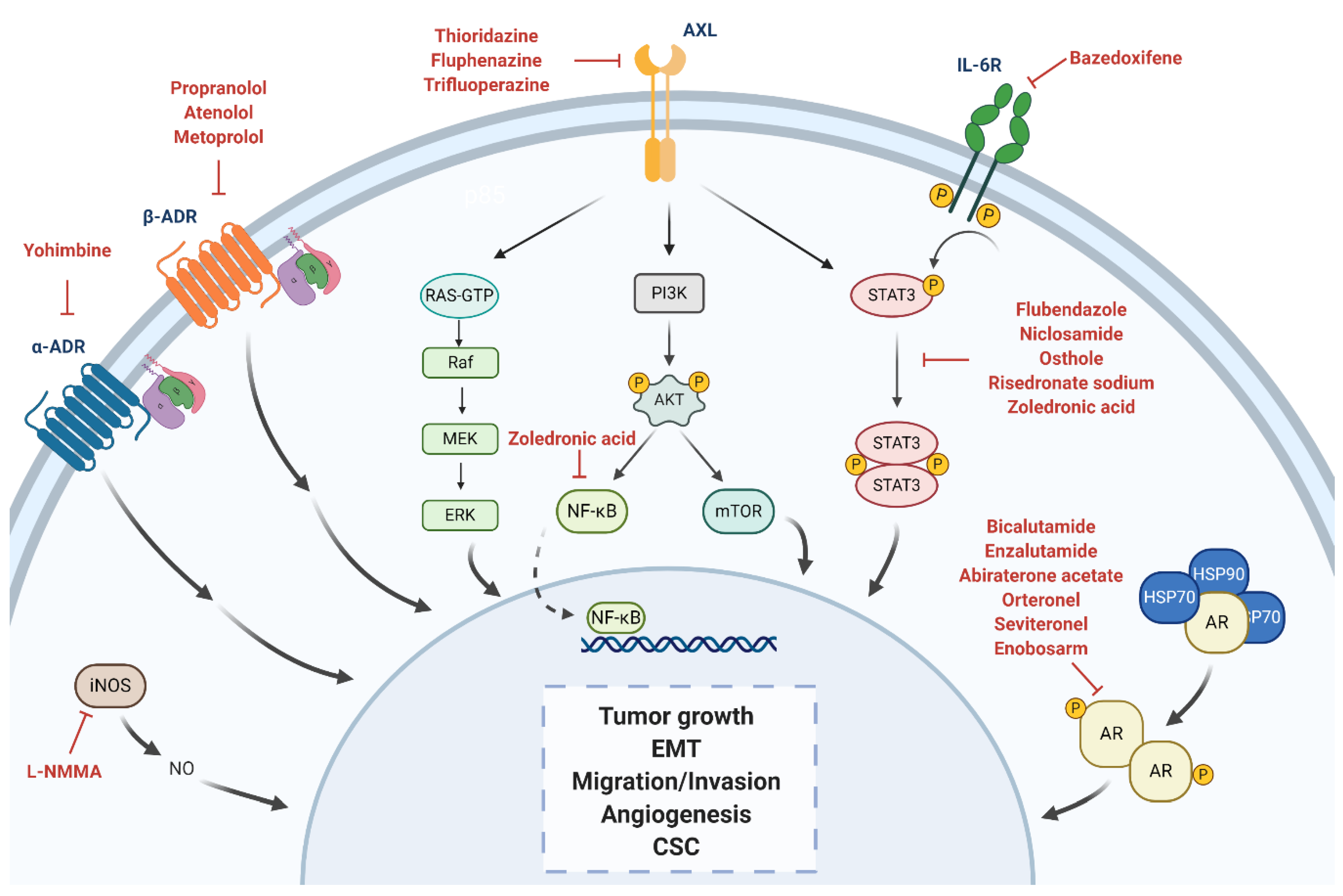
4.1. Androgen Receptor
4.2. Adrenergic Receptor
4.2.1. α-Adrenergic Receptor
4.2.2. β-Adrenergic Receptor
4.3. STAT3
4.4. Nitric Oxide Synthase
4.5. Anexelekto (AXL)
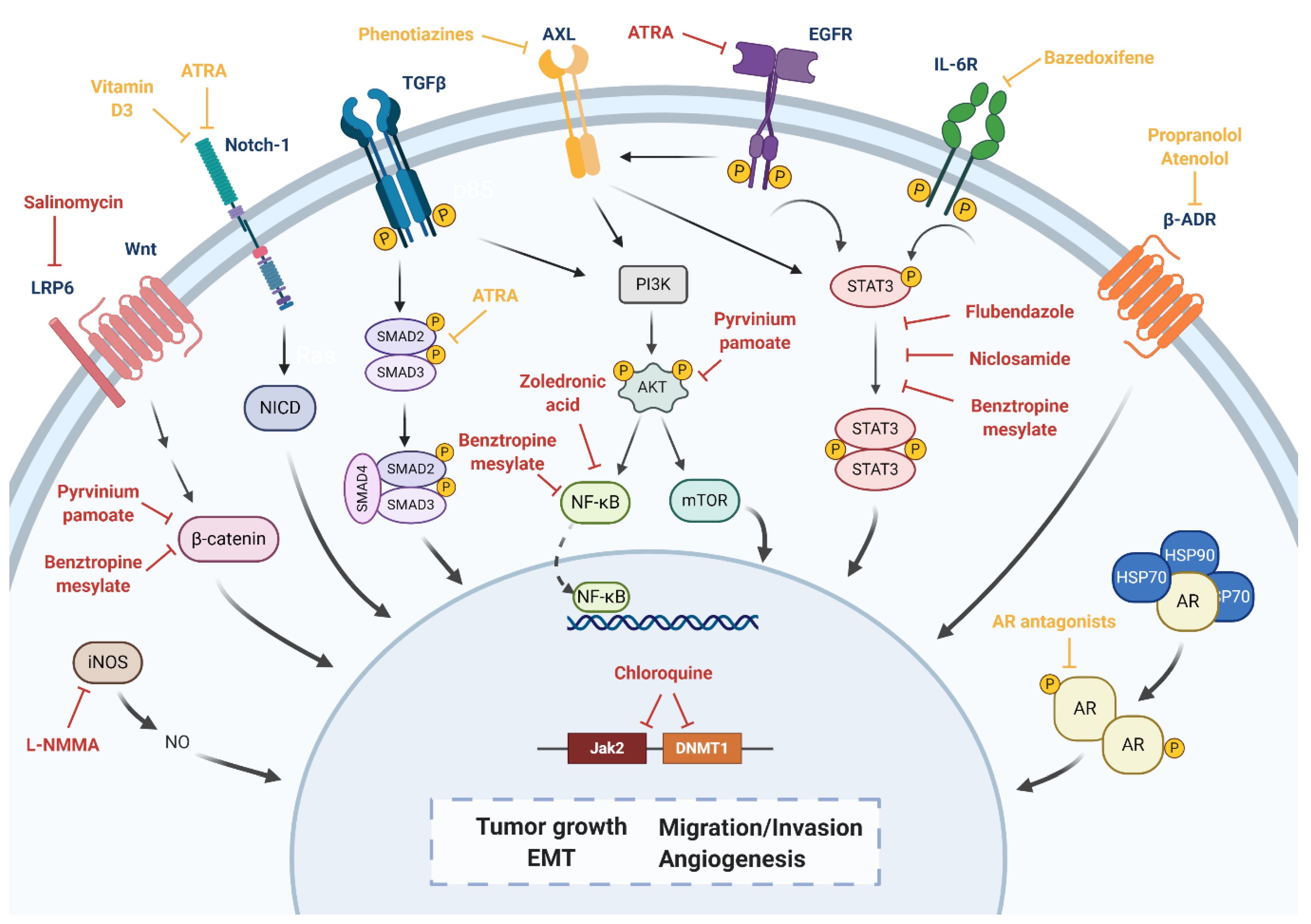
5. Drug Repositioning to Target Cancer Stem Cells in TNBC
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADC | Antibody drug conjugate |
| ADR | Adrenergic receptor |
| AE | Adverse events |
| Akt | Protein kinase B |
| ALDH1 | Aldehyde dehydrogenase 1 |
| AR | Androgen receptor |
| AREG | Amphiregulin |
| ATRA | All-trans retinoic acid |
| AXL | Anexelekto |
| BARK | β-adrenergic receptor kinase |
| BCSC | Breast cancer stem cells |
| BL1 | Basal-like 1 |
| BL2 | Basal-like 2 |
| cAMP | Cyclic AMP |
| CBN | Causal biological networks |
| CBR | Clinical benefit rate |
| CMap | Connectivity Map |
| CPIs | Checkpoint inhibitors |
| CSC | Cancer stem cells |
| CTLA-4 | Cytotoxic T-lymphocyte-associated antigen-4 |
| DFS | Disease free survival |
| DTI | Drug-target interaction |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial-to-mesenchymal transition |
| ER | Estrogen receptor |
| FAK | Focal adhesion kinase |
| FDA | Food and Drug Administration |
| FGF | Fibroblast growth factor |
| GEO | Gene Expression Omnibus |
| GPCR | G protein-coupled receptor |
| HER2 | Human epidermal growth factor receptor 2 |
| IGF | Insulin-like growth factor |
| IHC | Immunohistochemistry |
| IL-10 | Interleukin 10 |
| IL-6 | Interleukin 6 |
| IM | Immunomodulatory |
| ITT | Intent-to-treat population |
| JAKs | Janus kinases |
| LAR | Luminal androgen receptor |
| L-NMMA | NG-monomethyl-L-arginine |
| M | Mesenchymal |
| MAPK | Mitogen-activated protein kinase |
| mCRPC | Metastatic castration-resistant prostate cancer |
| MLF2 | Myeloid leukemia factor 2 |
| MSL | Mesenchymal stem–like |
| mTOR | Mammalian target of rapamycin |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| NOS1/nNOS | Neuronal nitric oxide synthase |
| NOS2/iNOS | Inducible nitric oxide synthase |
| NOS3/eNOS | Endothelial nitric oxide synthase |
| OS | Overall survival |
| PARP | Poly[adenosine diphosphate-ribose] polymerase |
| PDB | Protein Data Bank |
| pCR | Pathologic complete response |
| PD-1 | Programmed cell death 1 |
| PD-L1 | Programmed cell death-ligand 1 |
| PDX | Patient-derived xenografts |
| PFS | Progression free survival |
| PI3K | Phosphatidylinositol-3 kinase |
| PKA | Protein kinase A |
| PPI | Protein-protein interaction |
| PR | Progesterone receptor |
| PRISM | Protein Interactions by Structural Matching |
| ROS | Reactive oxygen species |
| RPL39 | Ribosomal protein L39 |
| RTK | Receptors tyrosine kinase |
| RXR | Retinoid X receptors |
| SARMs | Selective androgen receptor modulators |
| SAEs | Serious adverse events |
| STAT3 | Signal transducer and activator of transcription 3 |
| TAM | Tyro3, AXL and Mer |
| TCGA | The Cancer Genome Atlas |
| TGFβ | Transforming growth factor β |
| TILs | Tumor infiltrating lymphocytes |
| TNBC | Triple-negative breast cancer |
| Trop-2 | Trophoblast cell-surface antigen 2 |
| VDR | Vitamin D3 nuclear receptor |
| VEGF | Vascular endothelial growth factor |
| VHTS | Virtual high-throughput screening |
References
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Hon, J.D.C.; Singh, B.; Sahin, A.; Du, G.; Wang, J.; Wang, V.Y.; Deng, F.M.; Zhang, D.Y.; Monaco, M.E.; Lee, P. Breast cancer molecular subtypes: From TNBC to QNBC. Am. J. Cancer Res. 2016, 6, 1864–1872. [Google Scholar]
- Costa, R.L.B.; Gradishar, W.J. Triple-negative breast cancer: Current practice and future directions. J. Oncol. Pract. 2017, 13, 301–303. [Google Scholar] [CrossRef]
- Lee, A.; Djamgoz, M.B.A. Triple negative breast cancer: Emerging therapeutic modalities and novel combination therapies. Cancer Treat. Rev. 2018, 62, 110–122. [Google Scholar] [CrossRef]
- Jitariu, A.A.; Cîmpean, A.M.; Ribatti, D.; Raica, M. Triple negative breast cancer: The kiss of death. Oncotarget 2017, 8, 46652–46662. [Google Scholar] [CrossRef]
