
Personalized Medicine to Improve Treatment of Dopa-Responsive Dystonia—A Focus on Tyrosine Hydroxylase Deficiency


 , ,
, ,
Abstract
1. Introduction
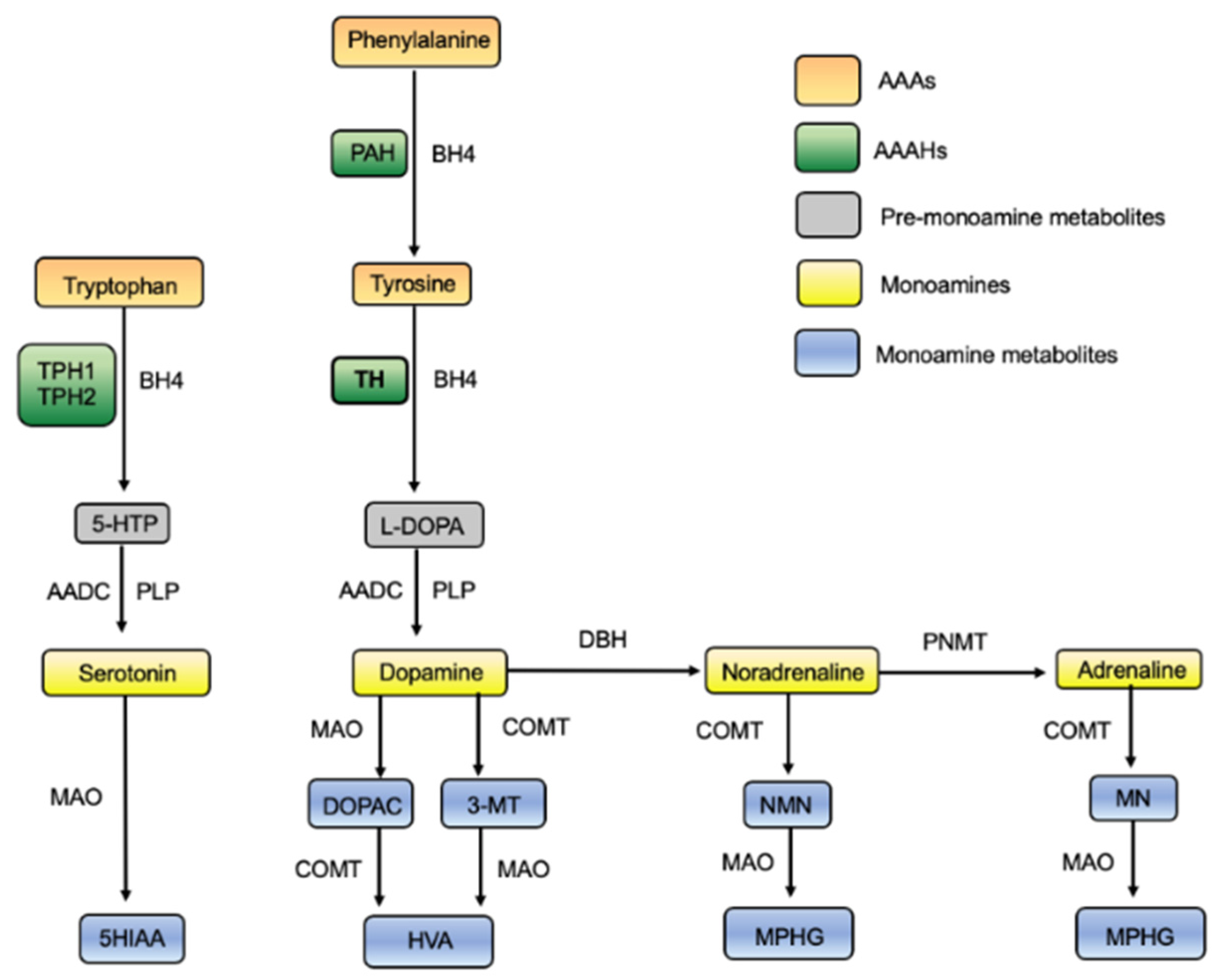
1.1. The Role of TH
1.2. The Clinical Manifestations of THD
1.3. Diagnosing THD
1.4. THD—An Orphan with Challenges Common for Rare Disorders
1.5. Tyrosine Hydroxylase
1.5.1. The Aromatic Amino Acid Hydroxylases
1.5.2. TH Location and Isoforms
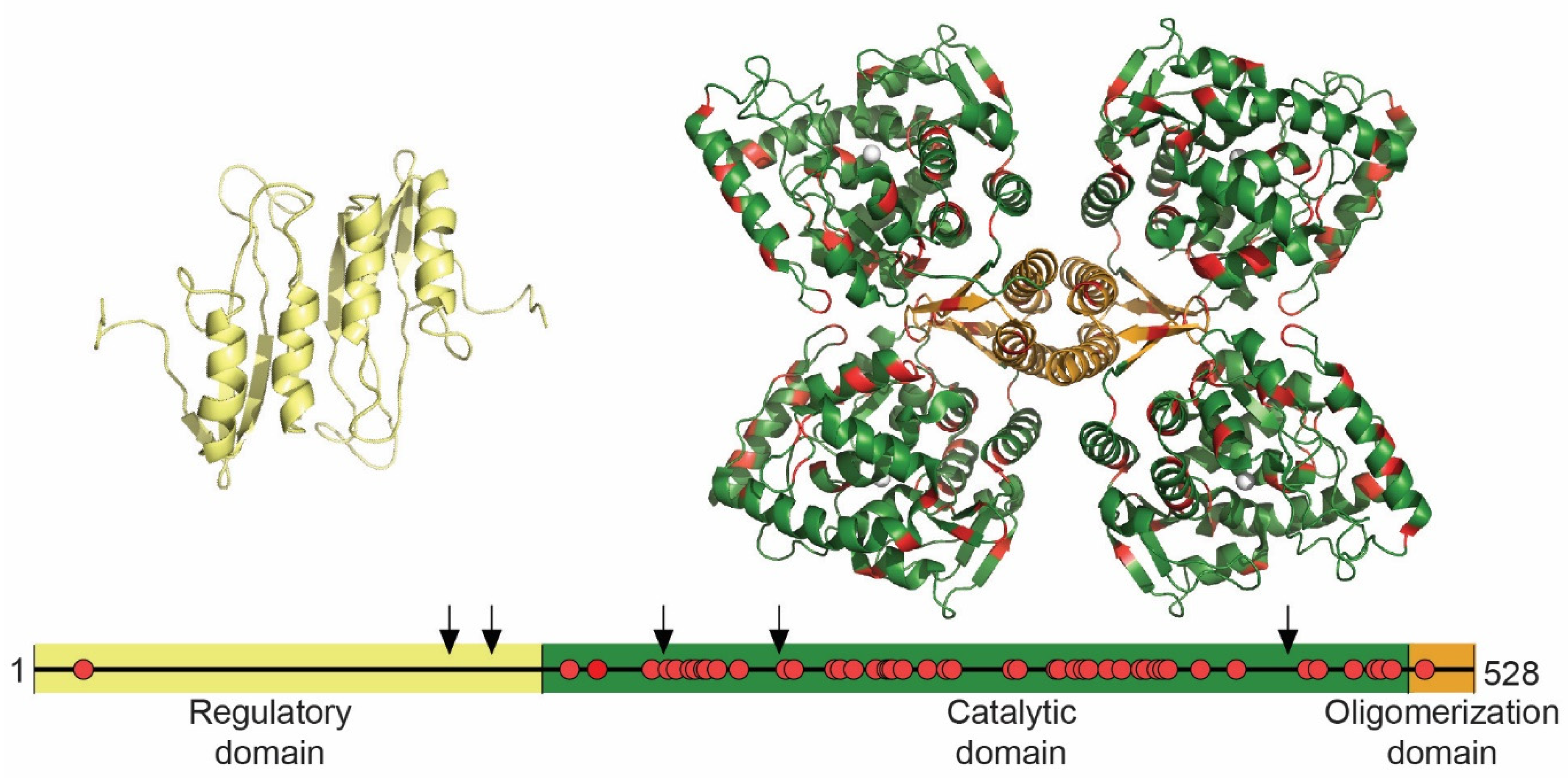
1.5.3. TH Structure and THD Mutations
1.5.4. Regulation of TH Activity
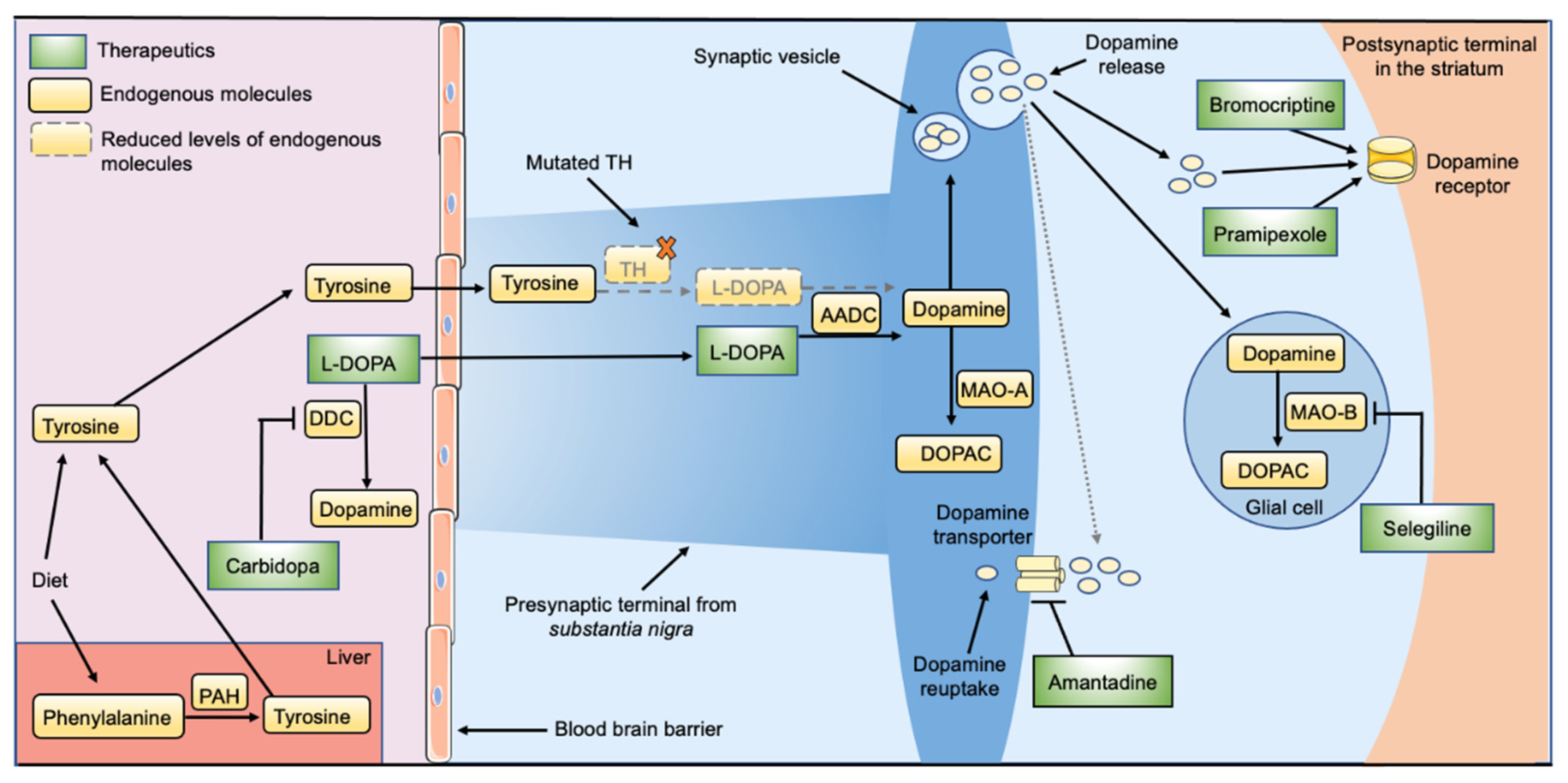
2. Current Treatment Options for THD
3. Promising and Future Treatment Opportunities for THD
3.1. Enzyme Replacement Therapy
3.2. Pharmacological Chaperones
3.3. Gene Therapy
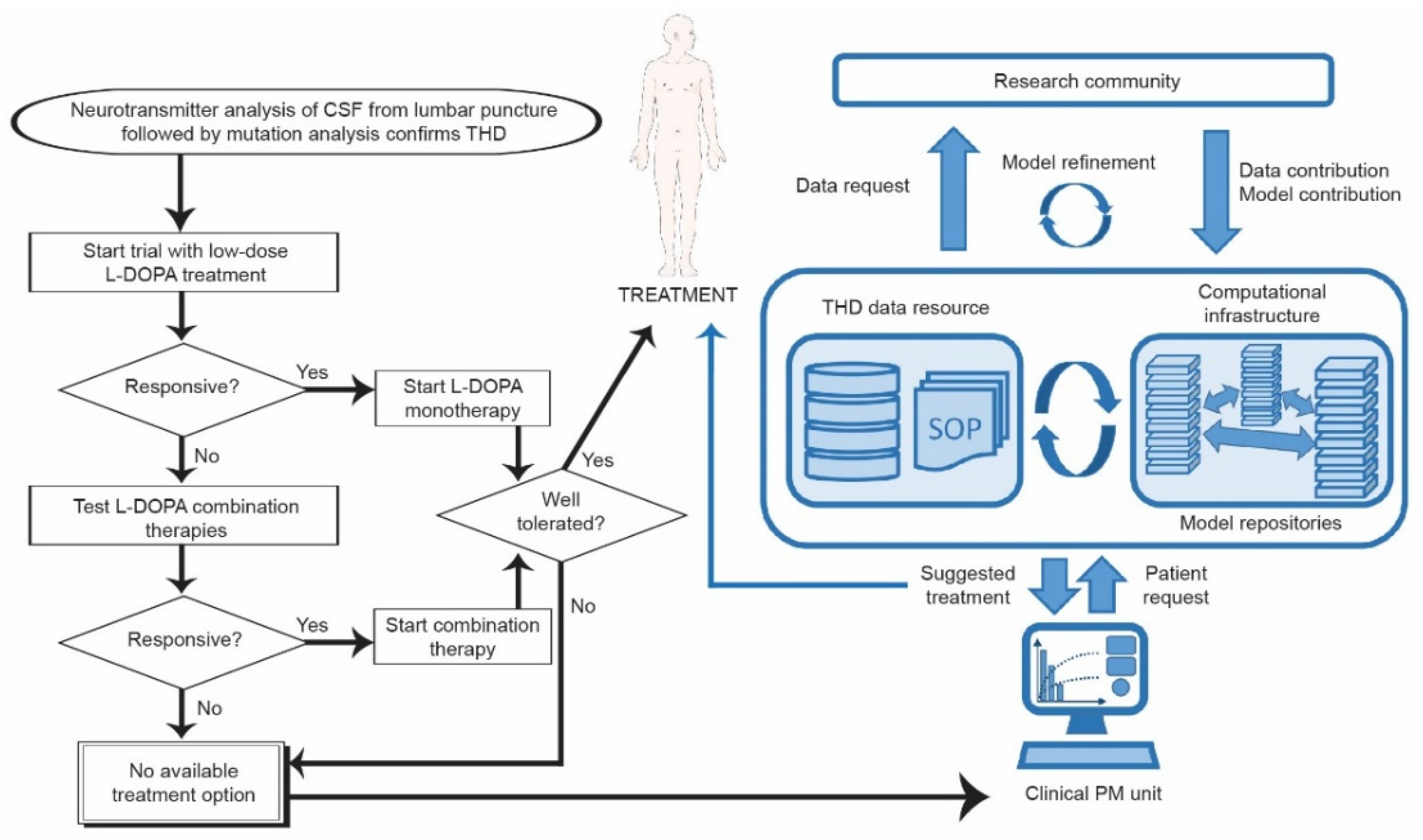
4. What Is Needed for a Personalized Medicine Approach in DRD?
4.1. Generating Relevant Data about Possible THD Mutations
4.1.1. Biochemical Assays Using Purified Proteins
4.1.2. Cell Assays to Assess Changes in Proteostasis
4.1.3. Complex Multi-Cell Cultures and Patient Derived iPSC
4.2. Computational Modeling for a Personalized Medicine Approach in THD
4.2.1. Systems Modeling of DA Synthesis and Metabolism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Model Type | Pathways | Objectives | Comments |
|---|---|---|---|---|
| Justice et al., 1988 [206] | ODE, MM | DA synthesis, release, metabolism | First of its kind—initial computational study | Compartmentalization of cytosolic DA |
| Kaushik et al., 2007 [200] | ODE, MM | DA synthesis, TH regulation by S40 phosphorylation/BH4 levels/Fe oxidation | Impact of TH regulation on DA levels | Focused on TH regulation, PKA phosphorylation, dephosphorylation and impact of α-Syn on dephosphorylation |
| Qi et al., 2008 [196,197] | ODE, PL | DA synthesis and metabolism | Analysis of presynaptic DA homeostasis | Detailed on DA metabolites, catecholamine auto-oxidation, melanin formation |
| Best et al., 2009 [207] | ODE, MM | DA synthesis, metabolism and release | Analysis of homeo-static mechanisms in DA synthesis and release | Models effect of substrate inhibition on DA homeostasis. Models regulation of TH by auto-receptors |
| Reed et al., 2012 [208] | ODE, MM | DA and serotonin synthesis and metabolism | Modeling the impact of L-DOPA treatment | Models DA synthesis in serotonergic neurons during L-DOPA treatment |
| Nijhout et al., 2014 [199] | ODE, MM | Several are discussed including DA synthesis and metabolism | Models impact of protein variants on DA homeostasis | Discusses homeostasis and robustness and illustrates system tolerance, e.g., towards enzyme variants of TH and DAT that have altered Vmax |
| Cullen & Wong-Lin 2015 [209] | ODE | Reduced model of Best et al., 2009 | Increase computational efficiency | Similar predictions as Best et al., 2009. Less intuitive to implement alterations in kinetic parameters |
| Véronneau-Veilleux et al., 2021 [198] | ODE | Pharmacokinetic model of levodopa, synaptic DA, and impact of DA on basal ganglia circuit activity | Models L-DOPA treatment in Parkinson’s disease | Hybrid model of three different modeling approaches illustrates how different models can be combined to bridge interactions between different subsystems and obtain clinically relevant predictions |
4.2.2. Implementing Systems Medicine in Personalized Medicine for DRD
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 3-MT | 3-methoxytyramine |
| 5-HTP | 5-hydroxytryptophan |
| 5HIAA | 5-hydroxyindolacetic acid |
| 5HIAA | 5-hydroxyindolacetic acid |
| AAAHs | aromatic amino acid hydroxylases |
| AADC | aromatic acid decarboxylase |
| BBB | blood–brain barrier |
| BH4 | Tetrahydrobiopterin |
| CA | Catecholamines |
| CD | catalytic domain |
| CNS | central nervous system |
| COMT | catechol-O-methyltransferase |
| CSF | cerebrospinal fluid |
| D2R | Dopamine receptor 2 |
| DBH | dopamine beta-hydroxylase |
| DOPAC | 3,4-dihydroxyphenylacetic acid |
| DRD | dopa-responsive dystonia |
| ERT | enzyme replacement therapy |
| GTPCHI | GTP cyclohydrolase I |
| hATTR | hereditary transthyretin |
| HVA | homovanilic acid |
| L-DOPA | 3,4-dihydroxyphenylalanine |
| LAT1 | large neutral amino acid transporter |
| LNAAs | large neutral amino acids |
| MHPG | 3-methoxy-4-hydroxyphenylethylene glycol |
| MN | Metanephrine |
| MAO-A | monoamine oxidase A |
| MAO-B | monoamine oxidase type B |
| MPHG | 3-methoxy-4-hydroxyphenylethylene glycol |
| NMN | Normetanephrine |
| NP | Nanoparticle |
| NT5DC2 | 5′-nucleotidase domain-containing protein 2 |
| OD | oligomerization domain |
| PAH | phenylalanine hydroxylase |
| PC | pharmacological chaperone |
| PD | Parkinson’s disorder |
| PKU | Phenylketonuria |
| PLP | pyridoxal phosphate |
| PM | personalized medicine |
| PNMT | phenylethanolamine N-methyltransferase |
| PP2A | protein phosphatase 2A |
| pSiNPs | porous silicon nanoparticles |
| PTPS | 6-pyruvoyl tetrahydrobiopterin synthase |
| RD | regulatory domain |
| SOP | standard operating procedure |
| SR | sepiapterin reductase |
| TH | tyrosine hydroxylase |
| THD | tyrosine hydroxylase deficiency |
| TPH1 | tryptophan hydroxylase 1 |
| TPH2 | tryptophan hydroxylase 2 |
| TTR | Transthyretin |
| Tyr | Tyrosine |
| VMAT1 | vesicular monoamine transporter 1 |
| VMAT2 | vesicular monoamine transporter 2 |
| VTA | ventral tegmental area |
| WT | wild type |
References
- Nygaard, T.G.; Marsden, C.D.; Duvoisin, R.C. Dopa-responsive dystonia. Adv. Neurol. 1988, 50, 377–384. [Google Scholar] [PubMed]
- Wijemanne, S.; Jankovic, J. Dopa-responsive dystonia--clinical and genetic heterogeneity. Nat. Rev. Neurol. 2015, 11, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Antelmi, E.; Stamelou, M.; Liguori, R.; Bhatia, K.P. Nonmotor Symptoms in Dopa-Responsive Dystonia. Mov. Disord. Clin. Pract. 2015, 2, 347–356. [Google Scholar] [CrossRef]
- Charlesworth, G.; Mohire, M.D.; Schneider, S.A.; Stamelou, M.; Wood, N.W.; Bhatia, K.P. Ataxia telangiectasia presenting as dopa-responsive cervical dystonia. Neurology 2013, 81, 1148–1151. [Google Scholar] [CrossRef]
- Nygaard, T.G. Dopa-responsive dystonia. Delineation of the clinical syndrome and clues to pathogenesis. Adv. Neurol. 1993, 60, 577–585. [Google Scholar]
- Segawa, M.; Hosaka, A.; Miyagawa, F.; Nomura, Y.; Imai, H. Hereditary progressive dystonia with marked diurnal fluctuation. Adv. Neurol. 1976, 14, 215–233. [Google Scholar] [PubMed]
- Bonafe, L.; Thony, B.; Leimbacher, W.; Kierat, L.; Blau, N. Diagnosis of dopa-responsive dystonia and other tetrahydrobiopterin disorders by the study of biopterin metabolism in fibroblasts. Clin. Chem. 2001, 47, 477–485. [Google Scholar] [CrossRef]
- Hanihara, T.; Inoue, K.; Kawanishi, C.; Sugiyama, N.; Miyakawa, T.; Onishi, H.; Yamada, Y.; Osaka, H.; Kosaka, K.; Iwabuchi, K.; et al. 6-Pyruvoyl-tetrahydropterin synthase deficiency with generalized dystonia and diurnal fluctuation of symptoms: A clinical and molecular study. Mov. Disord. 1997, 12, 408–411. [Google Scholar] [CrossRef]
