
Precision Medicine in Phaeochromocytoma and Paraganglioma


Abstract
:
1. Introduction
2. Genomic Landscape of PPGL
3. Mechanisms of Tumourigenesis in PPGL and Molecular Clusters
4. Clinical Phenotype According to Molecular Clusters
5. Biochemistry
6. Immunohistochemistry
7. The Influence of Genotype on Molecular Imaging Modality Selection in PPGL
7.1. Radiotracers Targeting Ligands Involved in Catecholamine Synthesis and Storage
7.2. Tracers Targeting Glucose Metabolism
7.3. Tracers Targeting Somatostatin Receptors
8. In Vivo Detection of Oncometabolites in PPGL
9. Precision Medicine for the Treatment of PPGL
9.1. Surgery
9.2. Radionuclide Therapies
9.3. Cytotoxic Chemotherapy
9.4. Alkylating Agents
9.5. Tyrosine Kinase Inhibitors
10. Emerging Targeted Therapies for PPGL
10.1. Cluster 1 PPGL
10.1.1. PARP Inhibitors
10.1.2. Immunotherapy
10.1.3. Demethylating Agents
10.1.4. HIF2α Antagonists
10.1.5. Targeting Metabolic Reprogramming and Redox Imbalance
10.2. Cluster 2 Mutated PPGL
New Kinase Inhibitors
11. Future Considerations for Precision Medicine in PPGL
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sutton, M.G.; Sheps, S.G.; Lie, J.T. Prevalence of clinically unsuspected pheochromocytoma. Review of a 50-year autopsy series. Mayo Clin. Proc. 1981, 56, 354–360. [Google Scholar] [CrossRef]
- Beard, C.M.; Sheps, S.G.; Kurland, L.T.; Carney, J.A.; Lie, J.T. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin. Proc. 1983, 58, 802–804. [Google Scholar] [PubMed]
- Baguet, J.P.; Hammer, L.; Mazzuco, T.L.; Chabre, O.; Mallion, J.M.; Sturm, N.; Chaffanjon, P. Circumstances of discovery of phaeochromocytoma: A retrospective study of 41 consecutive patients. Eur. J. Endocrinol. 2004, 150, 681–686. [Google Scholar] [CrossRef] [Green Version]
- Geroula, A.; Deutschbein, T.; Langton, K.; Masjkur, J.; Pamporaki, C.; Peitzsch, M.; Fliedner, S.; Timmers, H.; Bornstein, S.R.; Beuschlein, F.; et al. Pheochromocytoma and paraganglioma: Clinical feature-based disease probability in relation to catecholamine biochemistry and reason for disease suspicion. Eur. J. Endocrinol. 2019, 181, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Kakoki, K.; Miyata, Y.; Shida, Y.; Hakariya, T.; Takehara, K.; Izumida, S.; Sekino, M.; Kinoshita, N.; Igawa, T.; Fukuoka, J.; et al. Pheochromocytoma multisystem crisis treated with emergency surgery: A case report and literature review. BMC Res. Notes 2015, 8, 758. [Google Scholar] [CrossRef] [Green Version]
- Lam, A.K. Update on Adrenal Tumours in 2017 World Health Organization (WHO) of Endocrine Tumours. Endocr. Pathol. 2017, 28, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Amar, L.; Servais, A.; Gimenez-Roqueplo, A.P.; Zinzindohoue, F.; Chatellier, G.; Plouin, P.F. Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. J. Clin. Endocrinol. Metab. 2005, 90, 2110–2116. [Google Scholar] [CrossRef]
- Elder, E.E.; Elder, G.; Larsson, C. Pheochromocytoma and functional paraganglioma syndrome: No longer the 10% tumor. J. Surg. Oncol. 2005, 89, 193–201. [Google Scholar] [CrossRef]