- Aggarwal, S.; Verma, S.S.; Aggarwal, S.; Gupta, S.C. Drug repurposing for breast cancer therapy: Old weapon for new battle. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Waks, A.G.; Winer, E.P. Breast cancer treatment: A review. J. Am. Med. Assoc. 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Cruz-Lozano, M.; González-González, A.; Muñoz-Muela, E.; Cara, F.E.; Granados-Principal, S. Targeted therapies in triple negative breast cancer: Current knowledge and perspectives. Recent Stud. Adv. Breast Cancer 2019, 2, 1–17. [Google Scholar]
- Park, J.H.; Ahn, J.-H.; Kim, S.-B. How shall we treat early triple-negative breast cancer (TNBC): From the current standard to upcoming immuno-molecular strategies. ESMO Open 2018, 3, e000357. [Google Scholar] [CrossRef]
- Al-Mahmood, S.; Sapiezynski, J.; Garbuzenko, O.B.; Minko, T. Metastatic and triple-negative breast cancer: Challenges and treatment options. Drug Deliv. Transl. Res. 2018, 8, 1483–1507. [Google Scholar] [CrossRef]
- Timothy, C.J.; Kogularamanan, S.; Stephen, J.L. The next generation of platinum drugs: Targeted Pt(II) agents, nanoparticle delivery, and Pt(IV) prodrugs timothy. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef]
- Treatment of Triple-Negative Breast Cancer. Available online: https://www.cancer.org/cancer/breast-cancer/treatment/treatment-of-triple-negative.html (accessed on 14 October 2020).
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Oiseth, S.J.; Aziz, M.S. Cancer immunotherapy: A brief review of the history, possibilities, and challenges ahead. J. Cancer Metastasis Treat. 2017, 3, 250–261. [Google Scholar] [CrossRef]
- Kwa, M.J.; Adams, S. Checkpoint inhibitors in triple-negative breast cancer (TNBC): Where to go from here. Cancer 2018, 124, 2086–2103. [Google Scholar] [CrossRef]
- Soare, G.R.; Soare, C.A. Immunotherapy for breast cancer: First FDA approved regimen. Discoveries 2019, 7, e91. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Domchek, S.M.; Clark, A.S. Immunotherapy for breast cancer: What are we missing? Clin. Cancer Res. 2017, 23, 2640–2646. [Google Scholar] [CrossRef]
- Nagayama, A.; Vidula, N.; Ellisen, L.; Bardia, A. Novel antibody–drug conjugates for triple negative breast cancer. Ther. Adv. Med. Oncol. 2020, 12, 1–12. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Nosengo, N. Can you teach old drugs new tricks? Nature 2016, 534, 314–316. [Google Scholar] [CrossRef]
- Akhoon, B.A.; Tiwari, H.; Nargotra, A. In silico drug design methods for drug repurposing. In In Silico Drug Design; Academic Press: Cambridge, MA, USA, 2019; pp. 47–84. [Google Scholar]
- MacArron, R.; Banks, M.N.; Bojanic, D.; Burns, D.J.; Cirovic, D.A.; Garyantes, T.; Green, D.V.S.; Hertzberg, R.P.; Janzen, W.P.; Paslay, J.W.; et al. Impact of high-throughput screening in biomedical research. Nat. Rev. Drug Discov. 2011, 10, 188–195. [Google Scholar] [CrossRef]
- Hodos, R.A.; Kidd, B.A.; Shameer, K.; Readhead, B.P.; Dudley, J.T. In silico methods for drug repurposing and pharmacology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 186–210. [Google Scholar] [CrossRef]
- Karaman, B.; Sippl, W. Computational drug repurposing: Current trends. Curr. Med. Chem. 2018, 26, 5389–5409. [Google Scholar] [CrossRef]
- Alaimo, S.; Pulvirenti, A. Network-based drug repositioning: Approaches, resources, and research directions. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2019; Volume 1903, pp. 97–113. [Google Scholar]
- Gns, H.S.; Gr, S.; Murahari, M.; Krishnamurthy, M. An update on drug repurposing: Re-written saga of the drug’s fate. Biomed. Pharmacother. 2019, 110, 700–716. [Google Scholar] [CrossRef]
- Sohraby, F.; Bagheri, M.; Aryapour, H. Performing an in silico repurposing of existing drugs by combining virtual screening and molecular dynamics simulation. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2019; Volume 1903, pp. 23–43. [Google Scholar]
- Wang, Y.; Yella, J.; Jegga, A.G. Transcriptomic data mining and repurposing for computational drug discovery. Methods Mol. Biol. 2019, 1903, 73–95. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef]
- CLUE Connectopedia. Available online: https://clue.io/connectopedia/ (accessed on 30 July 2020).
