
Predictive Biomarkers of Age-Related Macular Degeneration Response to Anti-VEGF Treatment

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Population and Group Characteristics
2.2. PBMCs Isolation
2.3. RNA Purification
2.4. Next Generation Sequencing
2.5. Transcriptomic Analysis and Bioinformatics
2.6. Data Mining and Feature Selection
3. Results
3.1. Cohort Description
3.2. Messenger RNA Expression Differences between Good Responders and Poor Responders
3.3. Micro RNA Expression Differences between Good Responders and Poor Responders
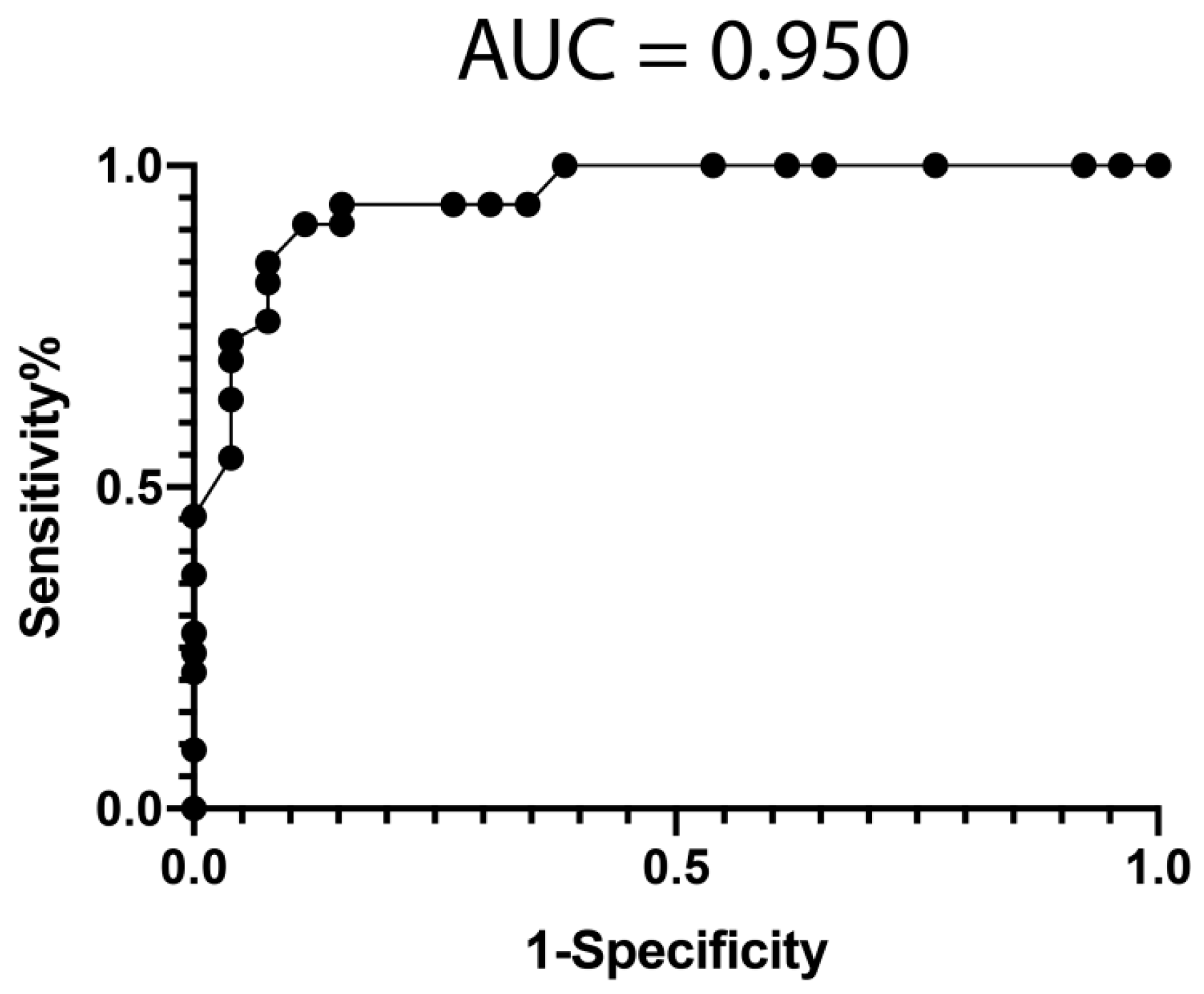
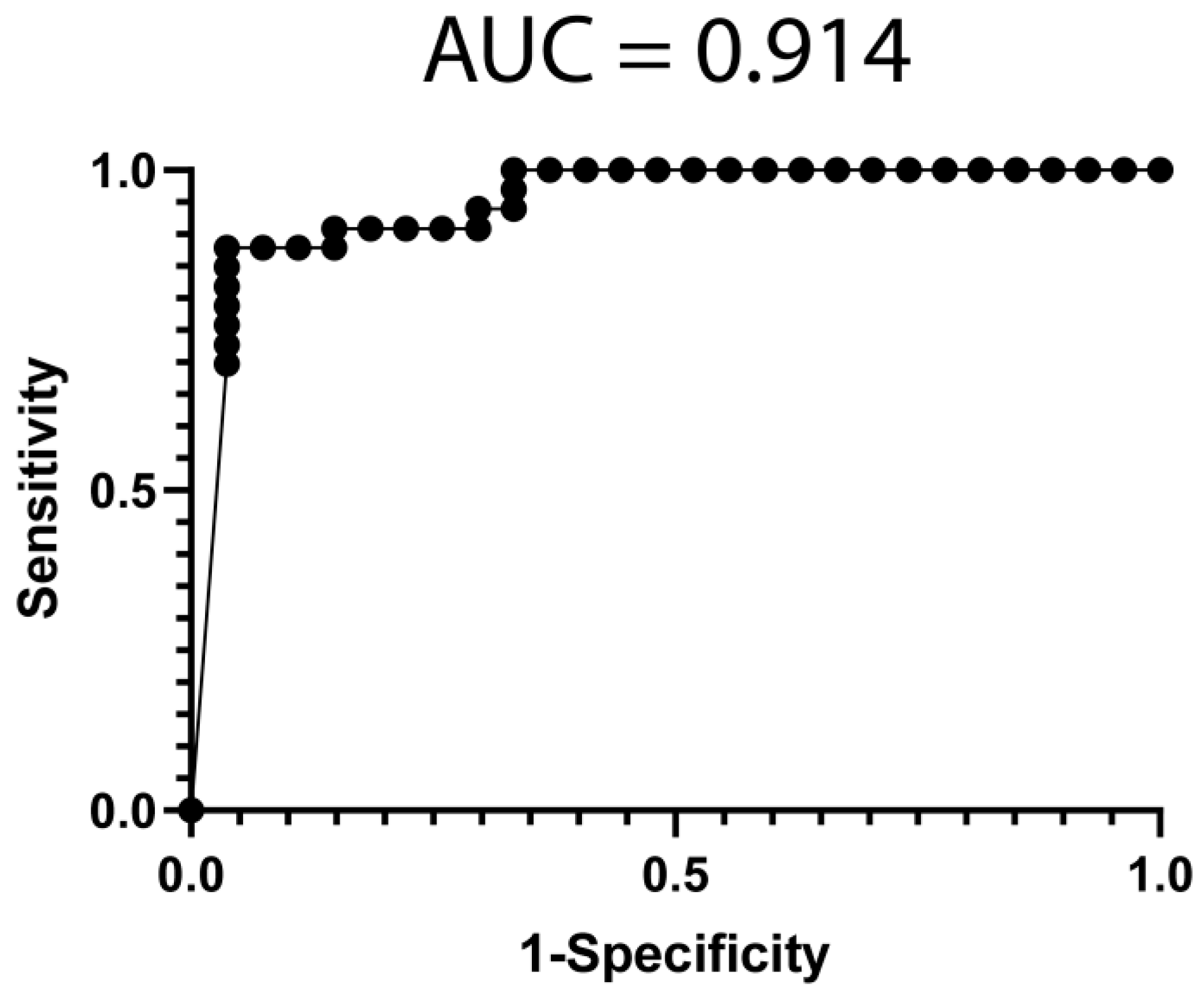
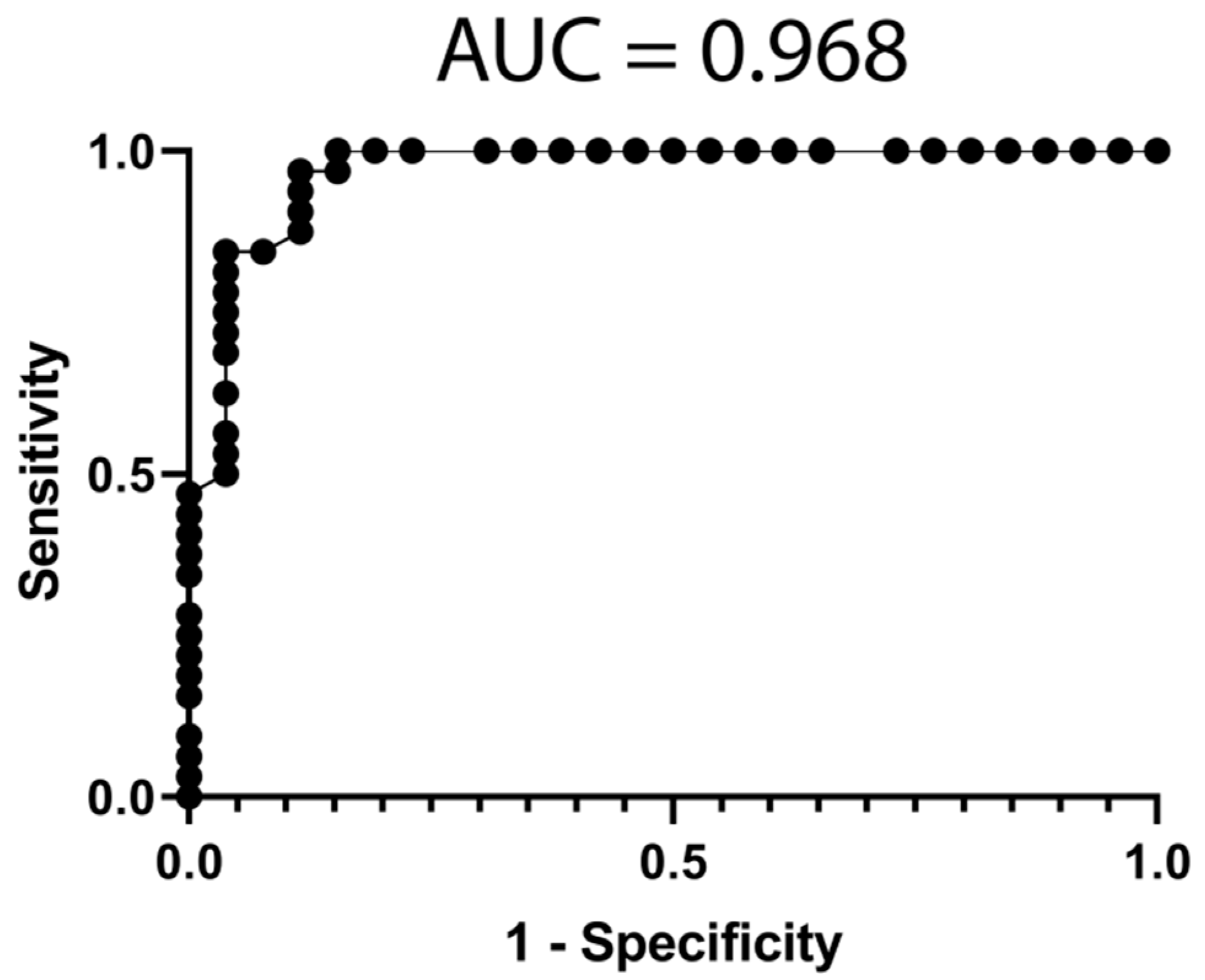
3.4. Classification Model from mRNA Data
3.5. Classification Model from miRNA Data
3.6. Classification Model from Combined mRNA and miRNA Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Curcio, C.A. Soft Drusen in Age-Related Macular Degeneration: Biology and Targeting Via the Oil Spill Strategies. Investig. Ophthalmol. Vis. Sci. 2018, 59, AMD160–AMD181. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-related macular degeneration. Nat. Rev. Dis. Prim. 2021, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.L.; Oh, L.J.; Wong, E.; Wei, J.; Chilov, M. Anti-vascular endothelial growth factor for neovascular age-related macular degeneration: A meta-analysis of randomized controlled trials. BMC Ophthalmol. 2018, 18, 130. [Google Scholar] [CrossRef]
- Ziemssen, F.; Eter, N.; Fauser, S.; Bopp, S.; Radermacher, M.; Hasanbasic, Z.; Holz, F.G. Retrospective investigation of anti-VEGF treatment reality and effectiveness in patients with neovascular age-related macular degeneration (AMD) in Germany: Treatment reality of ranibizumab for neovascular AMD in Germany. Ophthalmologe 2015, 112, 246–254. [Google Scholar] [CrossRef]
- Indaram, M.; Ma, W.; Zhao, L.; Fariss, R.N.; Rodriguez, I.R.; Wong, W.T. 7-Ketocholesterol increases retinal microglial migration, activation, and angiogenicity: A potential pathogenic mechanism underlying age-related macular degeneration. Sci. Rep. 2015, 5, 9144. [Google Scholar] [CrossRef] [PubMed]
- Lederman, M.; Weiss, A.; Chowers, I. Association of neovascular age-related macular degeneration with specific gene expression patterns in peripheral white blood cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 53–58. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Lauer, N.; Hartmann, A.; Stippa, S.; Keilhauer, C.N.; Oppermann, M.; Pandey, M.K.; Kohl, J.; Zipfel, P.F.; Weber, B.H.; et al. An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age-related macular degeneration (AMD). Hum. Mol. Genet. 2010, 19, 4694–4704. [Google Scholar] [CrossRef]
- Martinez-Barricarte, R.; Recalde, S.; Fernandez-Robredo, P.; Millan, I.; Olavarrieta, L.; Vinuela, A.; Perez-Perez, J.; Garcia-Layana, A.; Rodriguez de Cordoba, S. Relevance of complement factor H-related 1 (CFHR1) genotypes in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1087–1094. [Google Scholar] [CrossRef]
- Jabbarpoor Bonyadi, M.H.; Yaseri, M.; Soheilian, M. Association of combined complement factor H Y402H and ARMS2/LOC387715 A69S polymorphisms with age-related macular degeneration: An updated meta-analysis. Ophthalmic Genet. 2020, 41, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Romero-Vazquez, S.; Llorens, V.; Soler-Boronat, A.; Figueras-Roca, M.; Adan, A.; Molins, B. Interlink between Inflammation and Oxidative Stress in Age-Related Macular Degeneration: Role of Complement Factor H. Biomedicines 2021, 9, 763. [Google Scholar] [CrossRef]