- Roman-Gonzalez, A.; Jimenez, C. Malignant pheochromocytoma-paraganglioma: Pathogenesis, TNM staging, and current clinical trials. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 174–183. [Google Scholar] [CrossRef]
- Crona, J.; Taieb, D.; Pacak, K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocr. Rev. 2017, 38, 489–515. [Google Scholar] [CrossRef]
- Favier, J.; Amar, L.; Gimenez-Roqueplo, A.P. Paraganglioma and phaeochromocytoma: From genetics to personalized medicine. Nat. Rev. Endocrinol. 2015, 11, 101–111. [Google Scholar] [CrossRef]
- Nolting, S.; Bechmann, N.; Taieb, D.; Beuschlein, F.; Fassnacht, M.; Kroiss, M.; Eisenhofer, G.; Grossman, A.; Pacak, K. Personalized management of pheochromocytoma and paraganglioma. Endocr. Rev. 2021, 20, 1–41. [Google Scholar] [CrossRef]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buffet, A.; Ben Aim, L.; Leboulleux, S.; Drui, D.; Vezzosi, D.; Libe, R.; Ajzenberg, C.; Bernardeschi, D.; Cariou, B.; Chabolle, F.; et al. Positive Impact of Genetic Test on the Management and Outcome of Patients with Paraganglioma and/or Pheochromocytoma. J. Clin. Endocrinol. Metab. 2019, 104, 1109–1118. [Google Scholar] [CrossRef]
- Plouin, P.F.; Amar, L.; Dekkers, O.M.; Fassnacht, M.; Gimenez-Roqueplo, A.P.; Lenders, J.W.; Lussey-Lepoutre, C.; Steichen, O. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur. J. Endocrinol. 2016, 174, G1–G10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenders, J.W.M.; Kerstens, M.N.; Amar, L.; Prejbisz, A.; Robledo, M.; Taieb, D.; Pacak, K.; Crona, J.; Zelinka, T.; Mannelli, M.; et al. Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: A position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J. Hypertens 2020, 38, 1443–1456. [Google Scholar] [CrossRef]
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.P.; Grebe, S.K.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef] [PubMed]
- Buffet, A.; Burnichon, N.; Favier, J.; Gimenez-Roqueplo, A.P. An overview of 20 years of genetic studies in pheochromocytoma and paraganglioma. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101416. [Google Scholar] [CrossRef] [PubMed]
- Grubbs, E.G.; Halperin, D.M.; Waguespack, S.G.; Gagel, R.F. History of the multiple endocrine neoplasia workshops and overview of MEN2019. Endocr. Relat. Cancer 2020, 27, 1–5. [Google Scholar]
- Schlisio, S.; Kenchappa, R.S.; Vredeveld, L.C.; George, R.E.; Stewart, R.; Greulich, H.; Shahriari, K.; Nguyen, N.V.; Pigny, P.; Dahia, P.L.; et al. The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev. 2008, 22, 884–893. [Google Scholar] [CrossRef] [Green Version]
- Welander, J.; Andreasson, A.; Juhlin, C.C.; Wiseman, R.W.; Backdahl, M.; Hoog, A.; Larsson, C.; Gimm, O.; Soderkvist, P. Rare germline mutations identified by targeted next-generation sequencing of susceptibility genes in pheochromocytoma and paraganglioma. J. Clin. Endocrinol. Metab. 2014, 99, E1352–E1360. [Google Scholar] [CrossRef] [PubMed]