- Higurashi, M.; Ishida, T.; Kinoshita, K. Identification of transient hub proteins and the possible structural basis for their multiple interactions. Protein Sci. 2008, 17, 72–78. [Google Scholar] [CrossRef]
- Ozdemir, E.S.; Halakou, F.; Nussinov, R.; Gursoy, A.; Keskin, O. Methods for discovering and targeting druggable protein-protein interfaces and their application to repurposing. Methods Mol. Biol. 2019, 1903, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Su, E.W.; Sanger, T.M. Systematic drug repositioning through mining adverse event data in ClinicalTrials.gov. PeerJ 2017, 2017. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørile, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Ress, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Sørlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Fan, C.; Oh, D.S.; Marron, J.S.; He, X.; Qaqish, B.F.; Livasy, C.; Carey, L.A.; Reynolds, E.; Dressler, L.; et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genom. 2006, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- Bernard, P.S.; Parker, J.S.; Mullins, M.; Cheung, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Kreike, B.; van Kouwenhove, M.; Horlings, H.; Weigelt, B.; Peterse, H.; Bartelink, H.; van de Vijver, M.J. Gene expression profiling and histopathological characterization of triple-negative/basal-like breast carcinomas. Breast Cancer Res. 2007, 9, R65. [Google Scholar] [CrossRef]
- Prat, A.; Adamo, B.; Cheang, M.C.U.; Anders, C.K.; Carey, L.A.; Perou, C.M. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist 2013, 18, 123–133. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Pietenpol, J.A. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J. Pathol. 2014, 232, 142–150. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, J.; Gray, W.H.; Lehmann, B.D.; Bauer, J.A.; Shyr, Y.; Pietenpol, J.A. TNBCtype: A subtyping tool for triple-negative breast cancer. Cancer Inform. 2012, 11, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of triple-negative breast cancer molecular subtypes: Implications for neoadjuvant chemotherapy selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.W.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Le Du, F.; Eckhardt, B.L.; Lim, B.; Litton, J.K.; Moulder, S.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ueno, N.T. Is the future of personalized therapy in triple-negative breast cancer based on molecular subtype? Oncotarget 2015, 6, 12890–12908. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.R.; Jiang, Y.Z.; Xu, X.E.; Yu, K.D.; Jin, X.; Hu, X.; Zuo, W.J.; Hao, S.; Wu, J.; Liu, G.Y.; et al. Comprehensive transcriptome analysis identifies novel molecular subtypes and subtype-specific RNAs of triple-negative breast cancer. Breast Cancer Res. 2016, 18. [Google Scholar] [CrossRef] [PubMed]
- Ring, B.Z.; Hout, D.R.; Morris, S.W.; Lawrence, K.; Schweitzer, B.L.; Bailey, D.B.; Lehmann, B.D.; Pietenpol, J.A.; Seitz, R.S. Generation of an algorithm based on minimal gene sets to clinically subtype triple negative breast cancer patients. BMC Cancer 2016, 16, 143. [Google Scholar] [CrossRef]
- Espinosa Fernandez, J.R.; Eckhardt, B.L.; Lee, J.; Lim, B.; Pearson, T.; Seitz, R.S.; Hout, D.R.; Schweitzer, B.L.; Nielsen, T.J.; Lawrence, O.R.; et al. Identification of triple-negative breast cancer cell lines classified under the same molecular subtype using different molecular characterization techniques: Implications for translational research. PLoS ONE 2020, 15, e0231953. [Google Scholar] [CrossRef]
- Rampurwala, M.; Wisinski, K.B.; O’Regan, R. Role of the androgen receptor in triple-negative breast cancer. Clin. Adv. Hematol. Oncol. 2016, 14, 186–193. [Google Scholar]
- Caiazza, F.; Murray, A.; Madden, S.F.; Synnott, N.C.; Ryan, E.J.; O’Donovan, N.; Crown, J.; Duffy, M.J. Preclinical evaluation of the AR inhibitor enzalutamide in triple-negative breast cancer cells. Endocr. Relat. Cancer 2016, 23, 323–334. [Google Scholar] [CrossRef]
- Barton, V.N.; D’Amato, N.C.; Gordon, M.A.; Lind, H.T.; Spoelstra, N.S.; Babbs, B.L.; Heinz, R.E.; Elias, A.; Jedlicka, P.; Jacobsen, B.M.; et al. Multiple molecular subtypes of triple-negative breast cancer critically rely on androgen receptor and respond to enzalutamide in vivo. Mol. Cancer Ther. 2015, 14, 769–778. [Google Scholar] [CrossRef]
- Barton, V.N.; D’Amato, N.C.; Gordon, M.A.; Christenson, J.L.; Elias, A.; Richer, J.K. Androgen receptor biology in triple negative breast cancer: A case for classification as AR+ or quadruple negative disease. Horm. Cancer 2015, 6, 206–213. [Google Scholar] [CrossRef]
- Christenson, J.L.; Trepel, J.B.; Ali, H.Y.; Lee, S.; Eisner, J.R.; Baskin-Bey, E.S.; Elias, A.D.; Richer, J.K. Harnessing a different dependency: How to identify and target androgen receptor-positive versus quadruple-negative breast cancer. Horm. Cancer 2018, 9, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, R.; Mohler, M.L.; Bohl, C.E.; Miller, D.D.; Dalton, J.T. Selective androgen receptor modulators in preclinical and clinical development. Nucl. Recept. Signal. 2008, 6, e010. [Google Scholar] [CrossRef] [PubMed]
- Bird, I.M.; Abbott, D.H. The hunt for a selective 17,20 lyase inhibitor; learning lessons from nature. J. Steroid Biochem. Mol. Biol. 2016, 163, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.D.; Schellhammer, P.F.; McLeod, D.G.; Moul, J.W.; Higano, C.S.; Shore, N.; Denis, L.; Iversen, P.; Eisenberger, M.A.; Labrie, F. Androgen receptor targeted treatments of prostate cancer: 35 years of progress with antiandrogens. J. Urol. 2018, 200, 956–966. [Google Scholar] [CrossRef]
- Gucalp, A.; Traina, T.A. Targeting the androgen receptor in triple-negative breast cancer. Curr. Probl. Cancer 2016, 40, 141–150. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; De Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Chen, J.; Kim, J.; Dalton, J.T. Discovery and therapeutic promise of selective androgen receptor modulators. Mol. Interv. 2005, 5, 173–188. [Google Scholar] [CrossRef]
- Solomon, Z.J.; Mirabal, J.R.; Mazur, D.J.; Kohn, T.P.; Lipshultz, L.I.; Pastuszak, A.W. Selective androgen receptor modulators: Current knowledge and clinical applications. Sex. Med. Rev. 2019, 7, 84–94. [Google Scholar] [CrossRef]
- Masiello, D.; Cheng, S.; Bubley, G.J.; Lu, M.L.; Balk, S.P. Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. J. Biol. Chem. 2002, 277, 26321–26326. [Google Scholar] [CrossRef]
- Huang, R.; Han, J.; Liang, X.; Sun, S.; Jiang, Y.; Xia, B.; Niu, M.; Li, D.; Zhang, J.; Wang, S.; et al. Androgen receptor expression and bicalutamide antagonize androgen receptor inhibit β-catenin transcription complex in estrogen receptor-negative breast cancer. Cell. Physiol. Biochem. 2017, 43, 2212–2225. [Google Scholar] [CrossRef]