- Anand, A.; Sharma, N.K.; Gupta, A.; Prabhakar, S.; Sharma, S.K.; Singh, R.; Gupta, P.K. Single nucleotide polymorphisms in MCP-1 and its receptor are associated with the risk of age related macular degeneration. PLoS ONE 2012, 7, e49905. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Dang, G.; Yang, S.; Duan, L.; Zhang, Y. CX3CR1 polymorphisms and the risk of age-related macular degeneration. Int. J. Clin. Exp. Pathol. 2015, 8, 9592–9596. [Google Scholar] [PubMed]
- Yu, Y.; Bhangale, T.R.; Fagerness, J.; Ripke, S.; Thorleifsson, G.; Tan, P.L.; Souied, E.H.; Richardson, A.J.; Merriam, J.E.; Buitendijk, G.H.; et al. Common variants near FRK/COL10A1 and VEGFA are associated with advanced age-related macular degeneration. Hum. Mol. Genet. 2011, 20, 3699–3709. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Xu, Y.; Li, X.; Wang, W. Vascular endothelial growth factor A polymorphisms and age-related macular degeneration: A systematic review and meta-analysis. Mol. Vis. 2013, 19, 1211–1221. [Google Scholar] [PubMed]
- Ma, L.; Tang, F.Y.; Chu, W.K.; Young, A.L.; Brelen, M.E.; Pang, C.P.; Chen, L.J. Association of toll-like receptor 3 polymorphism rs3775291 with age-related macular degeneration: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 19718. [Google Scholar] [CrossRef]
- Rutar, M.; Provis, J.M. Role of Chemokines in Shaping Macrophage Activity in AMD. Adv. Exp. Med. Biol. 2016, 854, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.C.; Cui, J.; Gao, L.Q.; Zhang, C.; Dou, H.L.; Chen, D.N.; Wang, Y.X.; Jonas, J.B. Peripheral Monocyte Count and Age-Related Macular Degeneration. The Tongren Health Care Study. Am. J. Ophthalmol. 2021, 227, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Cinici, E.; Caglar, O.; Arslan, M.E.; Dilekmen, N.; Utlu, B.; Mardinoglu, A.; Turkez, H. Targeted Gene Candidates for Treatment and Early Diagnosis of Age-Related Macular Degeneration. Biomed. Res. Int. 2021, 2021, 6620900. [Google Scholar] [CrossRef] [PubMed]
- Daftarian, N.; Zandi, S.; Piryaie, G.; Nikougoftar Zarif, M.; Ranaei Pirmardan, E.; Yamaguchi, M.; Behzadian Nejad, Q.; Hasanpour, H.; Samiei, S.; Pfister, I.B.; et al. Peripheral blood CD163(+) monocytes and soluble CD163 in dry and neovascular age-related macular degeneration. FASEB J. 2020, 34, 8001–8011. [Google Scholar] [CrossRef]
- Behnke, V.; Wolf, A.; Langmann, T. The role of lymphocytes and phagocytes in age-related macular degeneration (AMD). Cell Mol. Life Sci. 2020, 77, 781–788. [Google Scholar] [CrossRef]
- Blasiak, J.; Watala, C.; Tuuminen, R.; Kivinen, N.; Koskela, A.; Uusitalo-Jarvinen, H.; Tuulonen, A.; Winiarczyk, M.; Mackiewicz, J.; Zmorzynski, S.; et al. Expression of VEGFA-regulating miRNAs and mortality in wet AMD. J. Cell Mol. Med. 2019, 23, 8464–8471. [Google Scholar] [CrossRef]
- Cascella, R.; Strafella, C.; Longo, G.; Ragazzo, M.; Manzo, L.; De Felici, C.; Errichiello, V.; Caputo, V.; Viola, F.; Eandi, C.M.; et al. Uncovering genetic and non-genetic biomarkers specific for exudative age-related macular degeneration: Significant association of twelve variants. Oncotarget 2018, 9, 7812–7821. [Google Scholar] [CrossRef] [PubMed]