- Currás-Freixes, M.; Piñeiro-Yañez, E.; Montero-Conde, C.; Apellániz-Ruiz, M.; Calsina, B.; Mancikova, V.; Remacha, L.; Richter, S.; Ercolino, T.; Rogowski-Lehmann, N.; et al. PheoSeq: A Targeted Next-Generation Sequencing Assay for Pheochromocytoma and Paraganglioma Diagnostics. J. Mol. Diagn. 2017, 19, 575–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Aim, L.; Pigny, P.; Castro-Vega, L.J.; Buffet, A.; Amar, L.; Bertherat, J.; Drui, D.; Guilhem, I.; Baudin, E.; Lussey-Lepoutre, C.; et al. Targeted next-generation sequencing detects rare genetic events in pheochromocytoma and paraganglioma. J. Med. Genet. 2019, 56, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Burnichon, N.; Vescovo, L.; Amar, L.; Libe, R.; de Reynies, A.; Venisse, A.; Jouanno, E.; Laurendeau, I.; Parfait, B.; Bertherat, J.; et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum. Mol. Genet. 2011, 20, 3974–3985. [Google Scholar] [CrossRef]
- Nolting, S.; Ullrich, M.; Pietzsch, J.; Ziegler, C.G.; Eisenhofer, G.; Grossman, A.; Pacak, K. Current Management of Pheochromocytoma/Paraganglioma: A Guide for the Practicing Clinician in the Era of Precision Medicine. Cancers 2019, 11, 1505. [Google Scholar] [CrossRef] [Green Version]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Eijkelenkamp, K.; Osinga, T.E.; Links, T.P.; van der Horst-Schrivers, A.N.A. Clinical implications of the oncometabolite succinate in SDHx-mutation carriers. Clin. Genet. 2020, 97, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Jochmanova, I.; Yang, C.; Zhuang, Z.; Pacak, K. Hypoxia-inducible factor signaling in pheochromocytoma: Turning the rudder in the right direction. J. Natl. Cancer Inst. 2013, 105, 1270–1283. [Google Scholar] [CrossRef]
- Favier, J.; Plouin, P.F.; Corvol, P.; Gasc, J.M. Angiogenesis and vascular architecture in pheochromocytomas: Distinctive traits in malignant tumors. Am. J. Pathol. 2002, 161, 1235–1246. [Google Scholar] [CrossRef]
- Schovanek, J.; Martucci, V.; Wesley, R.; Fojo, T.; del Rivero, J.; Huynh, T.; Adams, K.; Kebebew, E.; Frysak, Z.; Stratakis, C.A.; et al. The size of the primary tumor and age at initial diagnosis are independent predictors of the metastatic behavior and survival of patients with SDHB-related pheochromocytoma and paraganglioma: A retrospective cohort study. BMC Cancer 2014, 14, 523. [Google Scholar] [CrossRef] [Green Version]
- Crona, J.; Lamarca, A.; Ghosal, S.; Welin, S.; Skogseid, B.; Pacak, K. Genotype-phenotype correlations in pheochromocytoma and paraganglioma: A systematic review and individual patient meta-analysis. Endocr. Relat. Cancer 2019, 26, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Burnichon, N.; Rohmer, V.; Amar, L.; Herman, P.; Leboulleux, S.; Darrouzet, V.; Niccoli, P.; Gaillard, D.; Chabrier, G.; Chabolle, F.; et al. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J. Clin. Endocrinol. Metab. 2009, 94, 2817–2827. [Google Scholar] [CrossRef] [PubMed]
