
Tumor Inflammation, Obesity, and Proliferative Status as Biomarkers in Gastroesophageal Adenocarcinoma


 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Clinical Data
2.2. Quality Assessment of Clinical FFPE Tissue Specimens
2.3. Immunohistochemical Studies
2.4. Nucleic Acid Isolation and Gene Expression
2.5. Data Analyses
3. Results
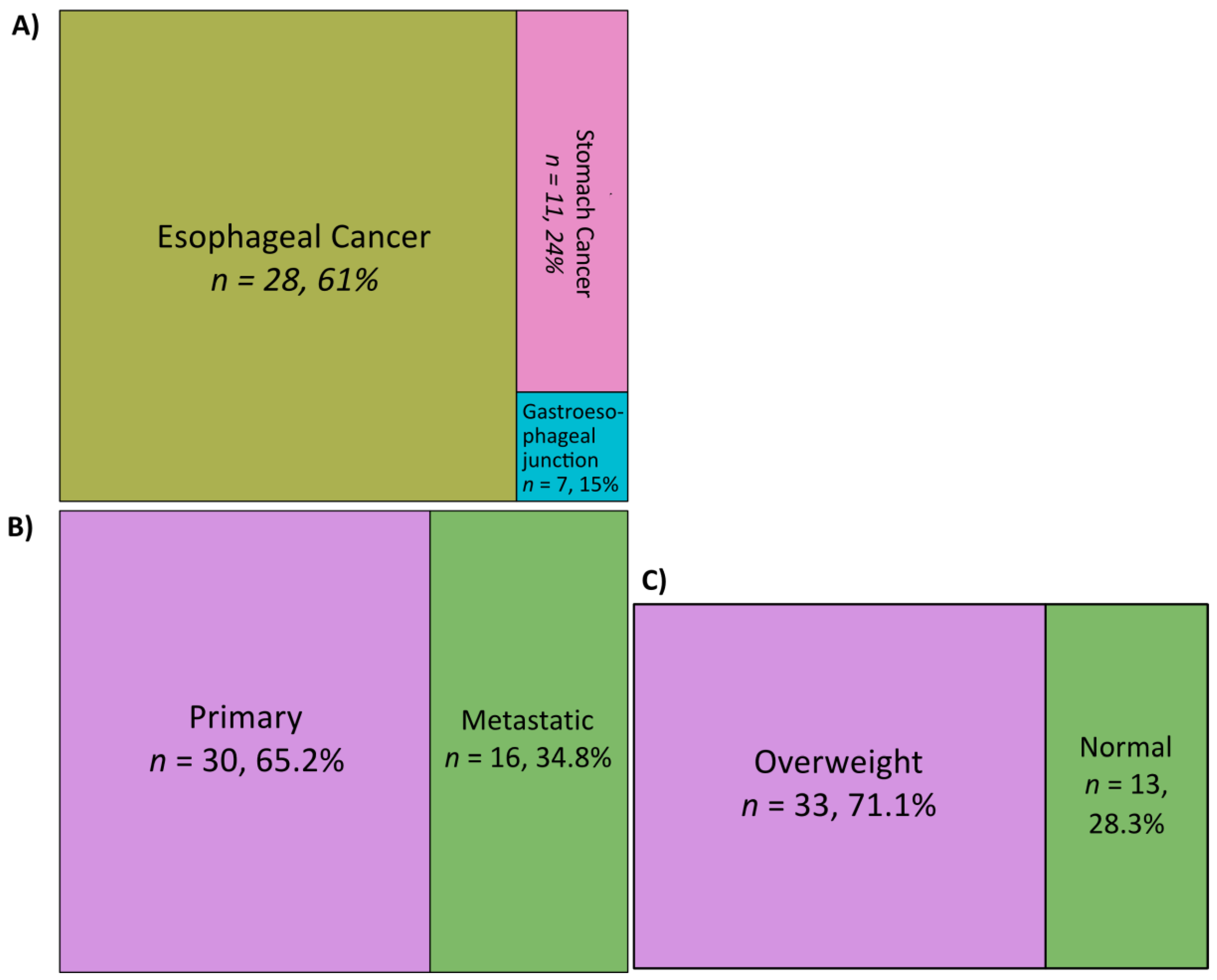
3.1. Patient Cohort
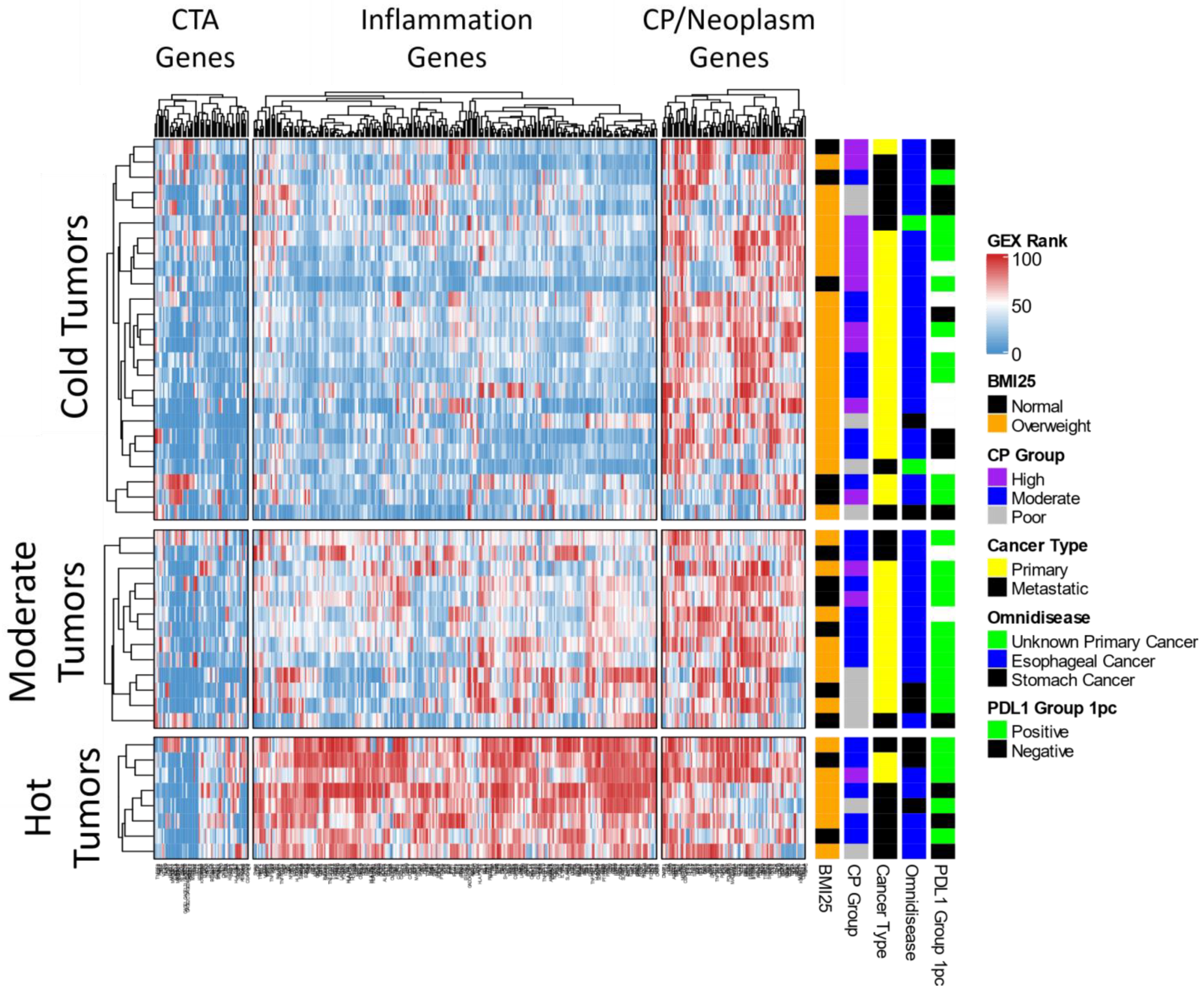
3.2. Unsupervised Gene Expression Analysis
3.3. BMI Status and Biomarkers
3.4. Overall Survival, BMI and Other Biomarkers
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Demeester, S.R. Epidemiology and biology of esophageal cancer. Gastrointest. Cancer Res. 2009, 3, S2–S5. [Google Scholar]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Bang, Y.-J.; Piha-Paul, S.A.; Razak, A.R.A.; Bennouna, J.; Soria, J.-C.; Rugo, H.S.; Cohen, R.B.; O’Neil, B.H.; Mehnert, J.M.; et al. T-Cell-Inflamed Gene-Expression Profile, Programmed Death Ligand 1 Expression, and Tumor Mutational Burden Predict Efficacy in Patients Treated with Pembrolizumab Across 20 Cancers: KEYNOTE-028. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 318–327. [Google Scholar] [CrossRef]
- Korkaya, H.; Kim, G.I.L.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 Inflammatory Loop Mediates Trastuzumab Resistance in HER2+ Breast Cancer by Expanding the Cancer Stem Cell Population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Arkenau, H.-T.; Calvo, E.; Bendell, J.C.; Penel, N.; Fuchs, C.S.; McNeely, S.; Rasmussen, E.R.; Wang, H.; Oliveira, J.M.; et al. Immune profiling and clinical outcomes in patients treated with ramucirumab and pembrolizumab in phase I study JVDF. J. Clin. Oncol. 2020, 38, 3089. [Google Scholar] [CrossRef]
- Calle, E.E.; Thun, M.J. Obesity and cancer. Oncogene 2004, 23, 6365–6378. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Aguilar, E.G.; Luna, J.I.; Dunai, C.; Khuat, L.T.; Le, C.T.; Mirsoian, A.; Minnar, C.M.; Stoffel, K.M.; Sturgill, I.R.; et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat. Med. 2019, 25, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, E.G.; Murphy, W.J. Obesity induced T cell dysfunction and implications for cancer immunotherapy. Curr. Opin. Immunol. 