- Brautigam, C.; Wevers, R.A.; Jansen, R.J.; Smeitink, J.A.; de Rijk-van Andel, J.F.; Gabreels, F.J.; Hoffmann, G.F. Biochemical hallmarks of tyrosine hydroxylase deficiency. Clin. Chem. 1998, 44, 1897–1904. [Google Scholar] [CrossRef]
- Nedelea, F.; Veduta, A.; Duta, S.; Vayna, A.M.; Panaitescu, A.; Peltecu, G.; Duba, H.C. Prenatal Genetic Testing for Dopa-Responsive Dystonia—Clinical Judgment in the Context of Next Generation Sequencing. J. Med. Life 2018, 11, 343–345. [Google Scholar]
- Wirth, T.; Mariani, L.L.; Bergant, G.; Baulac, M.; Habert, M.O.; Drouot, N.; Ollivier, E.; Hodzic, A.; Rudolf, G.; Nitschke, P.; et al. Loss-of-Function Mutations in NR4A2 Cause Dopa-Responsive Dystonia Parkinsonism. Mov. Disord. 2020, 35, 880–885. [Google Scholar] [CrossRef]
- Ng, J.; Papandreou, A.; Heales, S.J.; Kurian, M.A. Monoamine neurotransmitter disorders--Clinical advances and future perspectives. Nat. Rev. Neurol. 2015, 11, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Levitt, M.; Udenfriend, S. Tyrosine Hydroxylase. The Initial Step in Norepinephrine Biosynthesis. J. Biol. Chem. 1964, 239, 2910–2917. [Google Scholar] [CrossRef]
- Levitt, M.; Spector, S.; Sjoerdsma, A.; Udenfriend, S. Elucidation of the Rate-Limiting Step in Norepinephrine Biosynthesis in the Perfused Guinea-Pig Heart. J. Pharmacol. Exp. Ther. 1965, 148, 1–8. [Google Scholar]
- Eisenhofer, G.; Kopin, I.J.; Goldstein, D.S. Catecholamine metabolism: A contemporary view with implications for physiology and medicine. Pharmacol. Rev. 2004, 56, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Lepack, A.E.; Werner, C.T.; Stewart, A.F.; Fulton, S.L.; Zhong, P.; Farrelly, L.A.; Smith, A.C.W.; Ramakrishnan, A.; Lyu, Y.; Bastle, R.M.; et al. Dopaminylation of histone H3 in ventral tegmental area regulates cocaine seeking. Science 2020, 368, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Michaud-Soret, I.; Andersson, K.K.; Que, L., Jr.; Haavik, J. Resonance Raman studies of catecholate and phenolate complexes of recombinant human tyrosine hydroxylase. Biochemistry 1995, 34, 5504–5510. [Google Scholar] [CrossRef]
- Fossbakk, A.; Kleppe, R.; Knappskog, P.M.; Martinez, A.; Haavik, J. Functional studies of tyrosine hydroxylase missense variants reveal distinct patterns of molecular defects in Dopa-responsive dystonia. Hum. Mutat. 2014, 35, 880–890. [Google Scholar] [CrossRef]
- Ludecke, B.; Dworniczak, B.; Bartholome, K. A point mutation in the tyrosine hydroxylase gene associated with Segawa’s syndrome. Hum. Genet. 1995, 95, 123–125. [Google Scholar] [CrossRef]
- Willemsen, M.A.; Verbeek, M.M.; Kamsteeg, E.J.; de Rijk-van Andel, J.F.; Aeby, A.; Blau, N.; Burlina, A.; Donati, M.A.; Geurtz, B.; Grattan-Smith, P.J.; et al. Tyrosine hydroxylase deficiency: A treatable disorder of brain catecholamine biosynthesis. Brain 2010, 133, 1810–1822. [Google Scholar] [CrossRef]
- Kuseyri Hubschmann, O.; Horvath, G.; Cortes-Saladelafont, E.; Yildiz, Y.; Mastrangelo, M.; Pons, R.; Friedman, J.; Mercimek-Andrews, S.; Wong, S.N.; Pearson, T.S.; et al. Insights into the expanding phenotypic spectrum of inherited disorders of biogenic amines. Nat. Commun. 2021, 12, 5529. [Google Scholar] [CrossRef]
- Chen, Y.; Bao, X.; Wen, Y.; Wang, J.; Zhang, Q.; Yan, J. Clinical and Genetic Heterogeneity in a Cohort of Chinese Children With Dopa-Responsive Dystonia. Front. Pediatr. 2020, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Bijarnia-Mahay, S.; Jain, V.; Thony, B. Tyrosine hydroxylase deficiency-Clinical insights and a novel deletion in TH gene in an Indian patient. JIMD Rep. 2020, 53, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Haavik, J.; Blau, N.; Thony, B. Mutations in human monoamine-related neurotransmitter pathway genes. Hum. Mutat. 2008, 29, 891–902. [Google Scholar] [CrossRef]
- Moller, L.B.; Romstad, A.; Paulsen, M.; Hougaard, P.; Ormazabal, A.; Pineda, M.; Blau, N.; Guttler, F.; Artuch, R. Pre- and postnatal diagnosis of tyrosine hydroxylase deficiency. Prenat. Diagn. 2005, 25, 671–675. [Google Scholar] [CrossRef]
- Zhang, W.; Zhou, Z.; Li, X.; Huang, Y.; Li, T.; Lin, Y.; Shao, Y.; Hu, H.; Liu, H.; Liu, L. Dopa-responsive dystonia in Chinese patients: Including a novel heterozygous mutation in the GCH1 gene with an intermediate phenotype and one case of prenatal diagnosis. Neurosci. Lett. 2017, 644, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Pons, R.; Syrengelas, D.; Youroukos, S.; Orfanou, I.; Dinopoulos, A.; Cormand, B.; Ormazabal, A.; Garzia-Cazorla, A.; Serrano, M.; Artuch, R. Levodopa-induced dyskinesias in tyrosine hydroxylase deficiency. Mov. Disord. 2013, 28, 1058–1063. [Google Scholar] [CrossRef]
- Haavik, J.; Toska, K. Tyrosine hydroxylase and Parkinson’s disease. Mol. Neurobiol. 1998, 16, 285–309. [Google Scholar] [CrossRef]
- Nagatsu, T.; Nakashima, A.; Ichinose, H.; Kobayashi, K. Human tyrosine hydroxylase in Parkinson’s disease and in related disorders. J. Neural. Transm. 2019, 126, 397–409. [Google Scholar] [CrossRef]
- Kurian, M.A.; Gissen, P.; Smith, M.; Heales, S., Jr.; Clayton, P.T. The monoamine neurotransmitter disorders: An expanding range of neurological syndromes. Lancet Neurol. 2011, 10, 721–733. [Google Scholar] [CrossRef]
- Sawada, M.; Hirata, Y.; Arai, H.; Iizuka, R.; Nagatsu, T. Tyrosine hydroxylase, tryptophan hydroxylase, biopterin, and neopterin in the brains of normal controls and patients with senile dementia of Alzheimer type. J. Neurochem. 1987, 48, 760–764. [Google Scholar] [CrossRef]
- Priyadarshini, M.; Kamal, M.A.; Greig, N.H.; Reale, M.; Abuzenadah, A.M.; Chaudhary, A.G.; Damanhouri, G.A. Alzheimer’s disease and type 2 diabetes: Exploring the association to obesity and tyrosine hydroxylase. CNS Neurol. Disord. Drug Targets 2012, 11, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Cremers, S.; Aronson, J.K. Drugs for rare disorders. Br. J. Clin. Pharmacol. 2017, 83, 1607–1613. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, K.; Vernon, M.K.; Patrick, D.L.; Perfetto, E.; Nestler-Parr, S.; Burke, L. Patient-Reported Outcome and Observer-Reported Outcome Assessment in Rare Disease Clinical Trials: An ISPOR COA Emerging Good Practices Task Force Report. Value Health 2017, 20, 838–855. [Google Scholar] [CrossRef]
- Klein, C.; Gahl, W.A. Patients with rare diseases: From therapeutic orphans to pioneers of personalized treatments. EMBO Mol. Med. 2018, 10, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Cox, G.F. The art and science of choosing efficacy endpoints for rare disease clinical trials. Am. J. Med. Genet. A 2018, 176, 759–772. [Google Scholar] [CrossRef]
- Clarke, J.T.; Coyle, D.; Evans, G.; Martin, J.; Winquist, E. Toward a functional definition of a “rare disease” for regulatory authorities and funding agencies. Value Health 2014, 17, 757–761. [Google Scholar] [CrossRef]