- Hescot, S.; Curras-Freixes, M.; Deutschbein, T.; van Berkel, A.; Vezzosi, D.; Amar, L.; de la Fouchardiere, C.; Valdes, N.; Riccardi, F.; Do Cao, C.; et al. Prognosis of Malignant Pheochromocytoma and Paraganglioma (MAPP-Prono Study): A European Network for the Study of Adrenal Tumors Retrospective Study. J. Clin. Endocrinol. Metab. 2019, 104, 2367–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bechmann, N.; Moskopp, M.L.; Ullrich, M.; Calsina, B.; Wallace, P.W.; Richter, S.; Friedemann, M.; Langton, K.; Fliedner, S.M.J.; Timmers, H.; et al. HIF2alpha supports pro-metastatic behavior in pheochromocytomas/paragangliomas. Endocr. Relat. Cancer 2020, 27, 625–640. [Google Scholar] [CrossRef]
- Lenders, J.W.; Pacak, K.; Walther, M.M.; Linehan, W.M.; Mannelli, M.; Friberg, P.; Keiser, H.R.; Goldstein, D.S.; Eisenhofer, G. Biochemical diagnosis of pheochromocytoma: Which test is best? JAMA 2002, 287, 1427–1434. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Huynh, T.T.; Pacak, K.; Brouwers, F.M.; Walther, M.M.; Linehan, W.M.; Munson, P.J.; Mannelli, M.; Goldstein, D.S.; Elkahloun, A.G. Distinct gene expression profiles in norepinephrine- and epinephrine-producing hereditary and sporadic pheochromocytomas: Activation of hypoxia-driven angiogenic pathways in von Hippel-Lindau syndrome. Endocr. Relat. Cancer 2004, 11, 897–911. [Google Scholar] [CrossRef] [Green Version]
- Feldman, J.M.; Blalock, J.A.; Zern, R.T.; Shelburne, J.D.; Gaede, J.T.; Farrell, R.E.; Wells, S.A. Deficiency of dopamine-beta-hydroxylase. A new mechanism for normotensive pheochromocytomas. Am. J. Clin. Pathol. 1979, 72, 175–185. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Lenders, J.W.; Timmers, H.; Mannelli, M.; Grebe, S.K.; Hofbauer, L.C.; Bornstein, S.R.; Tiebel, O.; Adams, K.; Bratslavsky, G.; et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin. Chem. 2011, 57, 411–420. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Klink, B.; Richter, S.; Lenders, J.W.; Robledo, M. Metabologenomics of Phaeochromocytoma and Paraganglioma: An Integrated Approach for Personalised Biochemical and Genetic Testing. Clin. Biochem. Rev. 2017, 38, 69–100. [Google Scholar]
- Zuber, S.; Wesley, R.; Prodanov, T.; Eisenhofer, G.; Pacak, K.; Kantorovich, V. Clinical utility of chromogranin A in SDHx-related paragangliomas. Eur. J. Clin. Investig. 2014, 44, 365–371. [Google Scholar] [CrossRef]
- Hsiao, R.J.; Parmer, R.J.; Takiyyuddin, M.A.; O’Connor, D.T. Chromogranin A storage and secretion: Sensitivity and specificity for the diagnosis of pheochromocytoma. Medicine 1991, 70, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Bilek, R.; Vlcek, P.; Safarik, L.; Michalsky, D.; Novak, K.; Duskova, J.; Vaclavikova, E.; Widimsky, J., Jr.; Zelinka, T. Chromogranin A in the Laboratory Diagnosis of Pheochromocytoma and Paraganglioma. Cancers 2019, 11, 586. [Google Scholar] [CrossRef] [Green Version]
- Eisenhofer, G.; Lenders, J.W.; Siegert, G.; Bornstein, S.R.; Friberg, P.; Milosevic, D.; Mannelli, M.; Linehan, W.M.; Adams, K.; Timmers, H.J.; et al. Plasma methoxytyramine: A novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur. J. Cancer 2012, 48, 1739–1749. [Google Scholar] [CrossRef] [Green Version]