2018, 51, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Ibrahimi, S.; Zhang, Y.; Wang, J.; Kalinski, P. Impact of obesity on immunity in gastroesophageal adenocarcinoma [GEAC]. J. Immunother. Cancer 2019, 7 (Suppl. 1), 282. [Google Scholar] [CrossRef] [Green Version]
- Pabla, S.; Seager, R.J.; Van Roey, E.; Gao, S.; Hoefer, C.; Nesline, M.K.; DePietro, P.; Burgher, B.; Andreas, J.; Giamo, V.; et al. Integration of tumor inflammation, cell proliferation, and traditional biomarkers improves prediction of immunotherapy resistance and response. Biomark. Res. 2021, 9, 56. [Google Scholar] [CrossRef]
- Pabla, S.; Conroy, J.M.; Nesline, M.K.; Glenn, S.T.; Papanicolau-Sengos, A.; Burgher, B.; Hagen, J.; Giamo, V.; Andreas, J.; Lenzo, F.L.; et al. Proliferative potential and resistance to immune checkpoint blockade in lung cancer patients. J. Immunother. Cancer 2019, 7, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, N.; Gosain, R.; Lemini, R.; Wang, C.; Gabriel, E.; Mohammed, T.; Siromoni, B.; Mukherjee, S. Socio-Demographic Disparities in Gastric Adenocarcinoma: A Population-Based Study. Cancers 2020, 12, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallis, C.J.D.; Butaney, M.; Satkunasivam, R.; Freedland, S.J.; Patel, S.P.; Hamid, O.; Pal, S.K.; Klaassen, Z. Association of Patient Sex with Efficacy of Immune Checkpoint Inhibitors and Overall Survival in Advanced Cancers: A Systematic Review and Meta-analysis. JAMA Oncol. 2019, 5, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-J.; Du, Y.; Zhao, X.; Ma, L.-Y.; Cao, G.-W. Inflammation-related factors predicting prognosis of gastric cancer. World J. Gastroenterol. 2014, 20, 4586–4596. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.-P.; et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients with Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef] [PubMed]