- Cascella, R.; Strafella, C.; Caputo, V.; Errichiello, V.; Zampatti, S.; Milano, F.; Potenza, S.; Mauriello, S.; Novelli, G.; Ricci, F.; et al. Towards the application of precision medicine in Age-Related Macular Degeneration. Prog. Retin. Eye Res. 2018, 63, 132–146. [Google Scholar] [CrossRef]
- Grassmann, F.; Schoenberger, P.G.; Brandl, C.; Schick, T.; Hasler, D.; Meister, G.; Fleckenstein, M.; Lindner, M.; Helbig, H.; Fauser, S.; et al. A circulating microrna profile is associated with late-stage neovascular age-related macular degeneration. PLoS ONE 2014, 9, e107461. [Google Scholar] [CrossRef] [PubMed]
- Menard, C.; Rezende, F.A.; Miloudi, K.; Wilson, A.; Tetreault, N.; Hardy, P.; SanGiovanni, J.P.; De Guire, V.; Sapieha, P. MicroRNA signatures in vitreous humour and plasma of patients with exudative AMD. Oncotarget 2016, 7, 19171–19184. [Google Scholar] [CrossRef][Green Version]
- Wang, V.M.; Rosen, R.B.; Meyerle, C.B.; Kurup, S.K.; Ardeljan, D.; Agron, E.; Tai, K.; Pomykala, M.; Chew, E.Y.; Chan, C.C.; et al. Suggestive association between PLA2G12A single nucleotide polymorphism rs2285714 and response to anti-vascular endothelial growth factor therapy in patients with exudative age-related macular degeneration. Mol. Vis. 2012, 18, 2578–2585. [Google Scholar]
- Hagstrom, S.A.; Ying, G.S.; Pauer, G.J.T.; Sturgill-Short, G.M.; Huang, J.; Callanan, D.G.; Kim, I.K.; Klein, M.L.; Maguire, M.G.; Martin, D.F. Pharmacogenetics for genes associated with age-related macular degeneration in the Comparison of AMD Treatments Trials (CATT). Ophthalmology 2013, 120, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Noh, D.H.; Sagong, M.; Kim, I.T. Pharmacogenetic association with early response to intravitreal ranibizumab for age-related macular degeneration in a Korean population. Mol. Vis. 2013, 19, 702–709. [Google Scholar]
- Akiyama, M.; Takahashi, A.; Momozawa, Y.; Arakawa, S.; Miya, F.; Tsunoda, T.; Ashikawa, K.; Oshima, Y.; Yasuda, M.; Yoshida, S.; et al. Genome-wide association study suggests four variants influencing outcomes with ranibizumab therapy in exudative age-related macular degeneration. J. Hum. Genet. 2018, 63, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Lores-Motta, L.; de Jong, E.K.; den Hollander, A.I. Exploring the Use of Molecular Biomarkers for Precision Medicine in Age-Related Macular Degeneration. Mol. Diagn. Ther. 2018, 22, 315–343. [Google Scholar] [CrossRef]
- Amoaku, W.M.; Chakravarthy, U.; Gale, R.; Gavin, M.; Ghanchi, F.; Gibson, J.; Harding, S.; Johnston, R.L.; Kelly, S.P.; Lotery, A.; et al. Defining response to anti-VEGF therapies in neovascular AMD. Eye 2015, 29, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lai, T.Y.Y. Baseline Predictors of Visual Acuity Outcome in Patients with Wet Age-Related Macular Degeneration. Biomed. Res. Int. 2018, 2018, 9640131. [Google Scholar] [CrossRef] [PubMed]
- Larrayoz, I.M.; de Luis, A.; Rua, O.; Velilla, S.; Cabello, J.; Martinez, A. Molecular effects of doxycycline treatment on pterygium as revealed by massive transcriptome sequencing. PLoS ONE 2012, 7, e39359. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Pariente, A.; Perez-Sala, A.; Ochoa, R.; Pelaez, R.; Larrayoz, I.M. Genome-Wide Transcriptomic Analysis Identifies Pathways Regulated by Sterculic Acid in Retinal Pigmented Epithelium Cells. Cells 2020, 9, 1187. [Google Scholar] [CrossRef] [PubMed]