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic breast cancer. Clin. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, D.R.; Bernales, S.; Jacobsen, B.M.; Cittelly, D.M.; Howe, E.N.; D’Amato, N.C.; Spoelstra, N.S.; Edgerton, S.M.; Jean, A.; Guerrero, J.; et al. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 2014, 16. [Google Scholar] [CrossRef]
- Traina, T.A.; Miller, K.; Yardley, D.A.; O’Shaughnessy, J.; Cortes, J.; Awada, A.; Kelly, C.M.; Trudeau, M.E.; Schmid, P.; Gianni, L.; et al. Results from a phase 2 study of enzalutamide (ENZA), an androgen receptor (AR) inhibitor, in advanced AR+ triple-negative breast cancer (TNBC). J. Clin. Oncol. 2015, 33, 1003. [Google Scholar] [CrossRef]
- Yin, L.; Hu, Q. CYP17 inhibitors—Abiraterone, C17,20-lyase inhibitors and multi-targeting agents. Nat. Rev. Urol. 2014, 11, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoi, H.; Grellety, T.; Tredan, O.; Saghatchian, M.; Dalenc, F.; Mailliez, A.; Cottu, P.; Abadie-Lacourtoisie, S.; You, B.; Mousseau, M.; et al. A phase II trial of abiraterone acetate plus prednisone in patients with triple-negative androgen receptor positive locally advanced or metastatic breast cancer (UCBG 12-1) original articles Annals of Oncology. Ann. Oncol. 2016, 27, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Grellety, T.; Callens, C.; Richard, E.; Briaux, A.; Velasco, V.; Pulido, M.; Gonçalves, A.; Gestraud, P.; MacGrogan, G.; Bonnefoi, H.; et al. Enhancing abiraterone acetate efficacy in androgen receptor–positive triple-negative breast cancer: Chk1 as a potential target. Clin. Cancer Res. 2019, 25, 856–867. [Google Scholar] [CrossRef]
- Roviello, G.; Pacifico, C.; Chiriacò, G.; Generali, D. Is still there a place for orteronel in management of prostate cancer? Data from a literature based meta-analysis of randomized trials. Crit. Rev. Oncol. Hematol. 2017, 113, 18–21. [Google Scholar] [CrossRef]
- Reese, J.; Babbs, B.; Christenson, J.; Spoelstra, N.; Elias, A.; Eisner, J.; Baskin-Bey, E.; Gertz, J.; Richer, J. Abstract P5-05-05: Targeting the androgen receptor with seviteronel, a CYP17 lyase and AR inhibitor, in triple negative breast cancer. Cancer Res. 2019, 79. [Google Scholar] [CrossRef]
- Michmerhuizen, A.R.; Chandler, B.; Olsen, E.; Wilder-Romans, K.; Moubadder, L.; Liu, M.; Pesch, A.M.; Zhang, A.; Ritter, C.; Ward, S.T.; et al. Seviteronel, a novel CYP17 lyase inhibitor and androgen receptor antagonist, radiosensitizes ar-positive triple negative breast cancer cells. Front. Endocrinol. 2020, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Gucalp, A.; Danso, M.A.; Elias, A.D.; Bardia, A.; Ali, H.Y.; Potter, D.; Gabrail, N.Y.; Haley, B.B.; Khong, H.T.; Riley, E.C.; et al. Phase (Ph) 2 stage 1 clinical activity of seviteronel, a selective CYP17-lyase and androgen receptor (AR) inhibitor, in women with advanced AR+ triple-negative breast cancer (TNBC) or estrogen receptor (ER)+ BC: CLARITY-01. J. Clin. Oncol. 2017, 35, 1102. [Google Scholar] [CrossRef]
- Gao, W.; Dalton, J.T. Expanding the therapeutic use of androgens via selective androgen receptor modulators (SARMs). Drug Discov. Today 2007, 12, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, R.; Ahn, S.; Cheney, M.D.; Yepuru, M.; Miller, D.D.; Steiner, M.S.; Dalton, J.T. Selective Androgen Receptor Modulators (SARMs) negatively regulate triple-negative breast cancer growth and epithelial: Mesenchymal stem cell signaling. PLoS ONE 2014, 9, e103202. [Google Scholar] [CrossRef]
- Obeid, E.I.; Conzen, S.D. The role of adrenergic signaling in breast cancer biology. Cancer Biomark. 2013, 13, 161–169. [Google Scholar] [CrossRef]
- Powe, D.G.; Voss, M.J.; Zänker, K.S.; Habashy, H.O.; Green, A.R.; Ellis, I.O.; Entschladen, F. Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget 2010, 1, 628–638. [Google Scholar] [CrossRef]
- Melhem-Bertrandt, A.; Chavez-MacGregor, M.; Lei, X.; Brown, E.N.; Lee, R.T.; Meric-Bernstam, F.; Sood, A.K.; Conzen, S.D.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2011, 29, 2645–2652. [Google Scholar] [CrossRef]
- Kafetzopoulou, L.E.; Boocock, D.J.; Dhondalay, G.K.R.; Powe, D.G.; Ball, G.R. Biomarker identification in breast cancer: Beta-adrenergic receptor signaling and pathways to therapeutic response. Comput. Struct. Biotechnol. J. 2013, 6, e201303003. [Google Scholar] [CrossRef][Green Version]
- Vázquez, S.M.; Mladovan, A.G.; Pérez, C.; Bruzzone, A.; Baldi, A.; Lüthy, I.A. Human breast cell lines exhibit functional α2-adrenoceptors. Cancer Chemother. Pharmacol. 2006, 58, 50–61. [Google Scholar] [CrossRef]
- Pérez Piñero, C.; Bruzzone, A.; Sarappa, M.G.; Castillo, L.F.; Lüthy, I.A. Involvement of α2- and β2-adrenoceptors on breast cancer cell proliferation and tumour growth regulation. Br. J. Pharmacol. 2012, 166, 721–736. [Google Scholar] [CrossRef]
- Drell, T.L., IV; Joseph, J.; Lang, K.; Niggemann, B.; Zaenker, K.S.; Entschladen, F. Effects of neurotransmitters on the chemokinesis and chemotaxis of MDA-MB-468 human breast carcinoma cells. Breast Cancer Res. Treat. 2003, 80, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.; Drell, T.L.; Lindecke, A.; Niggemann, B.; Kaltschmidt, C.; Zaenker, K.S.; Entschladen, F. Induction of a metastatogenic tumor cell type by neurotransmitters and its pharmacological inhibition by established drugs. Int. J. Cancer 2004, 112, 231–238. [Google Scholar] [CrossRef]
- Strell, C.; Niggemann, B.; Voss, M.J.; Powe, D.G.; Zänker, K.S.; Entschladen, F. Norepinephrine promotes the β1-integrin-mediated adhesion of MDA-MB-231 cells to vascular endothelium by the induction of a GROα release. Mol. Cancer Res. 2012, 10, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, T.A.; Zhang, J.; Dancel, R.; Garcia, S.J.; Willis, C.; Seidler, F.J. β-adrenoceptor signaling and its control of cell replication in MDA-MB-231 human breast cancer cells. Breast Cancer Res. Treat. 2000, 60, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Barron, T.I.; Connolly, R.M.; Sharp, L.; Bennett, K.; Visvanathan, K. Beta blockers and breast cancer mortality: A population-based study. J. Clin. Oncol. 2011, 29, 2635–2644. [Google Scholar] [CrossRef] [PubMed]
- Botteri, E.; Munzone, E.; Rotmensz, N.; Cipolla, C.; De Giorgi, V.; Santillo, B.; Zanelotti, A.; Adamoli, L.; Colleoni, M.; Viale, G.; et al. Therapeutic effect of β-blockers in triple-negative breast cancer postmenopausal women. Breast Cancer Res. Treat. 2013, 140, 567–575. [Google Scholar] [CrossRef]
- Musini, V.M.; Pasha, P.; Gill, R.; Wright, J.M. Blood pressure lowering efficacy of clonidine for primary hypertension. Cochrane Database Syst. Rev. 2017, 2017. [Google Scholar] [CrossRef]
- Bruzzone, A.; Pinero, P.C.; Rojas, P.; Romanato, M.; Gass, H.; Lanari, C.; Luthy, A.I. α(2)-Adrenoceptors Enhance Cell Proliferation and Mammary Tumor Growth Acting Through both the Stroma and the Tumor Cells. Curr. Cancer Drug Targets 2011, 11, 763–774. [Google Scholar] [CrossRef]
- DrugBank Yohimbine. Available online: https://www.drugbank.ca/drugs/DB01392 (accessed on 2 July 2020).