- Mulberg, A.E.; Bucci-Rechtweg, C.; Giuliano, J.; Jacoby, D.; Johnson, F.K.; Liu, Q.; Marsden, D.; McGoohan, S.; Nelson, R.; Patel, N.; et al. Regulatory strategies for rare diseases under current global regulatory statutes: A discussion with stakeholders. Orphanet J. Rare Dis. 2019, 14, 36. [Google Scholar] [CrossRef]
- Schee Genannt Halfmann, S.; Mahlmann, L.; Leyens, L.; Reumann, M.; Brand, A. Personalized Medicine: What’s in it for Rare Diseases? Adv. Exp. Med. Biol. 2017, 1031, 387–404. [Google Scholar]
- Flydal, M.I.; Martinez, A. Phenylalanine hydroxylase: Function, structure, and regulation. IUBMB Life 2013, 65, 341–349. [Google Scholar] [CrossRef]
- Kapalka, G.M. Practical Resources for the Mental Health Professional, Nutritional and Herbal Therapies for Children and Adolescents; Academic Press: London, UK, 2010. [Google Scholar]
- Roberts, K.M.; Fitzpatrick, P.F. Mechanisms of tryptophan and tyrosine hydroxylase. IUBMB Life 2013, 65, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Daubner, S.C.; Le, T.; Wang, S. Tyrosine hydroxylase and regulation of dopamine synthesis. Arch. Biochem. Biophys. 2011, 508, 1–12. [Google Scholar] [CrossRef] [PubMed]
- van Spronsen, F.J.; Blau, N.; Harding, C.; Burlina, A.; Longo, N.; Bosch, A.M. Phenylketonuria. Nat. Rev. Dis. Primers 2021, 7, 36. [Google Scholar] [CrossRef]
- Pey, A.L.; Desviat, L.R.; Gamez, A.; Ugarte, M.; Perez, B. Phenylketonuria: Genotype-phenotype correlations based on expression analysis of structural and functional mutations in PAH. Hum. Mutat. 2003, 21, 370–378. [Google Scholar] [CrossRef]
- Gersting, S.W.; Kemter, K.F.; Staudigl, M.; Messing, D.D.; Danecka, M.K.; Lagler, F.B.; Sommerhoff, C.P.; Roscher, A.A.; Muntau, A.C. Loss of function in phenylketonuria is caused by impaired molecular motions and conformational instability. Am. J. Hum. Genet. 2008, 83, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Wettstein, S.; Underhaug, J.; Perez, B.; Marsden, B.D.; Yue, W.W.; Martinez, A.; Blau, N. Linking genotypes database with locus-specific database and genotype-phenotype correlation in phenylketonuria. Eur. J. Hum. Genet. 2015, 23, 302–309. [Google Scholar] [CrossRef]
- Aubi, O.; Prestegard, K.S.; Jung-Kc, K.; Shi, T.S.; Ying, M.; Grindheim, A.K.; Scherer, T.; Ulvik, A.; McCann, A.; Spriet, E.; et al. The Pah-R261Q mouse reveals oxidative stress associated with amyloid-like hepatic aggregation of mutant phenylalanine hydroxylase. Nat. Commun. 2021, 12, 2073. [Google Scholar] [CrossRef]
- Kleppe, R.; Haavik, J. Different stabilities and denaturation pathways for structurally related aromatic amino acid hydroxylases. FEBS Lett. 2004, 565, 155–159. [Google Scholar] [CrossRef][Green Version]
- Kitahama, K.; Pearson, J.; Denoroy, L.; Kopp, N.; Ulrich, J.; Maeda, T.; Jouvet, M. Adrenergic neurons in human brain demonstrated by immunohistochemistry with antibodies to phenylethanolamine-N-methyltransferase (PNMT): Discovery of a new group in the nucleus tractus solitarius. Neurosci. Lett. 1985, 53, 303–308. [Google Scholar] [CrossRef]
- Nagatsu, T.; Ichinose, H. Comparative studies on the structure of human tyrosine hydroxylase with those of the enzyme of various mammals. Comp. Biochem. Physiol. C Comp. Pharmacol. Toxicol. 1991, 98, 203–210. [Google Scholar]
- Pickel, V.M.; Joh, T.H.; Field, P.M.; Becker, C.G.; Reis, D.J. Cellular localization of tyrosine hydroxylase by immunohistochemistry. J. Histochem. Cytochem. 1975, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Han, V.K.; Snouweart, J.; Towle, A.C.; Lund, P.K.; Lauder, J.M. Cellular localization of tyrosine hydroxylase mRNA and its regulation in the rat adrenal medulla and brain by in situ hybridization with an oligodeoxyribonucleotide probe. J. Neurosci. Res. 1987, 17, 11–18. [Google Scholar] [CrossRef]
- Oomori, Y.; Iuchi, H.; Ishikawa, K.; Satoh, Y.; Ono, K. Immunocytochemical study of tyrosine hydroxylase and dopamine beta-hydroxylase immunoreactivities in the rat pancreas. Histochemistry 1994, 101, 313–323. [Google Scholar] [CrossRef]
- Mezey, E.; Eisenhofer, G.; Harta, G.; Hansson, S.; Gould, L.; Hunyady, B.; Hoffman, B.J. A novel nonneuronal catecholaminergic system: Exocrine pancreas synthesizes and releases dopamine. Proc. Natl. Acad. Sci. USA 1996, 93, 10377–10382. [Google Scholar] [CrossRef] [PubMed]
- Korner, J.; Cline, G.W.; Slifstein, M.; Barba, P.; Rayat, G.R.; Febres, G.; Leibel, R.L.; Maffei, A.; Harris, P.E. A role for foregut tyrosine metabolism in glucose tolerance. Mol. Metab. 2019, 23, 37–50. [Google Scholar] [CrossRef]
- Eisensmith, R.C.; Woo, S.L. Molecular basis of phenylketonuria and related hyperphenylalaninemias: Mutations and polymorphisms in the human phenylalanine hydroxylase gene. Hum. Mutat. 1992, 1, 13–23. [Google Scholar] [CrossRef]
- Xu, P.; Gildea, J.J.; Zhang, C.; Konkalmatt, P.; Cuevas, S.; Bigler Wang, D.; Tran, H.T.; Jose, P.A.; Felder, R.A. Stomach gastrin is regulated by sodium via PPAR-alpha and dopamine D1 receptor. J. Mol. Endocrinol. 2020, 64, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Aneman, A.; Friberg, P.; Hooper, D.; Fandriks, L.; Lonroth, H.; Hunyady, B.; Mezey, E. Substantial production of dopamine in the human gastrointestinal tract. J. Clin. Endocrinol. Metab. 1997, 82, 3864–3871. [Google Scholar] [CrossRef]
- Raj, B.; Blencowe, B.J. Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron 2015, 87, 14–27. [Google Scholar] [CrossRef]
- Barbosa-Morais, N.L.; Irimia, M.; Pan, Q.; Xiong, H.Y.; Gueroussov, S.; Lee, L.J.; Slobodeniuc, V.; Kutter, C.; Watt, S.; Colak, R.; et al. The evolutionary landscape of alternative splicing in vertebrate species. Science 2012, 338, 1587–1593. [Google Scholar] [CrossRef]
- Merkin, J.; Russell, C.; Chen, P.; Burge, C.B. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science 2012, 338, 1593–1599. [Google Scholar] [CrossRef]
- Haycock, J.W. Species differences in the expression of multiple tyrosine hydroxylase protein isoforms. J. Neurochem. 2002, 81, 947–953. [Google Scholar] [CrossRef][Green Version]
- Lewis, D.A.; Melchitzky, D.S.; Haycock, J.W. Four isoforms of tyrosine hydroxylase are expressed in human brain. Neuroscience 1993, 54, 477–492. [Google Scholar] [CrossRef]
- Grima, B.; Lamouroux, A.; Boni, C.; Julien, J.F.; Javoy-Agid, F.; Mallet, J. A single human gene encoding multiple tyrosine hydroxylases with different predicted functional characteristics. Nature 1987, 326, 707–711. [Google Scholar] [CrossRef]
- Nagatsu, T. Tyrosine hydroxylase: Human isoforms, structure and regulation in physiology and pathology. Essays Biochem. 1995, 30, 15–35. [Google Scholar]
- Kobayashi, K.; Kaneda, N.; Ichinose, H.; Kishi, F.; Nakazawa, A.; Kurosawa, Y.; Fujita, K.; Nagatsu, T. Structure of the human tyrosine hydroxylase gene: Alternative splicing from a single gene accounts for generation of four mRNA types. J. Biochem. 1988, 103, 907–912. [Google Scholar] [CrossRef]