- Larrayoz, I.M.; Rua, O.; Velilla, S.; Martinez, A. Transcriptomic profiling explains racial disparities in pterygium patients treated with doxycycline. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7553–7561. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Friedlander, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef] [PubMed]
- Frank, E.; Hall, M.; Trigg, L.; Holmes, G.; Witten, I.H. Data mining in bioinformatics using Weka. Bioinformatics 2004, 20, 2479–2481. [Google Scholar] [CrossRef] [PubMed]
- Bowes Rickman, C.; Farsiu, S.; Toth, C.A.; Klingeborn, M. Dry age-related macular degeneration: Mechanisms, therapeutic targets, and imaging. Investig. Ophthalmol. Vis. Sci. 2013, 54, ORSF68–ORSF80. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Erfurth, U.; Chong, V.; Loewenstein, A.; Larsen, M.; Souied, E.; Schlingemann, R.; Eldem, B.; Mones, J.; Richard, G.; Bandello, F. Guidelines for the management of neovascular age-related macular degeneration by the European Society of Retina Specialists (EURETINA). Br. J. Ophthalmol. 2014, 98, 1144–1167. [Google Scholar] [CrossRef]
- Dilley, J.; Taylor, S. The response to ranibizumab therapy after three injections predicts 12-month outcomes in DME in the absence of rescue laser therapy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2116. [Google Scholar]
- Lux, A.; Llacer, H.; Heussen, F.M.; Joussen, A.M. Non-responders to bevacizumab (Avastin) therapy of choroidal neovascular lesions. Br. J. Ophthalmol. 2007, 91, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Habbu, K.; Ehlers, J.P.; Lansang, M.C.; Hill, L.; Stoilov, I. The Impact of Systemic Factors on Clinical Response to Ranibizumab for Diabetic Macular Edema. Ophthalmology 2016, 123, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Grob, S.; Avery, R.; Kimura, A.; Pieramici, D.; Lee, J.; Rabena, M.; Ortiz, S.; Quach, J.; Cao, G.; et al. Common variant in VEGFA and response to anti-VEGF therapy for neovascular age-related macular degeneration. Curr. Mol. Med. 2013, 13, 929–934. [Google Scholar] [CrossRef]
- Piermarocchi, S.; Miotto, S.; Colavito, D.; Leon, A.; Segato, T. Combined effects of genetic and non-genetic risk factors affect response to ranibizumab in exudative age-related macular degeneration. Acta Ophthalmol. 2015, 93, e451–e457. [Google Scholar] [CrossRef] [PubMed]
- Ying, G.S.; Huang, J.; Maguire, M.G.; Jaffe, G.J.; Grunwald, J.E.; Toth, C.; Daniel, E.; Klein, M.; Pieramici, D.; Wells, J.; et al. Baseline predictors for one-year visual outcomes with ranibizumab or bevacizumab for neovascular age-related macular degeneration. Ophthalmology 2013, 120, 122–129. [Google Scholar] [CrossRef]
- Ying, G.S.; Maguire, M.G. Analyses Comparing Visual Acuity Between Ranibizumab and Bevacizumab in the Comparison of Age-Related Macular Degeneration Treatments Trials. JAMA Ophthalmol. 2015, 133, 726. [Google Scholar] [CrossRef]
- Jaffe, G.J.; Ying, G.S.; Toth, C.A.; Daniel, E.; Grunwald, J.E.; Martin, D.F.; Maguire, M.G. Macular Morphology and Visual Acuity in Year Five of the Comparison of Age-related Macular Degeneration Treatments Trials. Ophthalmology 2019, 126, 252–260. [Google Scholar] [CrossRef]
- Rua, O.; Larrayoz, I.M.; Barajas, M.T.; Velilla, S.; Martinez, A. Oral doxycycline reduces pterygium lesions; results from a double blind, randomized, placebo controlled clinical trial. PLoS ONE 2012, 7, e52696. [Google Scholar] [CrossRef]