- Udager, A.M.; Magers, M.J.; Goerke, D.M.; Vinco, M.L.; Siddiqui, J.; Cao, X.; Lucas, D.R.; Myers, J.L.; Chinnaiyan, A.M.; McHugh, J.B.; et al. The utility of SDHB and FH immunohistochemistry in patients evaluated for hereditary paraganglioma-pheochromocytoma syndromes. Hum. Pathol. 2018, 71, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Van Nederveen, F.H.; Gaal, J.; Favier, J.; Korpershoek, E.; Oldenburg, R.A.; de Bruyn, E.M.; Sleddens, H.F.; Derkx, P.; Riviere, J.; Dannenberg, H.; et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: A retrospective and prospective analysis. Lancet Oncol. 2009, 10, 764–771. [Google Scholar] [CrossRef] [Green Version]
- Wallace, P.W.; Conrad, C.; Bruckmann, S.; Pang, Y.; Caleiras, E.; Murakami, M.; Korpershoek, E.; Zhuang, Z.; Rapizzi, E.; Kroiss, M.; et al. Metabolomics, machine learning and immunohistochemistry to predict succinate dehydrogenase mutational status in phaeochromocytomas and paragangliomas. J. Pathol. 2020, 251, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Papathomas, T.G.; Oudijk, L.; Persu, A.; Gill, A.J.; van Nederveen, F.; Tischler, A.S.; Tissier, F.; Volante, M.; Matias-Guiu, X.; Smid, M.; et al. SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: A multicenter interobserver variation analysis using virtual microscopy: A Multinational Study of the European Network for the Study of Adrenal Tumors (ENS@T). Mod. Pathol. 2015, 28, 807–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardella, C.; El-Bahrawy, M.; Frizzell, N.; Adam, J.; Ternette, N.; Hatipoglu, E.; Howarth, K.; O’Flaherty, L.; Roberts, I.; Turner, G.; et al. Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J. Pathol. 2011, 225, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Favier, J.; Meatchi, T.; Robidel, E.; Badoual, C.; Sibony, M.; Nguyen, A.T.; Gimenez-Roqueplo, A.P.; Burnichon, N. Carbonic anhydrase 9 immunohistochemistry as a tool to predict or validate germline and somatic VHL mutations in pheochromocytoma and paraganglioma-a retrospective and prospective study. Mod. Pathol. 2020, 33, 57–64. [Google Scholar] [CrossRef]
- Richter, S.; Gieldon, L.; Pang, Y.; Peitzsch, M.; Huynh, T.; Leton, R.; Viana, B.; Ercolino, T.; Mangelis, A.; Rapizzi, E.; et al. Metabolome-guided genomics to identify pathogenic variants in isocitrate dehydrogenase, fumarate hydratase, and succinate dehydrogenase genes in pheochromocytoma and paraganglioma. Genet. Med. 2019, 21, 705–717. [Google Scholar] [CrossRef]
- Taieb, D.; Pacak, K. New Insights into the Nuclear Imaging Phenotypes of Cluster 1 Pheochromocytoma and Paraganglioma. Trends Endocrinol. Metab. 2017, 28, 807–817. [Google Scholar] [CrossRef]
- Taieb, D.; Hicks, R.J.; Hindie, E.; Guillet, B.A.; Avram, A.; Ghedini, P.; Timmers, H.J.; Scott, A.T.; Elojeimy, S.; Rubello, D.; et al. European Association of Nuclear Medicine Practice Guideline/Society of Nuclear Medicine and Molecular Imaging Procedure Standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2112–2137. [Google Scholar] [CrossRef]