- Le Bourdelles, B.; Horellou, P.; Le Caer, J.P.; Denefle, P.; Latta, M.; Haavik, J.; Guibert, B.; Mayaux, J.F.; Mallet, J. Phosphorylation of human recombinant tyrosine hydroxylase isoforms 1 and 2: An additional phosphorylated residue in isoform 2, generated through alternative splicing. J. Biol. Chem. 1991, 266, 17124–17130. [Google Scholar] [CrossRef]
- Royo, M.; Daubner, S.C.; Fitzpatrick, P.F. Effects of mutations in tyrosine hydroxylase associated with progressive dystonia on the activity and stability of the protein. Proteins 2005, 58, 14–21. [Google Scholar] [CrossRef]
- Calvo, A.C.; Scherer, T.; Pey, A.L.; Ying, M.; Winge, I.; McKinney, J.; Haavik, J.; Thony, B.; Martinez, A. Effect of pharmacological chaperones on brain tyrosine hydroxylase and tryptophan hydroxylase 2. J. Neurochem 2010, 114, 853–863. [Google Scholar] [CrossRef]
- Shehadeh, J.; Double, K.L.; Murphy, K.E.; Bobrovskaya, L.; Reyes, S.; Dunkley, P.R.; Halliday, G.M.; Dickson, P.W. Expression of tyrosine hydroxylase isoforms and phosphorylation at serine 40 in the human nigrostriatal system in Parkinson’s disease. Neurobiol. Dis. 2019, 130, 104524. [Google Scholar] [CrossRef]
- Haavik, J.; Le Bourdelles, B.; Martinez, A.; Flatmark, T.; Mallet, J. Recombinant human tyrosine hydroxylase isozymes. Reconstitution with iron and inhibitory effect of other metal ions. Eur. J. Biochem. 1991, 199, 371–378. [Google Scholar] [CrossRef]
- Nasrin, S.; Ichinose, H.; Hidaka, H.; Nagatsu, T. Recombinant human tyrosine hydroxylase types 1-4 show regulatory kinetic properties for the natural (6R)-tetrahydrobiopterin cofactor. J. Biochem. 1994, 116, 393–398. [Google Scholar] [CrossRef]
- Arturo, E.C.; Gupta, K.; Heroux, A.; Stith, L.; Cross, P.J.; Parker, E.J.; Loll, P.J.; Jaffe, E.K. First structure of full-length mammalian phenylalanine hydroxylase reveals the architecture of an autoinhibited tetramer. Proc. Natl. Acad. Sci. USA 2016, 113, 2394–2399. [Google Scholar] [CrossRef]
- Flydal, M.I.; Alcorlo-Pages, M.; Johannessen, F.G.; Martinez-Caballero, S.; Skjaerven, L.; Fernandez-Leiro, R.; Martinez, A.; Hermoso, J.A. Structure of full-length human phenylalanine hydroxylase in complex with tetrahydrobiopterin. Proc. Natl. Acad. Sci. USA 2019, 116, 11229–11234. [Google Scholar] [CrossRef]
- Meisburger, S.P.; Taylor, A.B.; Khan, C.A.; Zhang, S.; Fitzpatrick, P.F.; Ando, N. Domain Movements upon Activation of Phenylalanine Hydroxylase Characterized by Crystallography and Chromatography-Coupled Small-Angle X-ray Scattering. J. Am. Chem. Soc. 2016, 138, 6506–6516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Huang, T.; Ilangovan, U.; Hinck, A.P.; Fitzpatrick, P.F. The solution structure of the regulatory domain of tyrosine hydroxylase. J. Mol. Biol. 2014, 426, 1483–1497. [Google Scholar] [CrossRef]
- Goodwill, K.E.; Sabatier, C.; Marks, C.; Raag, R.; Fitzpatrick, P.F.; Stevens, R.C. Crystal structure of tyrosine hydroxylase at 2.3 A and its implications for inherited neurodegenerative diseases. Nat. Struct. Biol. 1997, 4, 578–585. [Google Scholar] [CrossRef]
- Goodwill, K.E.; Sabatier, C.; Stevens, R.C. Crystal structure of tyrosine hydroxylase with bound cofactor analogue and iron at 2.3 A resolution: Self-hydroxylation of Phe300 and the pterin-binding site. Biochemistry 1998, 37, 13437–13445. [Google Scholar] [CrossRef]
- Grant, G.A. The ACT domain: A small molecule binding domain and its role as a common regulatory element. J. Biol. Chem. 2006, 281, 33825–33829. [Google Scholar] [CrossRef]
- Bartholome, K. Deficiency of tyrosine hydroxylase or tryptophan hydroxylase: A possible cause of two hypothetical metabolic diseases. Acta Paediatr. Scand. 1983, 72, 921–922. [Google Scholar] [CrossRef]
- Gorke, W.; Bartholome, K. Biochemical and neurophysiological investigations in two forms of Segawa’s disease. Neuropediatrics 1990, 21, 3–8. [Google Scholar] [CrossRef]
- Cai, C.; Shi, W.; Zeng, Z.; Zhang, M.; Ling, C.; Chen, L.; Cai, C.; Zhang, B.; Li, W.D. GTP cyclohydrolase I and tyrosine hydroxylase gene mutations in familial and sporadic dopa-responsive dystonia patients. PLoS ONE 2013, 8, e65215. [Google Scholar] [CrossRef] [PubMed]
- Kumer, S.C.; Vrana, K.E. Intricate regulation of tyrosine hydroxylase activity and gene expression. J. Neurochem. 1996, 67, 443–462. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T. The catecholamine system in health and disease—Relation to tyrosine 3-monooxygenase and other catecholamine-synthesizing enzymes. Proc. Jpn. Acad Ser. B Phys. Biol. Sci. 2007, 82, 388–415. [Google Scholar] [CrossRef]
- Tank, A.W.; Xu, L.; Chen, X.; Radcliffe, P.; Sterling, C.R. Post-transcriptional regulation of tyrosine hydroxylase expression in adrenal medulla and brain. Ann. N. Y. Acad. Sci. 2008, 1148, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Lenartowski, R.; Goc, A. Epigenetic, transcriptional and posttranscriptional regulation of the tyrosine hydroxylase gene. Int. J. Dev. Neurosci. 2011, 29, 873–883. [Google Scholar] [CrossRef]
- Kappock, T.J.; Caradonna, J.P. Pterin-Dependent Amino Acid Hydroxylases. Chem. Rev. 1996, 96, 2659–2756. [Google Scholar] [CrossRef]
- Nakashima, A.; Mori, K.; Suzuki, T.; Kurita, H.; Otani, M.; Nagatsu, T.; Ota, A. Dopamine inhibition of human tyrosine hydroxylase type 1 is controlled by the specific portion in the N-terminus of the enzyme. J. Neurochem. 1999, 72, 2145–2153. [Google Scholar] [CrossRef]
- Flatmark, T. Catecholamine biosynthesis and physiological regulation in neuroendocrine cells. Acta Physiol. Scand. 2000, 168, 1–17. [Google Scholar] [CrossRef]
- Ota, A.; Nakashima, A.; Mori, K.; Nagatsu, T. Effects of dopamine on N-terminus-deleted human tyrosine hydroxylase type 1 expressed in Escherichia coli. Neurosci. Lett. 1997, 229, 57–60. [Google Scholar] [CrossRef]
- Nakashima, A.; Hayashi, N.; Mori, K.; Kaneko, Y.S.; Nagatsu, T.; Ota, A. Positive charge intrinsic to Arg(37)-Arg(38) is critical for dopamine inhibition of the catalytic activity of human tyrosine hydroxylase type 1. FEBS Lett. 2000, 465, 59–63. [Google Scholar] [CrossRef]
- Dunkley, P.R.; Bobrovskaya, L.; Graham, M.E.; von Nagy-Felsobuki, E.I.; Dickson, P.W. Tyrosine hydroxylase phosphorylation: Regulation and consequences. J. Neurochem. 2004, 91, 1025–1043. [Google Scholar] [CrossRef] [PubMed]
- Dickson, P.W.; Briggs, G.D. Tyrosine hydroxylase: Regulation by feedback inhibition and phosphorylation. Adv. Pharmacol. 2013, 68, 13–21. [Google Scholar]
- Gordon, S.L.; Bobrovskaya, L.; Dunkley, P.R.; Dickson, P.W. Differential regulation of human tyrosine hydroxylase isoforms 1 and 2 in situ: Isoform 2 is not phosphorylated at Ser35. Biochim. Biophys. Acta 2009, 1793, 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Dunkley, P.R.; Dickson, P.W. Tyrosine hydroxylase phosphorylation in vivo. J. Neurochem. 2019, 149, 706–728. [Google Scholar] [CrossRef]
- Haavik, J.; Martinez, A.; Flatmark, T. pH-dependent release of catecholamines from tyrosine hydroxylase and the effect of phosphorylation of Ser-40. FEBS Lett. 1990, 262, 363–365. [Google Scholar] [CrossRef]