- Grunin, M.; Hagbi-Levi, S.; Rinsky, B.; Smith, Y.; Chowers, I. Transcriptome Analysis on Monocytes from Patients with Neovascular Age-Related Macular Degeneration. Sci. Rep. 2016, 6, 29046. [Google Scholar] [CrossRef] [PubMed]
- Litwinska, Z.; Sobus, A.; Luczkowska, K.; Grabowicz, A.; Mozolewska-Piotrowska, K.; Safranow, K.; Kawa, M.P.; Machalinski, B.; Machalinska, A. The Interplay Between Systemic Inflammatory Factors and MicroRNAs in Age-Related Macular Degeneration. Front. Aging Neurosci. 2019, 11, 286. [Google Scholar] [CrossRef]
- Ulanczyk, Z.; Sobus, A.; Luczkowska, K.; Grabowicz, A.; Mozolewska-Piotrowska, K.; Safranow, K.; Kawa, M.P.; Palucha, A.; Krawczyk, M.; Sikora, P.; et al. Associations of microRNAs, Angiogenesis-Regulating Factors and CFH Y402H Polymorphism-An Attempt to Search for Systemic Biomarkers in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2019, 20, 5750. [Google Scholar] [CrossRef]
- McDonel, P.; Guttman, M. Approaches for Understanding the Mechanisms of Long Noncoding RNA Regulation of Gene Expression. Cold Spring Harb Perspect Biol. 2019, 11, a032151. [Google Scholar] [CrossRef]
- Hofmans, M.; Lammens, T.; Depreter, B.; Wu, Y.; Erlacher, M.; Caye, A.; Cave, H.; Flotho, C.; de Haas, V.; Niemeyer, C.M.; et al. Long non-coding RNAs as novel therapeutic targets in juvenile myelomonocytic leukemia. Sci. Rep. 2021, 11, 2801. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xiao, Z.; Zhou, J.; Yang, M.; Feng, S.; Huang, Q.; Zou, J.; Zeng, T.; Li, Y.; Peng, L.; et al. ZNF382: A transcription inhibitor down-regulated in multiple tumors due to promoter methylation. Clin. Chim. Acta 2020, 500, 220–225. [Google Scholar] [CrossRef]
- Mansoor, N.; Wahid, F.; Azam, M.; Shah, K.; den Hollander, A.I.; Qamar, R.; Ayub, H. Molecular Mechanisms of Complement System Proteins and Matrix Metalloproteinases in the Pathogenesis of Age-Related Macular Degeneration. Curr. Mol. Med. 2019, 19, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Schmit, K.; Michiels, C. TMEM Proteins in Cancer: A Review. Front. Pharm. 2018, 9, 1345. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Fujimori-Tonou, N.; Yoshimura, Y.; Kishi, T.; Okamoto, H.; Masai, I. Mutation of DNA primase causes extensive apoptosis of retinal neurons through the activation of DNA damage checkpoInt. and tumor suppressor p53. Development 2008, 135, 1247–1257. [Google Scholar] [CrossRef]
- Farrar, G.J.; Carrigan, M.; Dockery, A.; Millington-Ward, S.; Palfi, A.; Chadderton, N.; Humphries, M.; Kiang, A.S.; Kenna, P.F.; Humphries, P. Toward an elucidation of the molecular genetics of inherited retinal degenerations. Hum. Mol. Genet. 2017, 26, R2–R11. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Jiang, Y.; Liu, R.; Cao, D.; Pan, J.; Luo, Y. Identification of key miRNAs and genes for mouse retinal development using a linear model. Mol. Med. Rep. 2020, 22, 494–506. [Google Scholar] [CrossRef]
- Greenberg, E.; Hajdu, S.; Nemlich, Y.; Cohen, R.; Itzhaki, O.; Jacob-Hirsch, J.; Besser, M.J.; Schachter, J.; Markel, G. Differential regulation of aggressive features in melanoma cells by members of the miR-17-92 complex. Open Biol. 2014, 4, 140030. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Skalsky, R.L.; Corcoran, D.L.; Gottwein, E.; Frank, C.L.; Kang, D.; Hafner, M.; Nusbaum, J.D.; Feederle, R.; Delecluse, H.J.; Luftig, M.A.; et al. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog. 2012, 8, e1002484. [Google Scholar] [CrossRef]