- Aloj, L.; Giger, O.; Mendichovszky, I.A.; Challis, B.G.; Ronel, M.; Harper, I.; Cheow, H.; Hoopen, R.T.; Pitfield, D.; Gallagher, F.A.; et al. The role of [(68) Ga]Ga-DOTATATE PET/CT in wild-type KIT/PDGFRA gastrointestinal stromal tumours (GIST). EJNMMI Res. 2021, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Taieb, D.; Pacak, K. Molecular imaging and theranostic approaches in pheochromocytoma and paraganglioma. Cell Tissue Res. 2018, 372, 393–401. [Google Scholar] [CrossRef]
- Casey, R.T.; McLean, M.A.; Madhu, B.; Challis, B.G.; Ten Hoopen, R.; Roberts, T.; Clark, G.R.; Pittfield, D.; Simpson, H.L.; Bulusu, V.R.; et al. Translating in vivo metabolomic analysis of succinate dehydrogenase deficient tumours into clinical utility. JCO Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [PubMed]
- Andronesi, O.C.; Rapalino, O.; Gerstner, E.; Chi, A.; Batchelor, T.T.; Cahill, D.P.; Sorensen, A.G.; Rosen, B.R. Detection of oncogenic IDH1 mutations using magnetic resonance spectroscopy of 2-hydroxyglutarate. J. Clin. Investig. 2013, 123, 3659–3663. [Google Scholar] [CrossRef] [Green Version]
- Casey, R.T.; McLean, M.A.; Challis, B.G.; McVeigh, T.P.; Warren, A.Y.; Mendil, L.; Houghton, R.; de Sanctis, S.; Kosmoliaptsis, V.; Sandford, R.N.; et al. Fumarate Metabolic Signature for the Detection of Reed Syndrome in Humans. Clin. Cancer Res. 2020, 26, 391–396. [Google Scholar] [CrossRef] [Green Version]
- Castinetti, F.; Taieb, D.; Henry, J.F.; Walz, M.; Guerin, C.; Brue, T.; Conte-Devolx, B.; Neumann, H.P.; Sebag, F. Management of endocrine disease: Outcome of adrenal sparing surgery in heritable pheochromocytoma. Eur. J. Endocrinol. 2016, 174, R9–R18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, H.P.H.; Tsoy, U.; Bancos, I.; Amodru, V.; Walz, M.K.; Tirosh, A.; Kaur, R.J.; McKenzie, T.; Qi, X.; Bandgar, T.; et al. Comparison of Pheochromocytoma-Specific Morbidity and Mortality Among Adults with Bilateral Pheochromocytomas Undergoing Total Adrenalectomy vs Cortical-Sparing Adrenalectomy. JAMA Netw. Open 2019, 2, e198898. [Google Scholar] [CrossRef]
- Jha, A.; Taieb, D.; Carrasquillo, J.A.; Pryma, D.A.; Patel, M.; Millo, C.; de Herder, W.W.; del Rivero, J.; Crona, J.; Shulkin, B.L.; et al. High-Specific-Activity-(131)I-MIBG versus (177)Lu-DOTATATE Targeted Radionuclide Therapy for Metastatic Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2021, 27, 2989–2995. [Google Scholar] [CrossRef]
- Niemeijer, N.D.; Alblas, G.; van Hulsteijn, L.T.; Dekkers, O.M.; Corssmit, E.P. Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: Systematic review and meta-analysis. Clin. Endocrinol. 2014, 81, 642–651. [Google Scholar] [CrossRef]
- He, J.; Makey, D.; Fojo, T.; Adams, K.T.; Havekes, B.; Eisenhofer, G.; Sullivan, P.; Lai, E.W.; Pacak, K. Successful chemotherapy of hepatic metastases in a case of succinate dehydrogenase subunit B-related paraganglioma. Endocrine 2009, 36, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Fishbein, L.; Ben-Maimon, S.; Keefe, S.; Cengel, K.; Pryma, D.A.; Loaiza-Bonilla, A.; Fraker, D.L.; Nathanson, K.L.; Cohen, D.L. SDHB mutation carriers with malignant pheochromocytoma respond better to CVD. Endocr. Relat. Cancer 2017, 24, L51–L55. [Google Scholar] [CrossRef] [PubMed]