- Luthy, I.; Bruzzone, A.; Pinero, C.; Castillo, L.; Chiesa, I.; Vazquez, S.; Sarappa, M. Adrenoceptors: Non conventional target for breast cancer? Curr. Med. Chem. 2009, 16, 1850–1862. [Google Scholar] [CrossRef]
- Flint, M.S.; Kim, G.; Hood, B.L.; Bateman, N.W.; Stewart, N.A.; Conrads, T.P. Stress hormones mediate drug resistance to paclitaxel in human breast cancer cells through a CDK-1-dependent pathway. Psychoneuroendocrinology 2009, 34, 1533–1541. [Google Scholar] [CrossRef]
- Cole, S.W.; Sood, A.K. Molecular pathways: Beta-adrenergic signaling in cancer. Clin. Cancer Res. 2012, 18, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.Y.; He, R.H.; Zhang, J.; He, Y.J.; Wan, Z.; Zhou, C.F.; Tang, Y.J.; Li, Z.; McLeod, H.L.; Liu, J. β-blockers inhibit the viability of breast cancer cells by regulating the ERK/COX-2 signaling pathway and the drug response is affected by ADRB2 single-nucleotide polymorphisms. Oncol. Rep. 2019, 41, 341–350. [Google Scholar] [CrossRef]
- Pasquier, E.; Ciccolini, J.; Carre, M.; Giacometti, S.; Fanciullino, R.; Pouchy, C.; Montero, M.P.; Serdjebi, C.; Kavallaris, M.; André, N. Propranolol potentiates the anti-angiogenic effects and antitumor efficacy of chemotherapy agents: Implication in breast cancer treatment. Oncotarget 2011, 2, 797–809. [Google Scholar] [CrossRef]
- Ganz, P.A.; Cole, S.W. Expanding our therapeutic options: Beta blockers for breast cancer? J. Clin. Oncol. 2011, 29, 2612–2616. [Google Scholar] [CrossRef] [PubMed]
- Cakir, Y.; Plummer, H.K.; Tithof, P.K.; Schuller, H.M. Beta-adrenergic and arachidonic acid-mediated growth regulation of human breast cancer cell lines. Int. J. Oncol. 2002, 21, 153–157. [Google Scholar] [CrossRef]
- Talarico, G.; Orecchioni, S.; Dallaglio, K.; Reggiani, F.; Mancuso, P.; Calleri, A.; Gregato, G.; Labanca, V.; Rossi, T.; Noonan, D.M.; et al. Aspirin and atenolol enhance metformin activity against breast cancer by targeting both neoplastic and microenvironment cells. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.H.; Qin, L.; Li, X. Role of STAT3 signaling pathway in breast cancer. Cell Commun. Signal. 2020, 18, 33. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Chen, X.; Lo, H.W.; Lin, J. Combined bazedoxifene and paclitaxel treatments inhibit cell viability, cell migration, colony formation, and tumor growth and induce apoptosis in breast cancer. Cancer Lett. 2019, 448, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xiao, H.; Lin, L.; Jou, D.; Kumari, V.; Lin, J.; Li, C. Drug design targeting protein-protein interactions (PPIs) using multiple ligand simultaneous docking (MLSD) and drug repositioning: Discovery of raloxifene and bazedoxifene as novel inhibitors of IL-6/GP130 interface. J. Med. Chem. 2014, 57, 632–641. [Google Scholar] [CrossRef]
- Tian, J.; Chen, X.; Fu, S.; Zhang, R.; Pan, L.; Cao, Y.; Wu, X.; Xiao, H.; Lin, H.J.; Lo, H.W.; et al. Bazedoxifene is a novel IL-6/GP130 inhibitor for treating triple-negative breast cancer. Breast Cancer Res. Treat. 2019, 175, 553–566. [Google Scholar] [CrossRef]
- Oh, E.; Kim, Y.J.; An, H.; Sung, D.; Cho, T.M.; Farrand, L.; Jang, S.; Seo, J.H.; Kim, J.Y. Flubendazole elicits anti-metastatic effects in triple-negative breast cancer via STAT3 inhibition. Int. J. Cancer 2018, 143, 1978–1993. [Google Scholar] [CrossRef]
- Hou, Z.J.; Luo, X.; Zhang, W.; Peng, F.; Cui, B.; Wu, S.J.; Zheng, F.M.; Xu, J.; Xu, L.Z.; Long, Z.J.; et al. Flubendazole, FDA-approved anthelmintic, targets breast cancer stem-like cells. Oncotarget 2015, 6, 6326–6340. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, X.; Ward, T.; Pegram, M.; Shen, K. Combined niclosamide with cisplatin inhibits epithelial-mesenchymal transition and tumor growth in cisplatin-resistant triple-negative breast cancer. Tumor Biol. 2016, 37, 9825–9835. [Google Scholar] [CrossRef]
- Wang, Y.C.; Chao, T.K.; Chang, C.C.; Yo, Y.T.; Yu, M.H.; Lai, H.C. Drug screening identifies niclosamide as an inhibitor of breast cancer stem-like cells. PLoS ONE 2013, 8, e74538. [Google Scholar] [CrossRef]
- Lu, L.; Dong, J.; Wang, L.; Xia, Q.; Zhang, D.; Kim, H.; Yin, T.; Fan, S.; Shen, Q. Activation of STAT3 and Bcl-2 and reduction of reactive oxygen species (ROS) promote radioresistance in breast cancer and overcome of radioresistance with niclosamide. Oncogene 2018, 37, 5292–5304. [Google Scholar] [CrossRef]
- Wang, L.; Peng, Y.; Shi, K.; Wang, H.; Lu, J.; Li, Y.; Ma, C. Osthole inhibits proliferation of human breast cancer cells by inducing cell cycle arrest and apoptosis. J. Biomed. Res. 2015, 29, 132–138. [Google Scholar] [CrossRef]
- Tang, D.Z.; Hou, W.; Zhou, Q.; Zhang, M.; Holz, J.; Sheu, T.J.; Li, T.F.; Cheng, S.D.; Shi, Q.; Harris, S.E.; et al. Osthole stimulates osteoblast differentiation and bone formation by activation of β-catenin-BMP signaling. J. Bone Miner. Res. 2010, 25, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Yin, C.; Zhang, Y.; Guo, G.; Zhao, C.; Wang, O.; Xiang, Y.; Zhang, X.; Liang, G. Osthole inhibits triple negative breast cancer cells by suppressing STAT3. J. Exp. Clin. Cancer Res. 2018, 37, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Palaka, B.K.; Venkatesan, R.; Ampasala, D.R.; Periyasamy, L. Identification of novel inhibitors of signal transducer and activator of transcription 3 over signal transducer and activator of transcription 1 for the treatment of breast cancer by in-silico and in-vitro approach. Process Biochem. 2019, 82, 153–166. [Google Scholar] [CrossRef]
- Schech, A.J.; Kazi, A.A.; Gilani, R.A.; Brodie, A.H. Zoledronic acid reverses the epithelial-mesenchymal transition and inhibits self-renewal of breast cancer cells through inactivation of NF- B. Mol. Cancer Ther. 2013, 12, 1356–1366. [Google Scholar] [CrossRef]
- Ottewell, P.D.; Mönkkönen, H.; Jones, M.; Lefley, D.V.; Coleman, R.E.; Holen, I. Antitumor effects of doxorubicin followed by zoledronic acid in a mouse model of breast cancer. J. Natl. Cancer Inst. 2008, 100, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Akazawa, K.; Hasegawa, Y.; Tanino, H.; Horiguchi, J.; Miura, D.; Hayashi, M.; Kohno, N. Survival outcomes of neoadjuvant chemotherapy with zoledronic acid for HER2-negative breast cancer. J. Surg. Res. 2017, 220, 46–51. [Google Scholar] [CrossRef]
- Burke, A.J.; Sullivan, F.J.; Giles, F.J.; Glynn, S.A. The yin and yang of nitric oxide in cancer progression. Carcinogenesis 2013, 34, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Glynn, S.A.; Boersma, B.J.; Dorsey, T.H.; Yi, M.; Yfantis, H.G.; Ridnour, L.A.; Martin, D.N.; Switzer, C.H.; Hudson, R.S.; Wink, D.A.; et al. Increased NOS2 predicts poor survival in estrogen receptor-negative breast cancer patients. J. Clin. Investig. 2010, 120, 3843–3854. [Google Scholar] [CrossRef] [PubMed]
- Granados-Principal, S.; Liu, Y.; Guevara, M.L.; Blanco, E.; Choi, D.S.; Qian, W.; Patel, T.; Rodriguez, A.A.; Cusimano, J.; Weiss, H.L.; et al. Inhibition of iNOS as a novel effective targeted therapy against triple-negative breast cancer. Breast Cancer Res. 2015, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Dave, B.; Granados-Principal, S.; Zhu, R.; Benz, S.; Rabizadeh, S.; Soon-Shiong, P.; Yu, K.D.; Shao, Z.; Li, X.; Gilcrease, M.; et al. Targeting RPL39 and MLF2 reduces tumor initiation and metastasis in breast cancer by inhibiting nitric oxide synthase signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 8838–8843. [Google Scholar] [CrossRef] [PubMed]