- Barbi, J.; Patnaik, S.K.; Pabla, S.; Zollo, R.; Smith, R.J.J.; Sass, S.N.; Srinivasan, A.; Petrucci, C.; Seager, R.; Conroy, J.; et al. Visceral Obesity Promotes Lung Cancer Progression-Toward Resolution of the Obesity Paradox in Lung Cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2021, 16, 1333–1348. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Conroy, J.M.; Pabla, S.; Glenn, S.T.; Burgher, B.; Nesline, M.; Papanicolau-Sengos, A.; Andreas, J.; Giamo, V.; Lenzo, F.L.; Hyland, F.C.L.; et al. Analytical Validation of a Next-Generation Sequencing Assay to Monitor Immune Responses in Solid Tumors. J. Mol. Diagn. 2018, 20, 95–109. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Pabla, S.; Lenzo, F.L.; Conroy, J.M.; Nesline, M.K.; Glenn, S.T.; Papanicolau-Sengos, A.; Burgher, B.; Giamo, V.; Andreas, J.; et al. Proliferative potential and response to nivolumab in clear cell renal cell carcinoma patients. Oncoimmunology 2020, 9, 1773200. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Variable | Category | Median/Freq |
|---|---|---|
| Dx Age | Med (SD) | 64 (10.8) years |
| Sex | Female N (%) | 5 (10.9%) |
| Male N (%) | 41 (89.1%) | |
| Inflammation status | Cold N (%) | 25 (54.3%) |
| Moderate N (%) | 13 (38.3%) | |
| Hot N (%) | 8 (17.4%) | |
| BMI group | Normal N (%) | 13 (28.3%) |
| Overweight N (%) | 33 (71.7%) | |
| Cell Proliferation | High N (%) | 14 (30.4%) |
| Moderate N (%) | 21 (45.7%) | |
| Poor N (%) | 11 (23.9%) | |
| Immune-modulating drugs | Yes N (%) | 23 (50%) |
| No N (%) | 23 (50%) |
| OS (Dx Date to Last Follow-Up/Death) n = 35 | ||||
|---|---|---|---|---|
| Model | Comparison | Hazard Ratio | 95% CI | p Value |
| Inflammation Status | Cold vs. Hot | 24.50 | 2.40–249.37 | <0.01 |
| Moderate vs. Hot | 11.41 | 1.11–117.37 | 0.041 | |
| Cold vs. Moderate | 2.146 | 0.44–10.58 | 0.348 | |
| BMI | Overweight vs. Normal | 0.73 | 0.19–2.88 | 0.653 |
| Cell Proliferation | High vs. Poor | 1.18 | 0.16–8.58 | 0.871 |
| Moderate vs. Poor | 1.32 | 0.23–7.44 | 0.754 | |
| Moderate vs. High | 1.12 | 0.21–5.86 | 0.894 | |
| Inflammation Status + BMI + Cell Proliferation | Cold vs. Hot | 49.16 | 3.67–658.10 | <0.01 |