- Andersson, K.K.; Vassort, C.; Brennan, B.A.; Que, L., Jr.; Haavik, J.; Flatmark, T.; Gros, F.; Thibault, J. Purification and characterization of the blue-green rat phaeochromocytoma (PC12) tyrosine hydroxylase with a dopamine-Fe(III) complex. Reversal of the endogenous feedback inhibition by phosphorylation of serine-40. Biochem. J. 1992, 284 Pt 3, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Almas, B.; Le Bourdelles, B.; Flatmark, T.; Mallet, J.; Haavik, J. Regulation of recombinant human tyrosine hydroxylase isozymes by catecholamine binding and phosphorylation. Structure/activity studies and mechanistic implications. Eur. J. Biochem. 1992, 209, 249–255. [Google Scholar] [CrossRef]
- Daubner, S.C.; Lauriano, C.; Haycock, J.W.; Fitzpatrick, P.F. Site-directed mutagenesis of serine 40 of rat tyrosine hydroxylase. Effects of dopamine and cAMP-dependent phosphorylation on enzyme activity. J. Biol. Chem. 1992, 267, 12639–12646. [Google Scholar] [CrossRef]
- Toska, K.; Kleppe, R.; Armstrong, C.G.; Morrice, N.A.; Cohen, P.; Haavik, J. Regulation of tyrosine hydroxylase by stress-activated protein kinases. J. Neurochem. 2002, 83, 775–783. [Google Scholar] [CrossRef]
- Martinez, A.; Haavik, J.; Flatmark, T.; Arrondo, J.L.; Muga, A. Conformational properties and stability of tyrosine hydroxylase studied by infrared spectroscopy. Effect of iron/catecholamine binding and phosphorylation. J. Biol. Chem. 1996, 271, 19737–19742. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, A.J.; Hillas, P.J.; Fitzpatrick, P.F. Characterization of the active site iron in tyrosine hydroxylase. Redox states of the iron. J. Biol. Chem. 1996, 271, 24395–24400. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, A.J.; Fitzpatrick, P.F. Effects of phosphorylation on binding of catecholamines to tyrosine hydroxylase: Specificity and thermodynamics. Biochemistry 2000, 39, 773–778. [Google Scholar] [CrossRef]
- Bueno-Carrasco, M.T.; Cuellar, J.; Flydal, M.I.; Santiago, C.; Krakenes, T.A.; Kleppe, R.; Lopez-Blanco, J.R.; Teigen, K.; Alvira, S.; Chacon, P.; et al. The structure of human tyrosine hydroxylase reveals the mechanism for feedback inhibition by dopamine. Res. Sq. 2020, preprint. [Google Scholar] [CrossRef]
- Wang, J.; Lou, H.; Pedersen, C.J.; Smith, A.D.; Perez, R.G. 14-3-3zeta contributes to tyrosine hydroxylase activity in MN9D cells: Localization of dopamine regulatory proteins to mitochondria. J. Biol. Chem. 2009, 284, 14011–14019. [Google Scholar] [CrossRef]
- Lindgren, N.; Xu, Z.Q.; Herrera-Marschitz, M.; Haycock, J.; Hokfelt, T.; Fisone, G. Dopamine D(2) receptors regulate tyrosine hydroxylase activity and phosphorylation at Ser40 in rat striatum. Eur. J. Neurosci. 2001, 13, 773–780. [Google Scholar] [CrossRef] [PubMed]
- el Mestikawy, S.; Glowinski, J.; Hamon, M. Presynaptic dopamine autoreceptors control tyrosine hydroxylase activation in depolarized striatal dopaminergic terminals. J. Neurochem. 1986, 46, 12–22. [Google Scholar] [CrossRef]
- Itagaki, C.; Isobe, T.; Taoka, M.; Natsume, T.; Nomura, N.; Horigome, T.; Omata, S.; Ichinose, H.; Nagatsu, T.; Greene, L.A.; et al. Stimulus-coupled interaction of tyrosine hydroxylase with 14-3-3 proteins. Biochemistry 1999, 38, 15673–15680. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, R.; Rosati, S.; Jorge-Finnigan, A.; Alvira, S.; Ghorbani, S.; Haavik, J.; Valpuesta, J.M.; Heck, A.J.; Martinez, A. Phosphorylation dependence and stoichiometry of the complex formed by tyrosine hydroxylase and 14-3-3gamma. Mol. Cell Proteom. 2014, 13, 2017–2030. [Google Scholar] [CrossRef]
- Kjarland, E.; Keen, T.J.; Kleppe, R. Does isoform diversity explain functional differences in the 14-3-3 protein family? Curr. Pharm. Biotechnol. 2006, 7, 217–223. [Google Scholar] [CrossRef]
- Ichimura, T.; Isobe, T.; Okuyama, T.; Yamauchi, T.; Fujisawa, H. Brain 14-3-3 protein is an activator protein that activates tryptophan 5-monooxygenase and tyrosine 3-monooxygenase in the presence of Ca2+,calmodulin-dependent protein kinase II. FEBS Lett. 1987, 219, 79–82. [Google Scholar] [CrossRef]
- Bowling, K.M.; Huang, Z.; Xu, D.; Ferdousy, F.; Funderburk, C.D.; Karnik, N.; Neckameyer, W.; O’Donnell, J.M. Direct binding of GTP cyclohydrolase and tyrosine hydroxylase: Regulatory interactions between key enzymes in dopamine biosynthesis. J. Biol. Chem. 2008, 283, 31449–31459. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Taira, T.; Niki, T.; Takahashi-Niki, K.; Maita, C.; Maita, H.; Ariga, H.; Iguchi-Ariga, S.M. Oxidative status of DJ-1-dependent activation of dopamine synthesis through interaction of tyrosine hydroxylase and 4-dihydroxy-L-phenylalanine (L-DOPA) decarboxylase with DJ-1. J. Biol. Chem. 2009, 284, 28832–28844. [Google Scholar] [CrossRef] [PubMed]
- Jorge-Finnigan, A.; Kleppe, R.; Jung-Kc, K.; Ying, M.; Marie, M.; Rios-Mondragon, I.; Salvatore, M.F.; Saraste, J.; Martinez, A. Phosphorylation at serine 31 targets tyrosine hydroxylase to vesicles for transport along microtubules. J. Biol. Chem. 2017, 292, 14092–14107. [Google Scholar] [CrossRef]
- Nakashima, A.; Yamaguchi, H.; Kondo, M.; Furumura, T.; Kodani, Y.; Kaneko, Y.S.; Kawata, M.; Nagasaki, H.; Nagatsu, T.; Ota, A. NT5DC2 affects the phosphorylation of tyrosine hydroxylase regulating its catalytic activity. J. Neural. Transm. 2020, 127, 1631–1640. [Google Scholar] [CrossRef]
- Perez, R.G.; Waymire, J.C.; Lin, E.; Liu, J.J.; Guo, F.; Zigmond, M.J. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 2002, 22, 3090–3099. [Google Scholar] [CrossRef]
- Parra, L.A.; Baust, T.B.; Smith, A.D.; Jaumotte, J.D.; Zigmond, M.J.; Torres, S.; Leak, R.K.; Pino, J.A.; Torres, G.E. The Molecular Chaperone Hsc70 Interacts with Tyrosine Hydroxylase to Regulate Enzyme Activity and Synaptic Vesicle Localization. J. Biol. Chem. 2016, 291, 17510–17522. [Google Scholar] [CrossRef]
- Anikster, Y.; Haack, T.B.; Vilboux, T.; Pode-Shakked, B.; Thony, B.; Shen, N.; Guarani, V.; Meissner, T.; Mayatepek, E.; Trefz, F.K.; et al. Biallelic Mutations in DNAJC12 Cause Hyperphenylalaninemia, Dystonia, and Intellectual Disability. Am. J. Hum. Genet. 2017, 100, 257–266. [Google Scholar] [CrossRef]
- Jung-Kc, K.; Himmelreich, N.; Prestegard, K.S.; Shi, T.S.; Scherer, T.; Ying, M.; Jorge-Finnigan, A.; Thony, B.; Blau, N.; Martinez, A. Phenylalanine hydroxylase variants interact with the co-chaperone DNAJC12. Hum. Mutat. 2019, 40, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, R.; Hao, Z.; Wang, G.; Mu, C.; Ding, J.; Sun, W.; Ren, H. DJ-1 regulates tyrosine hydroxylase expression through CaMKKbeta/CaMKIV/CREB1 pathway in vitro and in vivo. J. Cell Physiol. 2020, 235, 869–879. [Google Scholar] [CrossRef]
- Carlsson, A.; Lindqvist, M.; Magnusson, T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature 1957, 180, 1200. [Google Scholar] [CrossRef]
- Carlsson, A. The occurrence, distribution and physiological role of catecholamines in the nervous system. Pharmacol. Rev. 1959, 11, 490–493. [Google Scholar] [PubMed]