- González-González, A.; Muñoz-Muela, E.; Marchal, J.A.; Cara, F.E.; Molina, M.P.; Cruz-Lozano, M.; Jiménez, G.; Verma, A.; Ramírez, A.; Qian, W.; et al. Activating transcription factor 4 modulates TGFb-induced aggressiveness in triple-negative breast cancer via SMad2/3/4 and mTORC2 signaling. Clin. Cancer Res. 2018, 24, 5697–5709. [Google Scholar] [CrossRef]
- Davila-Gonzalez, D.; Choi, D.S.; Rosato, R.R.; Granados-Principal, S.M.; Kuhn, J.G.; Li, W.F.; Qian, W.; Chen, W.; Kozielski, A.J.; Wong, H.; et al. Pharmacological inhibition of NOS activates ASK1/JNK pathway augmenting docetaxel-mediated apoptosis in triple-negative breast cancer. Clin. Cancer Res. 2018, 24, 1152–1162. [Google Scholar] [CrossRef]
- Walsh, E.M.; Keane, M.M.; Wink, D.A.; Callagy, G.; Glynn, S.A. Review of triple negative breast cancer and the impact of inducible nitric oxide synthase on tumor biology and patient outcomes. Crit. Rev. Oncog. 2016, 21, 333–351. [Google Scholar] [CrossRef]
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013, 6, a009076. [Google Scholar] [CrossRef]
- Colavito, S.A. AXL as a target in breast cancer therapy. J. Oncol. 2020, 2020, 5291952. [Google Scholar] [CrossRef]
- Asiedu, M.K.; Beauchamp-perez, F.D.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. AXL induces epithelial to mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene 2014, 33, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Goyette, M.A.; Cusseddu, R.; Elkholi, I.; Abu-Thuraia, A.; El-Hachem, N.; Haibe-Kains, B.; Gratton, J.P.; Côté, J.F. AXL knockdown gene signature reveals a drug repurposing opportunity for a class of antipsychotics to reduce growth and metastasis of triple-negative breast cancer. Oncotarget 2019, 10, 2055–2067. [Google Scholar] [CrossRef]
- Meyer, A.S.; Miller, M.A.; Gertler, F.B.; Lauffenburger, D.A. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci. Signal. 2013, 6, ra66. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Kreso, A.; Dick, J.E. Cancer stem cells in solid tumors: An overview. Semin. Radiat. Oncol. 2009, 19, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Bomken, S.; Fišer, K.; Heidenreich, O.; Vormoor, J. Understanding the cancer stem cell. Br. J. Cancer 2010, 103, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Rosen, J.M.; Jordan, C.T. The increasing complexity of the cancer stem cell paradigm. Science 2009, 324, 1670–1673. [Google Scholar] [CrossRef]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef]
- Asiedu, M.K.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. TGFβ/TNFα-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. 2011, 71, 4707–4719. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.J.; Olsson, A.K.; Moustakas, A. Reprogramming during epithelial to mesenchymal transition under the control of TGFβ. Cell Adhes. Migr. 2015, 9, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Idowu, M.O.; Kmieciak, M.; Dumur, C.; Burton, R.S.; Grimes, M.M.; Powers, C.N.; Manjili, M.H. CD44+/CD24−/low cancer stem/progenitor cells are more abundant in triple-negative invasive breast carcinoma phenotype and are associated with poor outcome. Hum. Pathol. 2012, 43, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ma, F.; Wang, H.; Lin, C.; Fan, Y.; Zhang, X.; Qian, H.; Xu, B. Stem cell marker aldehyde dehydrogenase 1 (ALDH1)-expressing cells are enriched in triple-negative breast cancer. Int. J. Biol. Markers 2013, 28, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Palomeras, S.; Ruiz-Martínez, S.; Puig, T. Targeting breast cancer stem cells to overcome treatment resistance. Molecules 2018, 23, 2193. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, L.; Hu, C.; Liang, S.; Fei, X.; Yan, N.; Zhang, Y.; Zhang, F. WNT pathway inhibitor pyrvinium pamoate inhibits the self-renewal and metastasis of breast cancer stem cells. Int. J. Oncol. 2016, 48, 1175–1186. [Google Scholar] [CrossRef]
- Papi, A.; Orlandi, M. Role of nuclear receptors in breast cancer stem cells. World J. Stem Cells 2016, 8, 62–72. [Google Scholar] [CrossRef]
- Cui, J.; Hollmén, M.; Li, L.; Chen, Y.; Proulx, S.T.; Reker, D.; Schneider, G.; Detmar, M. New use of an old drug: Inhibition of breast cancer stem cells by benztropine mesylate. Oncotarget 2017, 8, 1007–1022. [Google Scholar] [CrossRef]
- Sogawa, C.; Eguchi, T.; Tran, M.T.; Ishige, M.; Trin, K.; Okusha, Y.; Taha, E.A.; Lu, Y.; Kawai, H.; Sogawa, N.; et al. Antiparkinson drug benztropine suppresses tumor growth, circulating tumor cells, and metastasis by acting on SLC6A3/dat and reducing STAT3. Cancers 2020, 12, 523. [Google Scholar] [CrossRef]
- Choi, D.S.; Blanco, E.; Kim, Y.; Rodriguez, A.A.; Zhao, H.; Huang, T.H.; Chen, C.; Jin, G.; Landis, M.D.; Lacey, A.; et al. Chloroquine eliminates cancer stem cells through deregulation of Jak2 and DNMT1. Stem Cells 2014, 32, 2309–2323. [Google Scholar] [CrossRef]
- Yang, L.; Wu, X.; Wang, Y.; Zhang, K.; Wu, J.; Yuan, Y.C.; Deng, X.; Chen, L.; Kim, C.C.H.; Lau, S.; et al. FZD7 has a critical role in cell proliferation in triple negative breast cancer. Oncogene 2011, 30, 4437–4446. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Prior, J.; Piwnica-Worms, D.; Bu, G. LRP6 overexpression defines a class of breast cancer subtype and is a target for therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 5136–5141. [Google Scholar] [CrossRef]
- King, T.D.; Suto, M.J.; Li, Y. The wnt/β-catenin signaling pathway: A potential therapeutic target in the treatment of triple negative breast cancer. J. Cell. Biochem. 2012, 113, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Kai, M.; Kanaya, N.; Wu, S.V.; Mendez, C.; Nguyen, D.; Luu, T.; Chen, S. Targeting breast cancer stem cells in triple-negative breast cancer using a combination of LBH589 and salinomycin. Breast Cancer Res. Treat. 2015, 151, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Thorne, C.A.; Hanson, A.J.; Schneider, J.; Tahinci, E.; Orton, D.; Cselenyi, C.S.; Jernigan, K.K.; Meyers, K.C.; Hang, B.I.; Waterson, A.G.; et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat. Chem. Biol. 2010, 6, 829–836. [Google Scholar] [CrossRef]
- Carrella, D.; Manni, I.; Tumaini, B.; Dattilo, R.; Papaccio, F.; Mutarelli, M.; Sirci, F.; Amoreo, C.A.; Mottolese, M.; Iezzi, M.; et al. Computational drugs repositioning identifies inhibitors of oncogenic PI3K/AKT/P70S6K-dependent pathways among FDAapproved compounds. Oncotarget 2016, 7, 58743–58758. [Google Scholar] [CrossRef]
- Ishii, I.; Harada, Y.; Kasahara, T. Reprofiling a classical anthelmintic, pyrvinium pamoate, as an anti-cancer drug targeting mitochondrial respiration. Front. Oncol. 2012, 2, 1–4. [Google Scholar] [CrossRef]
- Dattilo, R.; Mottini, C.; Camera, E.; Lamolinara, A.; Auslander, N.; Doglioni, G.; Muscolini, M.; Tang, W.; Planque, M.; Ercolani, C.; et al. Pyrvinium pamoate induces death of triple-negative breast cancer stem-like cells and reduces metastases through effects on lipid anabolism. Cancer Res. 2020. [Google Scholar] [CrossRef]
- So, J.Y.; Suh, N. Targeting cancer stem cells in solid tumors by vitamin D. J. Steroid Biochem. Mol. Biol. 2015, 148, 79–85. [Google Scholar] [CrossRef]