- Wang, M.; Gu, H.; Qian, H.; Zhu, W.; Zhao, C.; Zhang, X.; Tao, Y.; Zhang, L.; Xu, W. miR-17-5p/20a are important markers for gastric cancer and murine double minute 2 participates in their functional regulation. Eur. J. Cancer 2013, 49, 2010–2021. [Google Scholar] [CrossRef]
- Yang, Y.; Mei, Q. miRNA signature identification of retinoblastoma and the correlations between differentially expressed miRNAs during retinoblastoma progression. Mol. Vis. 2015, 21, 1307–1317. [Google Scholar]
- Pin, A.L.; Houle, F.; Guillonneau, M.; Paquet, E.R.; Simard, M.J.; Huot, J. miR-20a represses endothelial cell migration by targeting MKK3 and inhibiting p38 MAP kinase activation in response to VEGF. Angiogenesis 2012, 15, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Du, F.; Tan, Y.L.; Luo, J.; Xiong, D.; Song, W.T. VEGF-mediated angiogenesis in retinopathy of prematurity is co-regulated by miR-17-5p and miR-20a-5p. Biochem. Cell Biol. 2021, 99, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, M.; Munk, M.R.; Kurmann, T.; De Zanet, S.; Mosinska, A.; Karagoz, I.K.; Zinkernagel, M.S.; Wolf, S.; Sznitman, R. Machine Learning Can Predict Anti-VEGF Treatment Demand in a Treat-and-Extend Regimen for Patients with Neovascular AMD, DME, and RVO Associated Macular Edema. Ophthalmol. Retin. 2021, 5, 604–624. [Google Scholar] [CrossRef]
- Diack, C.; Schwab, D.; Cosson, V.; Buchheit, V.; Mazer, N.; Frey, N. A Baseline Score to Predict Response to Ranibizumab Treatment in Neovascular Age-Related Macular Degeneration. Transl. Vis. Sci. Technol. 2021, 10, 11. [Google Scholar] [CrossRef]
- Sharma, S.; Baweja, S.; Maras, J.S.; Shasthry, S.M.; Moreau, R.; Sarin, S.K. Differential blood transcriptome modules predict response to corticosteroid therapy in alcoholic hepatitis. JHEP Rep. 2021, 3, 100283. [Google Scholar] [CrossRef]
- Gadd, V.L.; Patel, P.J.; Jose, S.; Horsfall, L.; Powell, E.E.; Irvine, K.M. Altered Peripheral Blood Monocyte Phenotype and Function in Chronic Liver Disease: Implications for Hepatic Recruitment and Systemic Inflammation. PLoS ONE 2016, 11, e0157771. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Rouphael, N.; Duraisingham, S.; Romero-Steiner, S.; Presnell, S.; Davis, C.; Schmidt, D.S.; Johnson, S.E.; Milton, A.; Rajam, G.; et al. Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat. Immunol. 2014, 15, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Oosting, M.; Deelen, P.; Ricano-Ponce, I.; Smeekens, S.; Jaeger, M.; Matzaraki, V.; Swertz, M.A.; Xavier, R.J.; Franke, L.; et al. Inter-individual variability and genetic influences on cytokine responses to bacteria and fungi. Nat. Med. 2016, 22, 952–960. [Google Scholar] [CrossRef]
- Amoaku, W.; Balaskas, K.; Cudrnak, T.; Downey, L.; Groppe, M.; Mahmood, S.; Mehta, H.; Mohamed, Q.; Mushtaq, B.; Severn, P.; et al. Initiation and maintenance of a Treat-and-Extend regimen for ranibizumab therapy in wet age-related macular degeneration: Recommendations from the UK Retinal Outcomes Group. Clin. Ophthalmol. 2018, 12, 1731–1740. [Google Scholar] [CrossRef]
- Khanna, S.; Komati, R.; Eichenbaum, D.A.; Hariprasad, I.; Ciulla, T.A.; Hariprasad, S.M. Current and upcoming anti-VEGF therapies and dosing strategies for the treatment of neovascular AMD: A comparative review. BMJ Open Ophthalmol. 2019, 4, e000398. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| All | Poor Responders | Good Responders | p-Value | ||