- Carlsson, A. A paradigm shift in brain research. Science 2001, 294, 1021–1024. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Goel, P.; Kaeser, P.S. Spatial and temporal scales of dopamine transmission. Nat. Rev. Neurosci. 2021, 22, 345–358. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kish, S. Tyrosine Hydroxylase Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Lonlay, P.D.E.; Nassogne, M.C.; van Gennip, A.H.; van Cruchten, A.C.; Billatte de Villemeur, T.; Cretz, M.; Stoll, C.; Launay, J.M.; Steenberger-Spante, G.C.; van den Heuvel, L.P.; et al. Tyrosine hydroxylase deficiency unresponsive to L-dopa treatment with unusual clinical and biochemical presentation. J. Inherit. Metab. Dis. 2000, 23, 819–825. [Google Scholar] [CrossRef]
- Hoffmann, G.F.; Assmann, B.; Brautigam, C.; Dionisi-Vici, C.; Haussler, M.; de Klerk, J.B.; Naumann, M.; Steenbergen-Spanjers, G.C.; Strassburg, H.M.; Wevers, R.A. Tyrosine hydroxylase deficiency causes progressive encephalopathy and dopa-nonresponsive dystonia. Ann. Neurol. 2003, 54 (Suppl. 6), S56–S65. [Google Scholar] [CrossRef]
- Zafeiriou, D.I.; Willemsen, M.A.; Verbeek, M.M.; Vargiami, E.; Ververi, A.; Wevers, R. Tyrosine hydroxylase deficiency with severe clinical course. Mol. Genet. Metab. 2009, 97, 18–20. [Google Scholar] [CrossRef]
- Dionisi-Vici, C.; Hoffmann, G.F.; Leuzzi, V.; Hoffken, H.; Brautigam, C.; Rizzo, C.; Steebergen-Spanjers, G.C.; Smeitink, J.A.; Wevers, R.A. Tyrosine hydroxylase deficiency with severe clinical course: Clinical and biochemical investigations and optimization of therapy. J. Pediatr. 2000, 136, 560–562. [Google Scholar] [CrossRef]
- Yosunkaya, E.; Karaca, E.; Basaran, S.; Seven, M.; Yuksel, A. Marked improvement in Segawa syndrome after L-dopa and selegiline treatment. Pediatr. Neurol. 2010, 42, 348–350. [Google Scholar] [CrossRef] [PubMed]
- Yeung, W.L.; Wong, V.C.; Chan, K.Y.; Hui, J.; Fung, C.W.; Yau, E.; Ko, C.H.; Lam, C.W.; Mak, C.M.; Siu, S.; et al. Expanding phenotype and clinical analysis of tyrosine hydroxylase deficiency. J. Child. Neurol. 2011, 26, 179–187. [Google Scholar] [CrossRef]
- Chi, C.S.; Lee, H.F.; Tsai, C.R. Tyrosine hydroxylase deficiency in Taiwanese infants. Pediatr. Neurol. 2012, 46, 77–82. [Google Scholar] [CrossRef]
- Katus, L.E.; Frucht, S.J. An unusual presentation of tyrosine hydroxylase deficiency. J. Clin. Mov. Disord. 2017, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Thony, B.; Calvo, A.C.; Scherer, T.; Svebak, R.M.; Haavik, J.; Blau, N.; Martinez, A. Tetrahydrobiopterin shows chaperone activity for tyrosine hydroxylase. J. Neurochem. 2008, 106, 672–681. [Google Scholar] [CrossRef]
- Gonzalez-Carter, D.A.; Ong, Z.Y.; McGilvery, C.M.; Dunlop, I.E.; Dexter, D.T.; Porter, A.E. L-DOPA functionalized, multi-branched gold nanoparticles as brain-targeted nano-vehicles. Nanomedicine 2019, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Opladen, T.; Lopez-Laso, E.; Cortes-Saladelafont, E.; Pearson, T.S.; Sivri, H.S.; Yildiz, Y.; Assmann, B.; Kurian, M.A.; Leuzzi, V.; Heales, S.; et al. Consensus guideline for the diagnosis and treatment of tetrahydrobiopterin (BH4) deficiencies. Orphanet J. Rare Dis. 2020, 15, 126. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.W. From structural biology to designing therapy for inborn errors of metabolism. J. Inherit. Metab. Dis. 2016, 39, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Eavri, R.; Lorberboum-Galski, H. A novel approach for enzyme replacement therapy. The use of phenylalanine hydroxylase-based fusion proteins for the treatment of phenylketonuria. J. Biol. Chem. 2007, 282, 23402–23409. [Google Scholar] [CrossRef]
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Goetz, C.G.; Marin, C.; Kordower, J.H.; Rodriguez, M.; Hirsch, E.C.; Farrer, M.; Schapira, A.H.; Halliday, G. Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 2010, 16, 653–661. [Google Scholar] [CrossRef]
- Duncko, R.; Kiss, A.; Skultetyova, I.; Rusnak, M.; Jezova, D. Corticotropin-releasing hormone mRNA levels in response to chronic mild stress rise in male but not in female rats while tyrosine hydroxylase mRNA levels decrease in both sexes. Psychoneuroendocrinology 2001, 26, 77–89. [Google Scholar] [CrossRef]
- Dahoun, T.; Trossbach, S.V.; Brandon, N.J.; Korth, C.; Howes, O.D. The impact of Disrupted-in-Schizophrenia 1 (DISC1) on the dopaminergic system: A systematic review. Transl. Psychiatry 2017, 7, e1015. [Google Scholar] [CrossRef]
- Bezem, M.T.; Baumann, A.; Skjaerven, L.; Meyer, R.; Kursula, P.; Martinez, A.; Flydal, M.I. Stable preparations of tyrosine hydroxylase provide the solution structure of the full-length enzyme. Sci. Rep. 2016, 6, 30390. [Google Scholar] [CrossRef]
- Tosi, G.; Duskey, J.T.; Kreuter, J. Nanoparticles as carriers for drug delivery of macromolecules across the blood-brain barrier. Expert Opin. Drug Deliv. 2020, 17, 23–32. [Google Scholar] [CrossRef]
- Bezem, M.T.; Johannessen, F.G.; Jung-Kc, K.; Gundersen, E.T.; Jorge-Finnigan, A.; Ying, M.; Betbeder, D.; Herfindal, L.; Martinez, A. Stabilization of Human Tyrosine Hydroxylase in Maltodextrin Nanoparticles for Delivery to Neuronal Cells and Tissue. Bioconjug. Chem. 2018, 29, 493–502. [Google Scholar] [CrossRef]
- Bezem, M.T.; Johannessen, F.G.; Krakenes, T.A.; Sailor, M.J.; Martinez, A. Relevance of Electrostatics for the Interaction of Tyrosine Hydroxylase with Porous Silicon Nanoparticles. Mol. Pharm. 2021, 18, 976–985. [Google Scholar] [CrossRef]
- Bernier, V.; Lagace, M.; Bichet, D.G.; Bouvier, M. Pharmacological chaperones: Potential treatment for conformational diseases. Trends Endocrinol. Metab. 2004, 15, 222–228. [Google Scholar] [CrossRef]
- Ludecke, B.; Knappskog, P.M.; Clayton, P.T.; Surtees, R.A.; Clelland, J.D.; Heales, S.J.; Brand, M.P.; Bartholome, K.; Flatmark, T. Recessively inherited L-DOPA-responsive parkinsonism in infancy caused by a point mutation (L205P) in the tyrosine hydroxylase gene. Hum. Mol. Genet. 1996, 5, 1023–1028. [Google Scholar] [CrossRef]
- Hole, M.; Underhaug, J.; Diez, H.; Ying, M.; Rohr, A.K.; Jorge-Finnigan, A.; Fernandez-Castillo, N.; Garcia-Cazorla, A.; Andersson, K.K.; Teigen, K.; et al. Discovery of compounds that protect tyrosine hydroxylase activity through different mechanisms. Biochim. Biophys. Acta 2015, 1854, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Underhaug, J.; Aubi, O.; Martinez, A. Phenylalanine hydroxylase misfolding and pharmacological chaperones. Curr. Top. Med. Chem. 2012, 12, 2534–2545. [Google Scholar] [CrossRef] [PubMed]
- Marmolino, D.; Acquaviva, F. Friedreich’s Ataxia: From the (GAA)n repeat mediated silencing to new promising molecules for therapy. Cerebellum 2009, 8, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, F.E.; Al-Gazali, L.; Al-Jasmi, F.; Ali, B.R. Pharmaceutical Chaperones and Proteostasis Regulators in the Therapy of Lysosomal Storage Disorders: Current Perspective and Future Promises. Front. Pharmacol. 2017, 8, 448. [Google Scholar] [CrossRef]