- Wahler, J.; So, J.Y.; Cheng, L.C.; Maehr, H.; Uskokovic, M.; Suh, N. Vitamin D compounds reduce mammosphere formation and decrease expression of putative stem cell markers in breast cancer. J. Steroid Biochem. Mol. Biol. 2015, 148, 148–155. [Google Scholar] [CrossRef]
- Pickholtz, I.; Saadyan, S.; Keshet, G.I.; Wang, V.S.; Cohen, R.; Bouwman, P.; Jonkers, J.; Byers, S.W.; Papa, M.Z.; Yarden, R.I. Cooperation between BRCA1 and vitamin D is critical for histone acetylation of the p21waf1 promoter and for growth inhibition of breast cancer cells and cancer stem-like cells. Oncotarget 2014, 5, 11827–11846. [Google Scholar] [CrossRef][Green Version]
- Pervin, S.; Hewison, M.; Braga, M.; Tran, L.; Chun, R.; Karam, A.; Chaudhuri, G.; Norris, K.; Singh, R. Correction: Down-regulation of vitamin D receptor in mammospheres: Implications for vitamin D resistance in breast cancer and potential for combination therapy. PLoS ONE 2013, 8, e53287. [Google Scholar] [CrossRef]
- Zanetti, A.; Affatato, R.; Centritto, F.; Fratelli, M.; Kurosaki, M.; Barzago, M.M.; Bolis, M.; Terao, M.; Garattini, E.; Paroni, G. All-trans-retinoic acid modulates the plasticity and inhibits the motility of breast cancer cells role of notch1 and transforming growth factor (TGF β). J. Biol. Chem. 2015, 290, 17690–17709. [Google Scholar] [CrossRef]
- Bhat-Nakshatri, P.; Goswami, C.P.; Badve, S.; Sledge, G.W.; Nakshatri, H. Identification of FDA-approved drugs targeting breast cancer stem cells along with biomarkers of sensitivity. Sci. Rep. 2013, 3, 2530. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Wicinski, J.; Cervera, N.; Monville, F.; Finetti, P.; Bertucci, F.; Wicha, M.S.; Birnbaum, D.; Charafe-Jauffret, E. Retinoid signaling regulates breast cancer stem cell differentiation. Cell Cycle 2009, 8, 3297–3302. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.; Niravath, P.; Patel, T.; Ensor, J.; Rodriguez, A.; Boone, T.; Wong, S.T.; Chang, J.C. A Phase II study of the efficacy and safety of Chloroquine in combination with Taxanes in the treatment of patients with advanced or metastatic anthracycline-refractory breast cancer. Clin. Breast Cancer 2020. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Agent | Mechanism | Original Indication |
|---|---|---|---|
| Microtubule inhibitors | Paclitaxel Docetaxel | Disruption of microtubule dynamics leading to the end of cell division. | Ovarian cancer, atrial restenosis hormone-refractory prostate cancer |
| Anthracyclines | Doxorubicin, Epirubicin | Inhibition of DNA, RNA synthesis forming an anthracycline-DNA-topoisomerase II ternary complex. Harm of mitochondrial function. Generation of oxygen-free radicals. Activation of apoptosis and matrix metalloproteinase. Immune reactions. | Antibiotics from Streptomyces peucetius bacterium |
| Alkylating agents | Cyclophosphamide | Inhibition of DNA replication. | Immuno-modulator in autoimmune diseases. Immunosuppressant |
| Antimetabolites | Methotrexate | Antagonist of dihydrofolate reductase. Decrease the synthesis of purines and pyrimidines. | Leukemia |
| Capecitabine | 5-fluorouracil pro-drug. Inhibition of thymidylate synthetase. | Colon cancer | |
| Gemcitabine | Analogue of cytidine. Irreparable errors that inhibit DNA replication. | Anti-viral drug | |
| Platinum | Carboplatin, Cisplatin | Damage of genetic material | Testicular, ovarian, and bladder cancers |
| Class | Agent | Mechanism | Original Indication |
|---|---|---|---|
| PARP inhibitors | Olaparib Talazoparib | Inhibition of PARP. Cell death due to accumulation of irreparable DNA damage. | Ovarian cancer with BRCA mutation |
| PD-L1 inhibitor | Atezolizumab | Block interaction with receptors PD-1 and reverse T-cell suppression. | Non-small cell lung cancer Bladder cancer |
| ADC | Sacituzumab govitecan | Targeted to Trop-2 and conjugated with SN-38, a DNA damaging agent. | - |
| Mechanism | Compound | Pre-Clinical Effects | Original Indication | Repurposing Method | References |
|---|---|---|---|---|---|
| α-ADR antagonist | α -yohimbine | Reduction of tumor growth in vitro. Development of resistance to paclitaxel when treated in combination with catecholamines and/or cortisol in vitro. Reversion of tumor growth after stimulation with clonidine in vivo. | Impotence | Non computational: target-based | [59,60,61] |
| Non-selective β1/β2-blocker | Propranolol | Inhibition of cell proliferation, arrest of the cell cycle at G0/G1 and S, and induction of cell apoptosis in vitro. Inhibition of tumor growth in vivo. Combination of propranolol with paclitaxel increased the anti-tumor efficacy of paclitaxel in vivo. Associated with less advanced disease at diagnosis and decreased risk of metastasis and mortality. Reverted isoproterenol-induced cell inhibition. | Hypertension | Non computational: target-based | [61,62,63,64,65] |
| Selective β1-blocker | Atenolol | Reduction of norepinephrine-induced cell migration in vitro. Inhibition of cell proliferation in vitro. Combination with metformin enhanced reduction of angiogenesis and metastasis in vivo. Not associated with differences tumor incidence, risk of metastasis and mortality rates. Associated with significantly lower recurrence but no significant OS. | Hypertension | Non computational: target-based | [63,66,67,68,69,70] |
| Metoprolol | Associated with significantly lower recurrence but no significant OS. | Hypertension | Non computational: target-based | [68] | |
| STAT3 inhibitor | Bazedoxifene | Decrease of cell viability, migration, colony formation. Increase cell apoptosis. Improvement of sensitivity to paclitaxel if combination. | Osteoporosis | Computational: structure-based | [71,72] |
| Flubendazole | Inhibition of cell proliferation in vitro and tumor growth in vivo. Reduction of CD44high/CD24low CSC population, mammosphere-forming ability and expression of stemness genes. Improvement of sensitivity to fluorouracil and doxorubicin if combination. | Anthelmintic | Non computational: target-based | [73] | |
| Niclosamide | Inhibition of cell proliferation in vitro and tumor growth in vivo. Reversion of EMT and inhibition of stem-like phenotype in cancer cells. Radiosensitizer in vitro and in vivo. | Anthelmintic | Non computational: screening | [74,75,76] | |
| Osthole | Induction of apoptosis in vitro. Reduction of tumor growth in vivo. | Osteoporosis | Non computational: literature-based | [77,78] | |
| Risedronate Sodium | Toxicity in TNBC cells in vitro. | Osteoporosis | Computational: structure-based | [79] | |
| AXL pathway modulator | Thioridazine Fluphenazine Trifluoperazine | Decrease of cell invasion, proliferation, and viability and increase of apoptosis in vitro. Reduction of tumor growth and metastasis in vivo. | Anti-psychotics | Computational: transcriptional signature-based | [80] |
| Mechanism | Compound | Preclinical and Clinical Effects | Clinical Trials 1 | Original Indication | Repurposing Method | References |
|---|---|---|---|---|---|---|
| AR antagonist | Bicalutamide | Reduction of cellular proliferation and colony formation, and induction cell apoptosis in vitro. Decreased cellular viability and induced apoptosis in vivo. CBR at 6 months of 19% and median PFS of 12 weeks (n = 26; AR expression higher than 10% by IHC). Grade 1–3 AEs included fatigue, limb edema, or hot flashes. | Phase II—completed (NCT00468715) Phase II—recruiting (NCT02605486) Phase III—recruiting (NCT03055312) | Prostate cancer | Non computational: target-based | [81,82] |