- Hadoux, J.; Favier, J.; Scoazec, J.Y.; Leboulleux, S.; Al Ghuzlan, A.; Caramella, C.; Deandreis, D.; Borget, I.; Loriot, C.; Chougnet, C.; et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int. J. Cancer 2014, 135, 2711–2720. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.V.; Siqueira, D.R.; Romitti, M.; Ceolin, L.; Brasil, B.A.; Meurer, L.; Capp, C.; Maia, A.L. Role of VEGF-A and its receptors in sporadic and MEN2-associated pheochromocytoma. Int. J. Mol. Sci. 2014, 15, 5323–5336. [Google Scholar] [CrossRef] [Green Version]
- O’Kane, G.M.; Ezzat, S.; Joshua, A.M.; Bourdeau, I.; Leibowitz-Amit, R.; Olney, H.J.; Krzyzanowska, M.; Reuther, D.; Chin, S.; Wang, L.; et al. A phase 2 trial of sunitinib in patients with progressive paraganglioma or pheochromocytoma: The SNIPP trial. Br. J. Cancer 2019, 120, 1113–1119. [Google Scholar] [CrossRef]
- Pang, Y.; Lu, Y.; Caisova, V.; Liu, Y.; Bullova, P.; Huynh, T.T.; Zhou, Y.; Yu, D.; Frysak, Z.; Hartmann, I.; et al. Targeting NAD(+)/PARP DNA Repair Pathway as a Novel Therapeutic Approach to SDHB-Mutated Cluster I Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2018, 24, 3423–3432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulkowski, P.L.; Oeck, S.; Dow, J.; Economos, N.G.; Mirfakhraie, L.; Liu, Y.; Noronha, K.; Bao, X.; Li, J.; Shuch, B.M.; et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 2020, 582, 586–591. [Google Scholar] [CrossRef]
- Chouaib, S.; Noman, M.Z.; Kosmatopoulos, K.; Curran, M.A. Hypoxic stress: Obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 2017, 36, 439–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letouze, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Yoo, C.B.; Jeong, S.; Egger, G.; Liang, G.; Phiasivongsa, P.; Tang, C.; Redkar, S.; Jones, P.A. Delivery of 5-aza-2’-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res. 2007, 67, 6400–6408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wedekind, M.F.; del Rivero, J.; Arnaldez, F.I.; Srinivasan, R.; Spencer, M.; Steinberg, S.M.; Peer, C.J.; Figg, W.D.; Killian, J.K.; Meltzer, P.S.; et al. A phase II trial of the DNA methyl transferase inhibitor, SGI-110 (Guadecitabine), in children and adults with SDH-deficient GIST, pheochromocytoma, and paraganglioma, and HLRCC-associated kidney cancer. J. Clin. Oncol. 2020, 38, 11540. [Google Scholar] [CrossRef]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A.; et al. On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature 2016, 539, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Lussey-Lepoutre, C.; Hollinshead, K.E.; Ludwig, C.; Menara, M.; Morin, A.; Castro-Vega, L.J.; Parker, S.J.; Janin, M.; Martinelli, C.; Ottolenghi, C.; et al. Loss of succinate dehydrogenase activity results in dependency on pyruvate carboxylation for cellular anabolism. Nat. Commun. 2015, 6, 8784. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pang, Y.; Zhu, B.; Uher, O.; Caisova, V.; Huynh, T.T.; Taieb, D.; Hadrava Vanova, K.; Ghayee, H.K.; Neuzil, J.; et al. Therapeutic Targeting of SDHB-Mutated Pheochromocytoma/Paraganglioma with Pharmacologic Ascorbic Acid. Clin. Cancer Res. 2020, 26, 3868–3880. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.Y.; Kim, T.W.; Park, Y.S.; Shin, S.J.; Shin, S.H.; Song, E.K.; Lee, H.J.; Lee, K.W.; Bang, Y.J. Phase 2 study of everolimus monotherapy in patients with nonfunctioning neuroendocrine tumors or pheochromocytomas/paragangliomas. Cancer 2012, 118, 6162–6170. [Google Scholar] [CrossRef]
- Dombi, E.; Baldwin, A.; Marcus, L.J.; Fisher, M.J.; Weiss, B.; Kim, A.; Whitcomb, P.; Martin, S.; Aschbacher-Smith, L.E.; Rizvi, T.A.; et al. Activity of Selumetinib in Neurofibromatosis Type 1-Related Plexiform Neurofibromas. N. Engl. J. Med. 2016, 375, 2550–2560. [Google Scholar] [CrossRef] [PubMed]
- Adalsteinsson, V.A.; Ha, G.; Freeman, S.S.; Choudhury, A.D.; Stover, D.G.; Parsons, H.A.; Gydush, G.; Reed, S.C.; Rotem, D.; Rhoades, J.; et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 2017, 8, 1324. [Google Scholar] [CrossRef] [Green Version]
- Pacak, K.; Kidd, M.; Meuter, L.; Modlin, I.M. A Novel Liquid Biopsy (NETest) identifies Paragangliomas and Pheochromocytomas with High Accuracy. Endocr. Relat. Cancer 2021, 28, 731–744. [Google Scholar] [CrossRef]
- MacFarlane, J.; Seong, K.C.; Bisambar, C.; Madhu, B.; Allinson, K.; Marker, A.; Warren, A.; Park, S.M.; Giger, O.; Challis, B.G.; et al. A review of the tumour spectrum of germline succinate dehydrogenase gene mutations: Beyond phaeochromocytoma and paraganglioma. Clin. Endocrinol. 2020, 93, 528–538. [Google Scholar] [CrossRef]
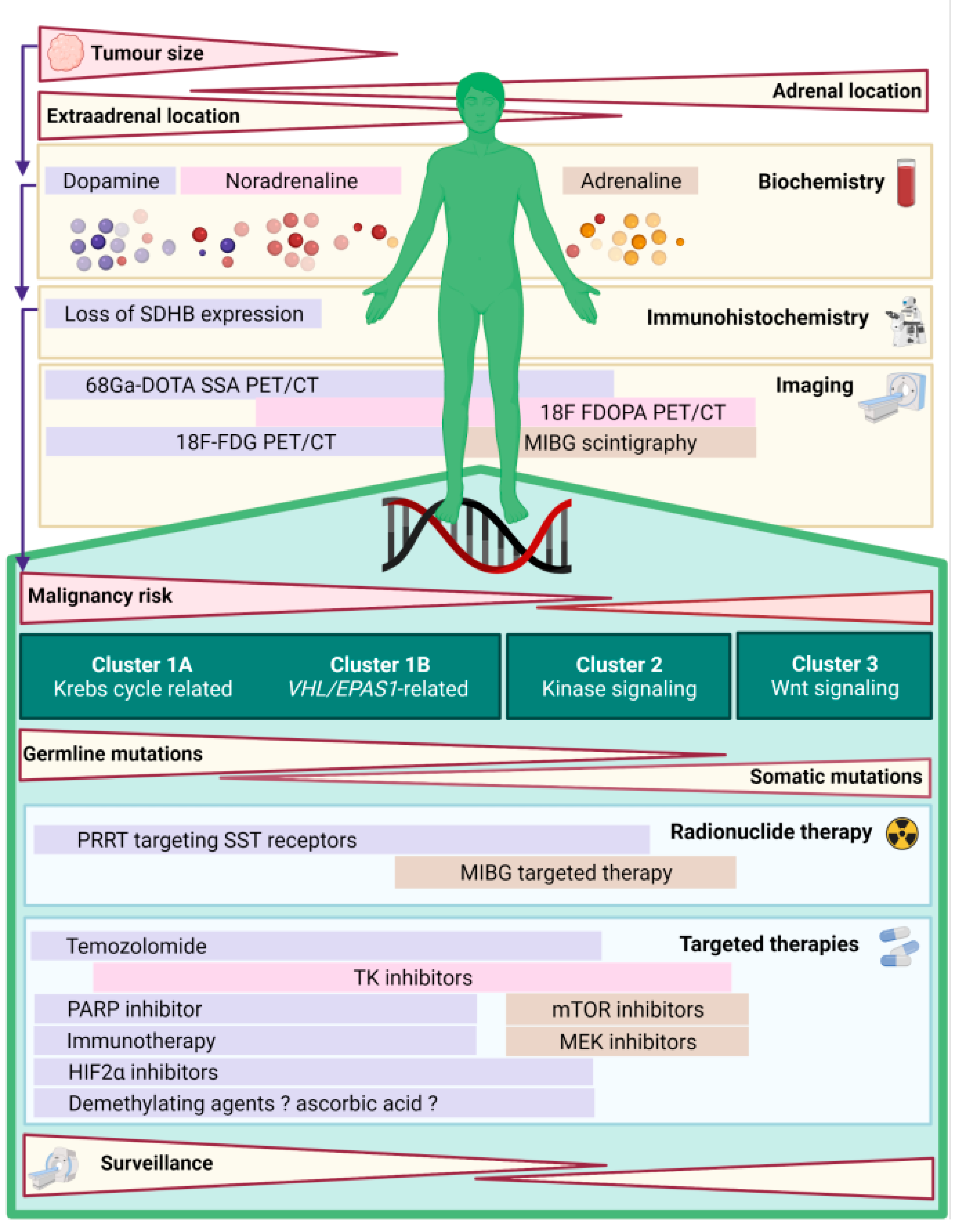

{kind=link}
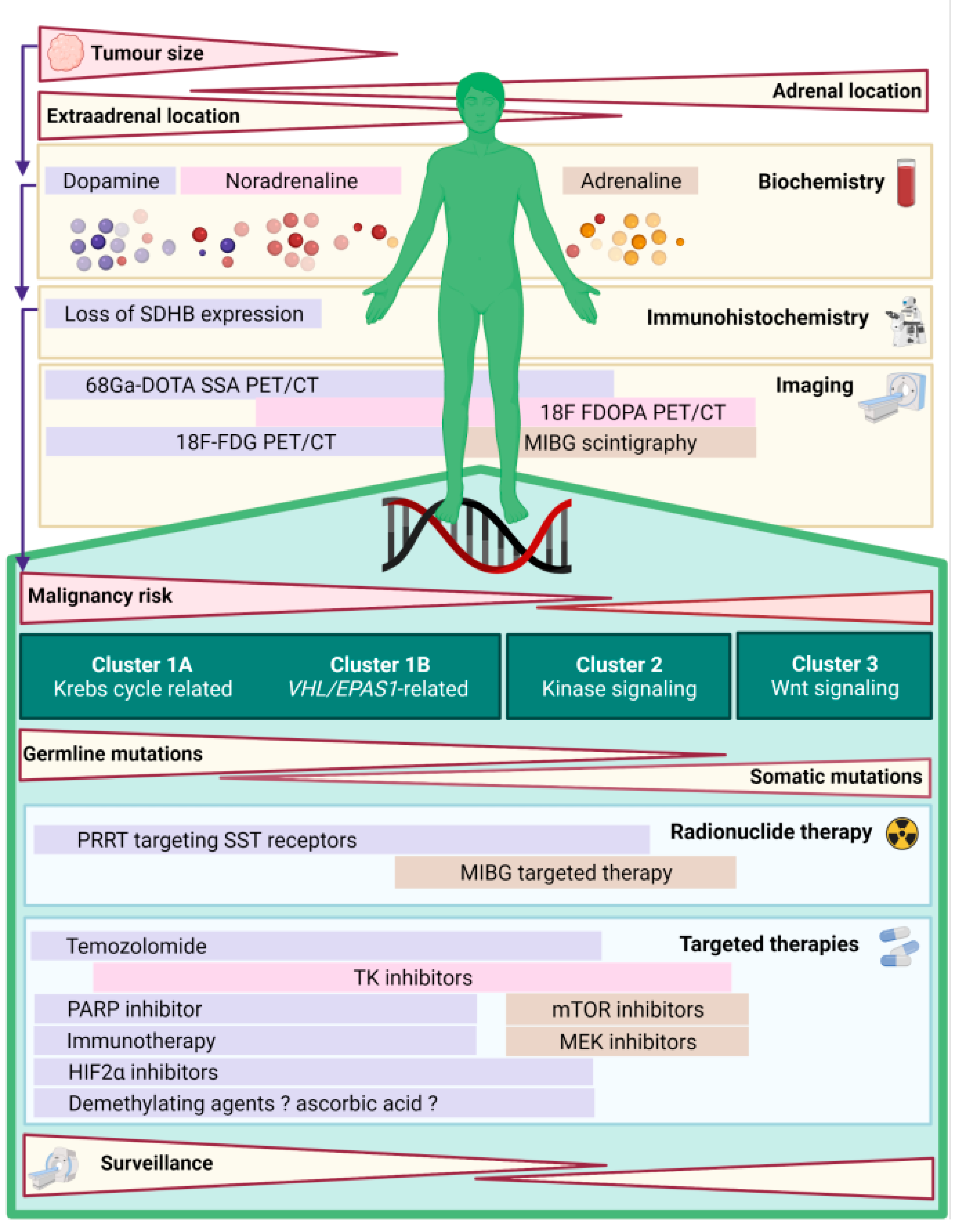{kind=link}
| Cluster 1 | Cluster 2 | Cluster 3 | |
|---|---|---|---|
| Genes | Cluster 1A: SDHx, FH, MDH2, IDH1/2, SLC25A11, DLST, GOT2, DNMT3A, EGLN1 | RET, NF1, TMEM127, MAX, HRAS, KRAS, FGFR1, NGRF, KIF1B, BRAF, MET, MERTK | MAML3, CSDE1 |
| Cluster 1B: VHL, EPAS1, EGLN1/2 IRP1 | |||
| Hallmarks of tumourigenesis | Pseudohypoxia | Increased Cell proliferation | Activated Wnt/β-catenine pathway |
| Angiogenesis | |||
| DNA and Histone methylation | Increased cell survival | ||
| Metabolic reprogramming |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Winzeler, B.; Challis, B.G.; Casey, R.T. Precision Medicine in Phaeochromocytoma and Paraganglioma. J. Pers. Med. 2021, 11, 1239. https://doi.org/10.3390/jpm11111239
Winzeler B, Challis BG, Casey RT. Precision Medicine in Phaeochromocytoma and Paraganglioma. Journal of Personalized Medicine. 2021; 11(11):1239. https://doi.org/10.3390/jpm11111239
Chicago/Turabian StyleWinzeler, Bettina, Benjamin G. Challis, and Ruth T. Casey. 2021. "Precision Medicine in Phaeochromocytoma and Paraganglioma" Journal of Personalized Medicine 11, no. 11: 1239. https://doi.org/10.3390/jpm11111239
APA StyleWinzeler, B., Challis, B. G., & Casey, R. T. (2021). Precision Medicine in Phaeochromocytoma and Paraganglioma. Journal of Personalized Medicine, 11(11), 1239. https://doi.org/10.3390/jpm11111239