- Parenti, G.; Andria, G.; Valenzano, K.J. Pharmacological Chaperone Therapy: Preclinical Development, Clinical Translation, and Prospects for the Treatment of Lysosomal Storage Disorders. Mol. Ther. 2015, 23, 1138–1148. [Google Scholar] [CrossRef]
- Dugger, S.A.; Platt, A.; Goldstein, D.B. Drug development in the era of precision medicine. Nat. Rev. Drug Discov. 2018, 17, 183–196. [Google Scholar] [CrossRef]
- Urano, F.; Hayashi, N.; Arisaka, F.; Kurita, H.; Murata, S.; Ichinose, H. Molecular mechanism for pterin-mediated inactivation of tyrosine hydroxylase: Formation of insoluble aggregates of tyrosine hydroxylase. J. Biochem. 2006, 139, 625–635. [Google Scholar] [CrossRef]
- Pey, A.L.; Ying, M.; Cremades, N.; Velazquez-Campoy, A.; Scherer, T.; Thony, B.; Sancho, J.; Martinez, A. Identification of pharmacological chaperones as potential therapeutic agents to treat phenylketonuria. J. Clin. Investig. 2008, 118, 2858–2867. [Google Scholar] [CrossRef] [PubMed]
- Flydal, M.I.; Krakenes, T.A.; Tai, M.D.S.; Tran, M.P.A.; Teigen, K.; Martinez, A. Levalbuterol lowers the feedback inhibition by dopamine and delays misfolding and aggregation in tyrosine hydroxylase. Biochimie 2021, 183, 126–132. [Google Scholar] [CrossRef]
- Fletcher, E.J.R.; Jamieson, A.D.; Williams, G.; Doherty, P.; Duty, S. Targeted repositioning identifies drugs that increase fibroblast growth factor 20 production and protect against 6-hydroxydopamine-induced nigral cell loss in rats. Sci. Rep. 2019, 9, 8336. [Google Scholar] [CrossRef] [PubMed]
- Giorgianni, F.; Ernst, P.; Dell’Aniello, S.; Suissa, S.; Renoux, C. beta 2-Agonists and the Incidence of Parkinson Disease. Am. J. Epidemiol. 2020, 189, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Gronich, N.; Abernethy, D.R.; Auriel, E.; Lavi, I.; Rennert, G.; Saliba, W. beta2-adrenoceptor agonists and antagonists and risk of Parkinson’s disease. Mov. Disord. 2018, 33, 1465–1471. [Google Scholar] [CrossRef]
- Hopfner, F.; Hoglinger, G.U.; Kuhlenbaumer, G.; Pottegard, A.; Wod, M.; Christensen, K.; Tanner, C.M.; Deuschl, G. beta-adrenoreceptors and the risk of Parkinson’s disease. Lancet Neurol. 2020, 19, 247–254. [Google Scholar] [CrossRef]
- Townsend, D.J.; Mala, B.; Hughes, E.; Hussain, R.; Siligardi, G.; Fullwood, N.J.; Middleton, D.A. Circular Dichroism Spectroscopy Identifies the beta-Adrenoceptor Agonist Salbutamol As a Direct Inhibitor of Tau Filament Formation in Vitro. ACS Chem. Neurosci. 2020, 11, 2104–2116. [Google Scholar] [CrossRef] [PubMed]
- Uc, E.Y.; Lambert, C.P.; Harik, S.I.; Rodnitzky, R.L.; Evans, W.J. Albuterol improves response to levodopa and increases skeletal muscle mass in patients with fluctuating Parkinson disease. Clin. Neuropharmacol. 2003, 26, 207–212. [Google Scholar] [CrossRef]
- Alexander, G.M.; Schwartzman, R.J.; Nukes, T.A.; Grothusen, J.R.; Hooker, M.D. Beta 2-adrenergic agonist as adjunct therapy to levodopa in Parkinson’s disease. Neurology 1994, 44, 1511–1513. [Google Scholar] [CrossRef]
- Hishida, R.; Kurahashi, K.; Narita, S.; Baba, T.; Matsunaga, M. “Wearing-off” and beta 2-adrenoceptor agonist in Parkinson’s disease. Lancet 1992, 339, 870. [Google Scholar] [CrossRef]
- Baumann, A.; Jorge-Finnigan, A.; Jung-Kc, K.; Sauter, A.; Horvath, I.; Morozova-Roche, L.A.; Martinez, A. Tyrosine Hydroxylase Binding to Phospholipid Membranes Prompts Its Amyloid Aggregation and Compromises Bilayer Integrity. Sci. Rep. 2016, 6, 39488. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Komor, A.C.; Levy, J.M.; Packer, M.S.; Zhao, K.T.; Liu, D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Villiger, L.; Grisch-Chan, H.M.; Lindsay, H.; Ringnalda, F.; Pogliano, C.B.; Allegri, G.; Fingerhut, R.; Haberle, J.; Matos, J.; Robinson, M.D.; et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 2018, 24, 1519–1525. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef]
- Baruteau, J.; Waddington, S.N. Fetal gene therapy for neurodegenerative lysosomal storage diseases. J. Inherit. Metab. Dis. 2019, 42, 391–393. [Google Scholar] [CrossRef]
- Massaro, G.; Mattar, C.N.Z.; Wong, A.M.S.; Sirka, E.; Buckley, S.M.K.; Herbert, B.R.; Karlsson, S.; Perocheau, D.P.; Burke, D.; Heales, S.; et al. Fetal gene therapy for neurodegenerative disease of infants. Nat. Med. 2018, 24, 1317–1323. [Google Scholar] [CrossRef]
- Haavik, J.; Flatmark, T. Rapid and sensitive assay of tyrosine 3-monooxygenase activity by high-performance liquid chromatography using the native fluorescence of DOPA. J. Chromatogr. 1980, 198, 511–515. [Google Scholar] [CrossRef]
- Fossbakk, A.; Haavik, J. An oxygraphic method for determining kinetic properties and catalytic mechanism of aromatic amino acid hydroxylases. Anal. Biochem. 2005, 343, 100–105. [Google Scholar] [CrossRef]
- Vermeer, L.M.; Higgins, C.A.; Roman, D.L.; Doorn, J.A. Real-time monitoring of tyrosine hydroxylase activity using a plate reader assay. Anal. Biochem. 2013, 432, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Knappskog, P.M.; Flatmark, T.; Mallet, J.; Ludecke, B.; Bartholome, K. Recessively inherited L-DOPA-responsive dystonia caused by a point mutation (Q381K) in the tyrosine hydroxylase gene. Hum. Mol. Genet. 1995, 4, 1209–1212. [Google Scholar] [CrossRef]
- Swaans, R.J.; Rondot, P.; Renier, W.O.; Van Den Heuvel, L.P.; Steenbergen-Spanjers, G.C.; Wevers, R.A. Four novel mutations in the tyrosine hydroxylase gene in patients with infantile parkinsonism. Ann. Hum. Genet. 2000, 64, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Korner, G.; Noain, D.; Ying, M.; Hole, M.; Flydal, M.I.; Scherer, T.; Allegri, G.; Rassi, A.; Fingerhut, R.; Becu-Villalobos, D.; et al. Brain catecholamine depletion and motor impairment in a Th knock-in mouse with type B tyrosine hydroxylase deficiency. Brain 2015, 138, 2948–2963. [Google Scholar] [CrossRef]
- Tristan-Noguero, A.; Diez, H.; Jou, C.; Pineda, M.; Ormazabal, A.; Sanchez, A.; Artuch, R.; Garcia-Cazorla, A. Study of a fetal brain affected by a severe form of tyrosine hydroxylase deficiency, a rare cause of early parkinsonism. Metab. Brain Dis. 2016, 31, 705–709. [Google Scholar] [CrossRef]
- Szigetvari, P.D.; Muruganandam, G.; Kallio, J.P.; Hallin, E.I.; Fossbakk, A.; Loris, R.; Kursula, I.; Moller, L.B.; Knappskog, P.M.; Kursula, P.; et al. The quaternary structure of human tyrosine hydroxylase: Effects of dystonia-associated missense variants on oligomeric state and enzyme activity. J. Neurochem. 2019, 148, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Danecka, M.K.; Woidy, M.; Zschocke, J.; Feillet, F.; Muntau, A.C.; Gersting, S.W. Mapping the functional landscape of frequent phenylalanine hydroxylase (PAH) genotypes promotes personalised medicine in phenylketonuria. J. Med. Genet. 2015, 52, 175–185. [Google Scholar] [CrossRef]