| Enzalutamide | Reduction of cell proliferation, migration and invasion and increased apoptosis in vitro. Inhibition of tumor viability by inducing cell apoptosis in vivo. CBR at 16 weeks of 25%, median PFS of 2.9 months and median OS of 12.7 months (n = 118; AR expression higher than 0% by IHC). CBR at 16 weeks of 33%, median PFS of 3.3 months and median OS of 17.6 months (n = 78; AR expression higher than 10% by IHC). Grade 3 AEs included fatigue. | Phase II—completed (NCT01889238) Phase II—recruiting (NCT02689427) Phase Ib/II—active (NCT02457910) | Prostate cancer | Non computational: target-based | [55,56,83,84] | |
| Abiraterone acetate | Combination treatment with Chk1 inhibitors had an additive effect inhibiting cell apoptosis in vitro. Reduction of tumor growth, which was significantly higher with the combination treatment. CBR at 6 months of 20% and median PFS of 2.8 months (n = 30; AR expression higher than 10% by IHC). Grade 1/2 AEs included hypertension, fatigue, nausea, and hypokalemia. | Phase II—completed (NCT01842321) | Prostate cancer | Non computational: target-based | [85,86] | |
| Orteronel | Currently being investigated. | Phase II—active (NCT01990209) | Prostate cancer | Non computational: target-based | NCT01990209 | |
| Seviteronel | Inhibition of cellular growth in vitro. Inhibition of tumor volume in vivo. Induction of radiosensitization, both in vitro and in vivo. Early results: CBR at 16 weeks of 33% (n = 6). Grade 1/2 AEs included fatigue, nausea and decreased appetite. | Phase I/II—completed (NCT02580448) Phase II—completed (NCT02130700) | Prostate cancer | Non computational: target-based | [87,88,89] | |
| Enobosarm | Currently being investigated. | Phase II—terminated (NCT02368691) | Prostate cancer | Non computational: target-based | NCT02368691 | |
| STAT3 inhibitor | Zoledronic acid | Induction of cell cycle arrest, decrease of cell viability, cell proliferation, self-renewal and expression of EMT markers in vitro. Antitumor potential with doxorubicin in vivo. Improvement of pCR and DFS in combination with chemotherapy versus only chemotherapy. | Phase II—completed (UMIN000003261) Phase II—terminated (low accrual rate) (NCT02347163) Phase II—recruiting (NCT03358017) Phase III—active (NCT02595138) Phase unknown—recruiting (NCT04045522) | Osteoporosis | Computational: structure-based, Non computational: literature-based | [79,90,91,92] |
| NOS inhibitor | L-NMMA | Decrease of cell proliferation, migration, and CSC self-renewal in vitro. Decrease of growth, CSC self-renewal and tumor initiation in xenograft models of TNBC. Improvement of chemotherapy response in combination with docetaxel in PDX models of TNBC. | Phase Ib/II—recruiting (NCT02834403) | Septic shock | Non computational: target-based | [93,94] |
| Mechanism | Compound | Cellular and Molecular Effects | Original Indication | Repurposing Method | References |
|---|---|---|---|---|---|
| Wnt, LRP6 | Salinomycin | Decreased CD44+/CD24−/low population both in vitro and in vivo. Inhibition of tumor growth and expression of CSC genes in vivo. Combination with LBH589 induced apoptosis and cell cycle arrest and regulates EMT in BCSCs. | Antibiotic | Non computational: high-throughput screening | [147,148,149] |
| Wnt/β-catenin, PI3K dependent pathway, lipid anabolism | Pyrvinium pamoate | Reduction of CSC self-renewal. Reduction of CD44+/CD24−/low and ALDH+ populations. Reduction of expression of EMT markers (N-cadherin, Vimentin and Snail). Reduction of tumor growth in vivo. | Anthelmintic | Non computational: high-throughput screening | [142,150,151] |
| Notch-1, NF-κB1 | Vitamin D3 | Reduction of cell proliferation, CD44+/CD24−/low population and mammosphere formation in vitro. Relative insensitivity to vitamin D3 treatment, but combination therapy with DETA NONOate achieved a significant decrease in mammosphere formation in vitro and tumor growth in vivo. | Vitamin supplement | Non computational: target-based | [152,153,154] |
| Notch-1, TGF-β | ATRA | Inhibition of mammospheres formation and reduction of CSC self-renewal. Reduction of ALDH1 CSC subpopulation. | Dermatologic diseases, acute promyelocytic leukemia | Computational: transcriptional signature-based | [155,156] |
| STAT3, NF-κB, and β-catenin | Benztropine mesylate | Inhibition of mammospheres formation and reduction of CSC self-renewal. Reduction of ALDH and CD44+/CD24−/low populations. | Parkinson’s disease | Computational: cell-based phenotypic screening | [144] |
| Jak2, DNMT1 | Chloroquine | Inhibition of autophagy. Reduction of mammosphere formation efficiency and CD44+/CD24−/low population in vitro. Sensitization to paclitaxel through the inhibition of autophagy in vitro. Combination of paclitaxel significantly reduced tumor growth and CD44+/CD24−/low population in vivo. Phase II clinical trial for chloroquine in combination with taxanes: ORR of 45.16%, median PFS of 12.4 months and median OS of 25.4 months. 13.15% of patients experienced Grade ≥ 3 adverse events. | Antimalarial | Computational: transcriptional signature-based | [146,157] NCT01446016 |
| STAT3 | Flubendazole | Loss of CD44+/CD24−/low population. Decrease of mammosphere-forming ability. Suppression of stem cell genes expression. | Anthelmintic | Non computational: target-based | [73] |
| Niclosamide | Reversion of EMT. Inhibition of stem-like phenotype. | Anthelmintic | Non computational: high-throughput screening | [74] | |
| STAT3, NF- κB | Zoledronic acid | Induction of cell cycle arrest, decrease of cell viability, cell proliferation, self-renewal and expression of EMT markers in vitro. | Osteoporosis | Computational: structure-based. Non computational: literature-based | [91] |
| iNOS | L-NMMA | Decrease of mammosphere-forming ability. | Septic shock | Non computational: target-based | [93] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ávalos-Moreno, M.; López-Tejada, A.; Blaya-Cánovas, J.L.; Cara-Lupiañez, F.E.; González-González, A.; Lorente, J.A.; Sánchez-Rovira, P.; Granados-Principal, S. Drug Repurposing for Triple-Negative Breast Cancer. J. Pers. Med. 2020, 10, 200. https://doi.org/10.3390/jpm10040200
Ávalos-Moreno M, López-Tejada A, Blaya-Cánovas JL, Cara-Lupiañez FE, González-González A, Lorente JA, Sánchez-Rovira P, Granados-Principal S. Drug Repurposing for Triple-Negative Breast Cancer. Journal of Personalized Medicine. 2020; 10(4):200. https://doi.org/10.3390/jpm10040200
Chicago/Turabian StyleÁvalos-Moreno, Marta, Araceli López-Tejada, Jose L. Blaya-Cánovas, Francisca E. Cara-Lupiañez, Adrián González-González, Jose A. Lorente, Pedro Sánchez-Rovira, and Sergio Granados-Principal. 2020. "Drug Repurposing for Triple-Negative Breast Cancer" Journal of Personalized Medicine 10, no. 4: 200. https://doi.org/10.3390/jpm10040200
APA StyleÁvalos-Moreno, M., López-Tejada, A., Blaya-Cánovas, J. L., Cara-Lupiañez, F. E., González-González, A., Lorente, J. A., Sánchez-Rovira, P., & Granados-Principal, S. (2020). Drug Repurposing for Triple-Negative Breast Cancer. Journal of Personalized Medicine, 10(4), 200. https://doi.org/10.3390/jpm10040200