|---|---|---|---|---|---|
| Age (years, mean ± SD) | 78.03 ± 1.055 | 80.04 ± 1.379 | 76.45 ± 1.504 | 0.092 | |
| Sex (Male/Female) | 24/35 | 8/18 | 16/17 | 0.193 | |
| Tobacco (yes/no) | 23/36 | 8/18 | 15/18 | 0.292 | |
| ETDRS (letters) | 63.61 ± 1.131 | 62.73 ± 1.791 | 64.30 ± 1.464 | 0.495 | |
| OCT | Central Retinal Thickness, mean±SD (µm) | 329.18 ± 15.495 | 325.2 ± 28.09 | 332.4 ± 17.7 | 0.820 |
| Macular cube volume (µm3) | 10.333 ± 0.176 | 10.37 ± 0.3137 | 10.30 ± 0.2004 | 0.839 | |
| Retinal pigment epithelial detachment (yes/no) | 43/16 | 20/6 | 23/10 | 0.571 | |
| Intra-retinal fluid (yes/no) | 48/11 | 22/4 | 26/7 | 0.740 | |
| Sub-retinal fluid (yes/no) | 43/16 | 17/9 | 26/7 | 0.377 | |
| Intra-retinal cysts (yes/no) | 35/24 | 16/10 | 19/14 | 0.795 | |
| Fundoscopy | Hemorrhage (yes/no) | 28/31 | 15/11 | 13/20 | 0.196 |
| Exudation (yes/no) | 8/51 | 2/24 | 6/27 | 0.446 | |
| Atrophy (yes/no) | 13/46 | 10/16 | 3/30 | 0.011 | |
| Fibrosis (yes/no) | 4/55 | 2/24 | 2/31 | 1.000 | |
| AGF | Location (Sub-/Yuxta-/Extra-Foveal) | 16/35/5 | 9/16/0 | 7/19/5 | 0.085 |
| Size (mm) | 1.03 ± 0.130 | 1.193 ± 0.2181 | 0.9150 ± 0.1585 | 0.297 | |
| Pattern (O/PC/MC) | 26/22/8 | 14/9/3 | 12/13/5 | 0.577 |
| Ensembl_ID | BaseMean | Log2FC | p Value | Symbol | Name |
|---|---|---|---|---|---|
| ENSG00000273727 | 3.8 | −1.65 | 4.10 × 10−5 | U1 | U1 spliceosomal RNA |
| ENSG00000235621 | 9.8 | 0.82 | 1.16 × 10−4 | LINC00494 | long intergenic non-protein coding RNA 494 |
| ENSG00000158106 | 25.0 | 0.54 | 1.67 × 10−4 | RHPN1 | rhophilin Rho GTPase binding protein 1 |
| ENSG00000215012 | 75.3 | 0.25 | 2.13 × 10−4 | RTL10 | retrotransposon Gag like 10 |
| ENSG00000249572 | 1.8 | −1.31 | 2.41 × 10−4 | N/A | novel transcript |
| ENSG00000233913 | 23.6 | −1.95 | 3.18 × 10−4 | RPL10P9 | ribosomal protein L10 pseudogene 9 |
| ENSG00000260766 | 12.5 | 0.49 | 3.98 × 10−4 | N/A | N/A |
| ENSG00000160307 | 18.5 | −1.57 | 4.40 × 10−4 | S100B | S100 calcium binding protein B |
| ENSG00000226581 | 1.1 | 1.62 | 5.17 × 10−4 | LINC02848 | long intergenic non-protein coding RNA 2848 |
| ENSG00000204345 | 1.3 | −1.40 | 5.83 × 10−4 | CD300LD | CD300 molecule like family member d |
| ENSG00000174473 | 1.7 | 1.56 | 7.12 × 10−4 | GALNTL6 | polypeptide N-acetylgalactosaminyltransferase like 6 |
| ENSG00000211789 | 14.2 | 0.77 | 7.23 × 10−4 | TRAV12-2 | T cell receptor alpha variable 12-2 |
| ENSG00000205710 | 11.6 | −0.80 | 7.28 × 10−4 | C17orf107 | chromosome 17 open reading frame 107 |
| ENSG00000227309 | 3.3 | −0.95 | 7.79 × 10−4 | RPL31P19 | ribosomal protein L31 (RPL31) pseudogene |
| ENSG00000154723 | 43.8 | −0.26 | 7.79 × 10−4 | ATP5PF | ATP synthase peripheral stalk subunit F6 |
| ENSG00000107968 | 221.6 | −0.29 | 7.81 × 10−4 | MAP3K8 | mitogen-activated protein kinase kinase kinase 8 |
| ENSG00000211880 | 6.4 | 0.70 | 7.90 × 10−4 | TRAJ9 | T cell receptor alpha joining 9 |
| ENSG00000258511 | 2.1 | 1.13 | 7.95 × 10−4 | LINC02295 | long intergenic non-protein coding RNA 2295 |
| ENSG00000235361 | 0.8 | −1.67 | 9.34 × 10−4 | N/A | novel transcript, antisense to ABR |
| ENSG00000226430 | 1.7 | 1.29 | 1.03 × 10−3 | USP17L7 | ubiquitin specific peptidase 17 like family member 7 |
| ENSG00000197353 | 6.9 | −0.94 | 1.04 × 10−3 | LYPD2 | LY6/PLAUR domain containing 2 |
| ENSG00000180758 | 18.7 | 0.39 | 1.06 × 10−3 | GPR157 | G protein-coupled receptor 157 |