| Moderate vs. Hot | 24.50 | 1.61–373.30 | 0.02 | |
| Cold vs. Moderate | 2.01 | 0.35–11.40 | 0.432 | |
| Overweight vs Normal (BMI_Group) | 1.72 | 0.34–8.60 | 0.511 | |
| High vs. Poor | 0.94 | 0.09–9.58 | 0.960 | |
| Moderate vs. Poor | 2.40 | 0.33–17.42 | 0.386 | |
| Moderate vs. High | 2.54 | 0.40–16.30 | 0.324 | |
| OS (n = 23) | PFS (n = 23) | ||||||
|---|---|---|---|---|---|---|---|
| Model | Comparison | Hazard Ratio | 95% CI | p Value | Hazard Ratio | 95% CI | p Value |
| Inflammation Status | Cold vs. Hot | 14,003,350.00 | NA | 0.994 | 4.32 | 0.53–35.48 | 0.174 |
| Moderate vs. Hot | 11,292,320.00 | NA | 0.936 | 4.31 | 0.49–38.14 | 0.190 | |
| Cold vs. Moderate | 1.24 | 0.36–4.29 | 0.734 | 1 | 0.31–3.27 | 0.997 | |
| Inflammation Comparison | BMI | Cell Proliferation | Hazard Ratio | CI (Lower) | CI (Upper) |
|---|---|---|---|---|---|
| Cold vs. Hot | Overweight | Moderate | 31.129 | 2.117 | 457.83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukherjee, S.; Seager, R.J.; Lee, Y.H.; Conroy, J.M.; Kalinski, P.; Pabla, S. Tumor Inflammation, Obesity, and Proliferative Status as Biomarkers in Gastroesophageal Adenocarcinoma. J. Pers. Med. 2021, 11, 1324. https://doi.org/10.3390/jpm11121324
Mukherjee S, Seager RJ, Lee YH, Conroy JM, Kalinski P, Pabla S. Tumor Inflammation, Obesity, and Proliferative Status as Biomarkers in Gastroesophageal Adenocarcinoma. Journal of Personalized Medicine. 2021; 11(12):1324. https://doi.org/10.3390/jpm11121324
Chicago/Turabian StyleMukherjee, Sarbajit, R. J. Seager, Yong Hee Lee, Jeffrey M. Conroy, Pawel Kalinski, and Sarabjot Pabla. 2021. "Tumor Inflammation, Obesity, and Proliferative Status as Biomarkers in Gastroesophageal Adenocarcinoma" Journal of Personalized Medicine 11, no. 12: 1324. https://doi.org/10.3390/jpm11121324
APA StyleMukherjee, S., Seager, R. J., Lee, Y. H., Conroy, J. M., Kalinski, P., & Pabla, S. (2021). Tumor Inflammation, Obesity, and Proliferative Status as Biomarkers in Gastroesophageal Adenocarcinoma. Journal of Personalized Medicine, 11(12), 1324. https://doi.org/10.3390/jpm11121324