- Heintz, C.; Cotton, R.G.; Blau, N. Tetrahydrobiopterin, its mode of action on phenylalanine hydroxylase, and importance of genotypes for pharmacological therapy of phenylketonuria. Hum. Mutat. 2013, 34, 927–936. [Google Scholar] [CrossRef]
- Leandro, J.; Leandro, P.; Flatmark, T. Heterotetrameric forms of human phenylalanine hydroxylase: Co-expression of wild-type and mutant forms in a bicistronic system. Biochim. Biophys. Acta 2011, 1812, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Leandro, J.; Nascimento, C.; de Almeida, I.T.; Leandro, P. Co-expression of different subunits of human phenylalanine hydroxylase: Evidence of negative interallelic complementation. Biochim. Biophys. Acta 2006, 1762, 544–550. [Google Scholar] [CrossRef][Green Version]
- Kawahata, I.; Tokuoka, H.; Parvez, H.; Ichinose, H. Accumulation of phosphorylated tyrosine hydroxylase into insoluble protein aggregates by inhibition of an ubiquitin-proteasome system in PC12D cells. J. Neural. Transm. 2009, 116, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Park, T.E.; Mustafaoglu, N.; Herland, A.; Hasselkus, R.; Mannix, R.; FitzGerald, E.A.; Prantil-Baun, R.; Watters, A.; Henry, O.; Benz, M.; et al. Hypoxia-enhanced Blood-Brain Barrier Chip recapitulates human barrier function and shuttling of drugs and antibodies. Nat. Commun. 2019, 10, 2621. [Google Scholar] [CrossRef] [PubMed]
- Jung-Klawitter, S.; Blau, N.; Sebe, A.; Ebersold, J.; Gohring, G.; Opladen, T. Generation of an iPSC line from a patient with tyrosine hydroxylase (TH) deficiency: TH-1 iPSC. Stem Cell Res. 2016, 17, 580–583. [Google Scholar] [CrossRef][Green Version]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy-Promise and Challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef]
- Palsson, B.O. Systems Biology—Constraint-Based Reconstruction and Analysis; Cambridge University Press: Cambridge, UK, 2015. [Google Scholar]
- Tummler, K.; Lubitz, T.; Schelker, M.; Klipp, E. New types of experimental data shape the use of enzyme kinetics for dynamic network modeling. FEBS J. 2014, 281, 549–571. [Google Scholar] [CrossRef]
- Osterberg, L.; Domenzain, I.; Munch, J.; Nielsen, J.; Hohmann, S.; Cvijovic, M. A novel yeast hybrid modeling framework integrating Boolean and enzyme-constrained networks enables exploration of the interplay between signaling and metabolism. PLoS Comput. Biol. 2021, 17, e1008891. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Miller, G.W.; Voit, E.O. A mathematical model of presynaptic dopamine homeostasis: Implications for schizophrenia. Pharmacopsychiatry 2008, 41 (Suppl. 1), S89–S98. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Miller, G.W.; Voit, E.O. Computational systems analysis of dopamine metabolism. PLoS ONE 2008, 3, e2444. [Google Scholar] [CrossRef] [PubMed]
- Veronneau-Veilleux, F.; Robaey, P.; Ursino, M.; Nekka, F. An integrative model of Parkinson’s disease treatment including levodopa pharmacokinetics, dopamine kinetics, basal ganglia neurotransmission and motor action throughout disease progression. J. Pharmacokinet. Pharmacodyn. 2021, 48, 133–148. [Google Scholar] [CrossRef]
- Nijhout, H.F.; Best, J.; Reed, M.C. Escape from homeostasis. Math. Biosci. 2014, 257, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, P.; Gorin, F.; Vali, S. Dynamics of tyrosine hydroxylase mediated regulation of dopamine synthesis. J. Comput. Neurosci. 2007, 22, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.G.; Bhalla, U.S.; Hellgren Kotaleski, J. Role of DARPP-32 and ARPP-21 in the Emergence of Temporal Constraints on Striatal Calcium and Dopamine Integration. PLoS Comput. Biol. 2016, 12, e1005080. [Google Scholar] [CrossRef]
- Kotaleski, J.H.; Blackwell, K.T. Modelling the molecular mechanisms of synaptic plasticity using systems biology approaches. Nat. Rev. Neurosci. 2010, 11, 239–251. [Google Scholar] [CrossRef]
- Hjorth, J.J.J.; Kozlov, A.; Carannante, I.; Frost Nylen, J.; Lindroos, R.; Johansson, Y.; Tokarska, A.; Dorst, M.C.; Suryanarayana, S.M.; Silberberg, G.; et al. The microcircuits of striatum in silico. Proc. Natl. Acad. Sci. USA 2020, 117, 9554–9565. [Google Scholar] [CrossRef]
- Schroll, H.; Hamker, F.H. Basal Ganglia dysfunctions in movement disorders: What can be learned from computational simulations. Mov. Disord. 2016, 31, 1591–1601. [Google Scholar] [CrossRef]
- Drengstig, T.; Jolma, I.W.; Ni, X.Y.; Thorsen, K.; Xu, X.M.; Ruoff, P. A basic set of homeostatic controller motifs. Biophys. J. 2012, 103, 2000–2010. [Google Scholar] [CrossRef]
- Justice, J.B., Jr.; Nicolaysen, L.C.; Michael, A.C. Modeling the dopaminergic nerve terminal. J. Neurosci. Methods 1988, 22, 239–252. [Google Scholar] [CrossRef]
- Best, J.A.; Nijhout, H.F.; Reed, M.C. Homeostatic mechanisms in dopamine synthesis and release: A mathematical model. Theor. Biol. Med. Model. 2009, 6, 21. [Google Scholar] [CrossRef]
- Reed, M.C.; Nijhout, H.F.; Best, J.A. Mathematical insights into the effects of levodopa. Front. Integr Neurosci 2012, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Cullen, M.; Wong-Lin, K. Integrated dopaminergic neuronal model with reduced intracellular processes and inhibitory autoreceptors. IET Syst. Biol. 2015, 9, 245–258. [Google Scholar] [CrossRef]
- Apweiler, R.; Beissbarth, T.; Berthold, M.R.; Bluthgen, N.; Burmeister, Y.; Dammann, O.; Deutsch, A.; Feuerhake, F.; Franke, A.; Hasenauer, J.; et al. Whither systems medicine? Exp. Mol. Med. 2018, 50, e453. [Google Scholar] [CrossRef] [PubMed]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Drager, A.; Mih, N.; Gatto, F.; Nilsson, A.; Preciat Gonzalez, G.A.; Aurich, M.K.; et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018, 36, 272–281. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nygaard, G.; Szigetvari, P.D.; Grindheim, A.K.; Ruoff, P.; Martinez, A.; Haavik, J.; Kleppe, R.; Flydal, M.I. Personalized Medicine to Improve Treatment of Dopa-Responsive Dystonia—A Focus on Tyrosine Hydroxylase Deficiency. J. Pers. Med. 2021, 11, 1186. https://doi.org/10.3390/jpm11111186
Nygaard G, Szigetvari PD, Grindheim AK, Ruoff P, Martinez A, Haavik J, Kleppe R, Flydal MI. Personalized Medicine to Improve Treatment of Dopa-Responsive Dystonia—A Focus on Tyrosine Hydroxylase Deficiency. Journal of Personalized Medicine. 2021; 11(11):1186. https://doi.org/10.3390/jpm11111186
Chicago/Turabian StyleNygaard, Gyrid, Peter D. Szigetvari, Ann Kari Grindheim, Peter Ruoff, Aurora Martinez, Jan Haavik, Rune Kleppe, and Marte I. Flydal. 2021. "Personalized Medicine to Improve Treatment of Dopa-Responsive Dystonia—A Focus on Tyrosine Hydroxylase Deficiency" Journal of Personalized Medicine 11, no. 11: 1186. https://doi.org/10.3390/jpm11111186
APA StyleNygaard, G., Szigetvari, P. D., Grindheim, A. K., Ruoff, P., Martinez, A., Haavik, J., Kleppe, R., & Flydal, M. I. (2021). Personalized Medicine to Improve Treatment of Dopa-Responsive Dystonia—A Focus on Tyrosine Hydroxylase Deficiency. Journal of Personalized Medicine, 11(11), 1186. https://doi.org/10.3390/jpm11111186