| ENSG00000211575 | 0.9 | −1.60 | 1.19 × 10−3 | MIR760 | microRNA 760 |
| ENSG00000272666 | 12.3 | −0.75 | 1.26 × 10−3 | KLHDC7B-DT | KLHDC7B divergent transcript |
| ENSG00000156510 | 15.3 | 0.55 | 1.29 × 10−3 | HKDC1 | hexokinase domain containing 1 |
| ENSG00000007038 | 5.5 | 0.88 | 1.32 × 10−3 | PRSS21 | serine protease 21 |
| ENSG00000254088 | 5.4 | 0.61 | 1.33 × 10−3 | SLC2A3P4 | solute carrier family 2 member 3 pseudogene 4 |
| ENSG00000281106 | 23.3 | 0.71 | 1.58 × 10−3 | TMEM272 | transmembrane protein 272 |
| ENSG00000180822 | 67.2 | 0.28 | 1.58 × 10−3 | PSMG4 | proteasome assembly chaperone 4 |
| ENSG00000236709 | 7.6 | −1.10 | 1.61 × 10−3 | DAPK1-IT1 | DAPK1 intronic transcript 1 |
| miRNA | BaseMean | Log2FC | p Value |
|---|---|---|---|
| hsa-miR-3614-5p | 11.3 | 3.49 | 1.42 × 10−3 |
| hsa-miR-423-5p | 1779.1 | 10.80 | 2.37 × 10−3 |
| hsa-miR-20a-5p | 865.9 | 9.76 | 3.16 × 10−3 |
| hsa-miR-30c-1-3p | 24.6 | 4.62 | 5.19 × 10−3 |
| hsa-miR-1249-5p | 3.0 | 1.58 | 5.32 × 10−3 |
| hsa-miR-3605-3p | 7.5 | 2.91 | 6.96 × 10−3 |
| hsa-miR-320b | 22.2 | 4.48 | 7.55 × 10−3 |
| hsa-miR-320b-2 | 22.7 | 4.51 | 8.09 × 10−3 |
| hsa-miR-25-5p | 11.8 | 3.56 | 9.93 × 10−3 |
| hsa-miR-34c-5p | 4.7 | 2.22 | 1.36 × 10−2 |
| hsa-miR-6813-5p | 2.8 | 1.48 | 1.67 × 10−2 |
| hsa-miR-30a-5p | 225.9 | 7.82 | 1.73 × 10−2 |
| hsa-miR-132-3p | 25.0 | 4.64 | 1.76 × 10−2 |
| hsa-miR-642a-5p | 4.3 | 2.10 | 1.77 × 10−2 |
| hsa-miR-212-3p | 6.9 | 2.78 | 1.97 × 10−2 |
| hsa-miR-181b-5p | 1721.2 | 10.75 | 1.98 × 10−2 |
| hsa-miR-3127-5p | 2.8 | 1.47 | 2.00 × 10−2 |
| hsa-miR-1273h-5p | 6.4 | 2.68 | 2.11 × 10−2 |
| hsa-miR-181b-5p-2 | 1795.9 | 10.81 | 2.19 × 10−2 |
| hsa-miR-652-5p | 24.7 | 4.63 | 2.51 × 10−2 |
| hsa-let-7i-5p | 5009.0 | 12.29 | 2.51 × 10−2 |
| hsa-miR-369-3p | 20.0 | 4.32 | 2.59 × 10−2 |
| hsa-miR-6511a-3p | 4.7 | 2.24 | 2.62 × 10−2 |
| hsa-miR-6511a-3p-2 | 4.7 | 2.24 | 2.62 × 10−2 |
| hsa-miR-6511a-3p-3 | 4.7 | 2.24 | 2.62 × 10−2 |
| hsa-miR-6511a-3p-4 | 4.7 | 2.24 | 2.62 × 10−2 |
| hsa-miR-223-5p | 93.8 | 6.55 | 2.69 × 10−2 |
| hsa-miR-2110 | 14.5 | 3.85 | 2.84 × 10−2 |
| hsa-miR-190a-5p | 32.2 | 5.01 | 2.91 × 10−2 |
| hsa-miR-487a-3p | 9.7 | 3.28 | 3.27 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oca, A.I.; Pérez-Sala, Á.; Pariente, A.; Ochoa, R.; Velilla, S.; Peláez, R.; Larráyoz, I.M. Predictive Biomarkers of Age-Related Macular Degeneration Response to Anti-VEGF Treatment. J. Pers. Med. 2021, 11, 1329. https://doi.org/10.3390/jpm11121329
Oca AI, Pérez-Sala Á, Pariente A, Ochoa R, Velilla S, Peláez R, Larráyoz IM. Predictive Biomarkers of Age-Related Macular Degeneration Response to Anti-VEGF Treatment. Journal of Personalized Medicine. 2021; 11(12):1329. https://doi.org/10.3390/jpm11121329
Chicago/Turabian StyleOca, Ana I., Álvaro Pérez-Sala, Ana Pariente, Rodrigo Ochoa, Sara Velilla, Rafael Peláez, and Ignacio M. Larráyoz. 2021. "Predictive Biomarkers of Age-Related Macular Degeneration Response to Anti-VEGF Treatment" Journal of Personalized Medicine 11, no. 12: 1329. https://doi.org/10.3390/jpm11121329
APA StyleOca, A. I., Pérez-Sala, Á., Pariente, A., Ochoa, R., Velilla, S., Peláez, R., & Larráyoz, I. M. (2021). Predictive Biomarkers of Age-Related Macular Degeneration Response to Anti-VEGF Treatment. Journal of Personalized Medicine, 11(12), 1329. https://doi.org/10.3390/jpm11121329