
MEDTEC Students against Coronavirus: Investigating the Role of Hemostatic Genes in the Predisposition to COVID-19 Severity

 , , , ,
, , , ,  , , , , , , ,
, , , , , , ,  ,
,  add
Show full author list
add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Cohorts for Genetic Analyses
2.2. Imputation
2.3. SNP Selection
2.4. Statistical Analysis
2.5. Polygenic Risk Score (PRS)
2.6. Meta-Analysis
2.7. Role of MEDTEC Students
3. Results
3.1. Hemostatic Gene Variants and the Predisposition to COVID-19 Severity
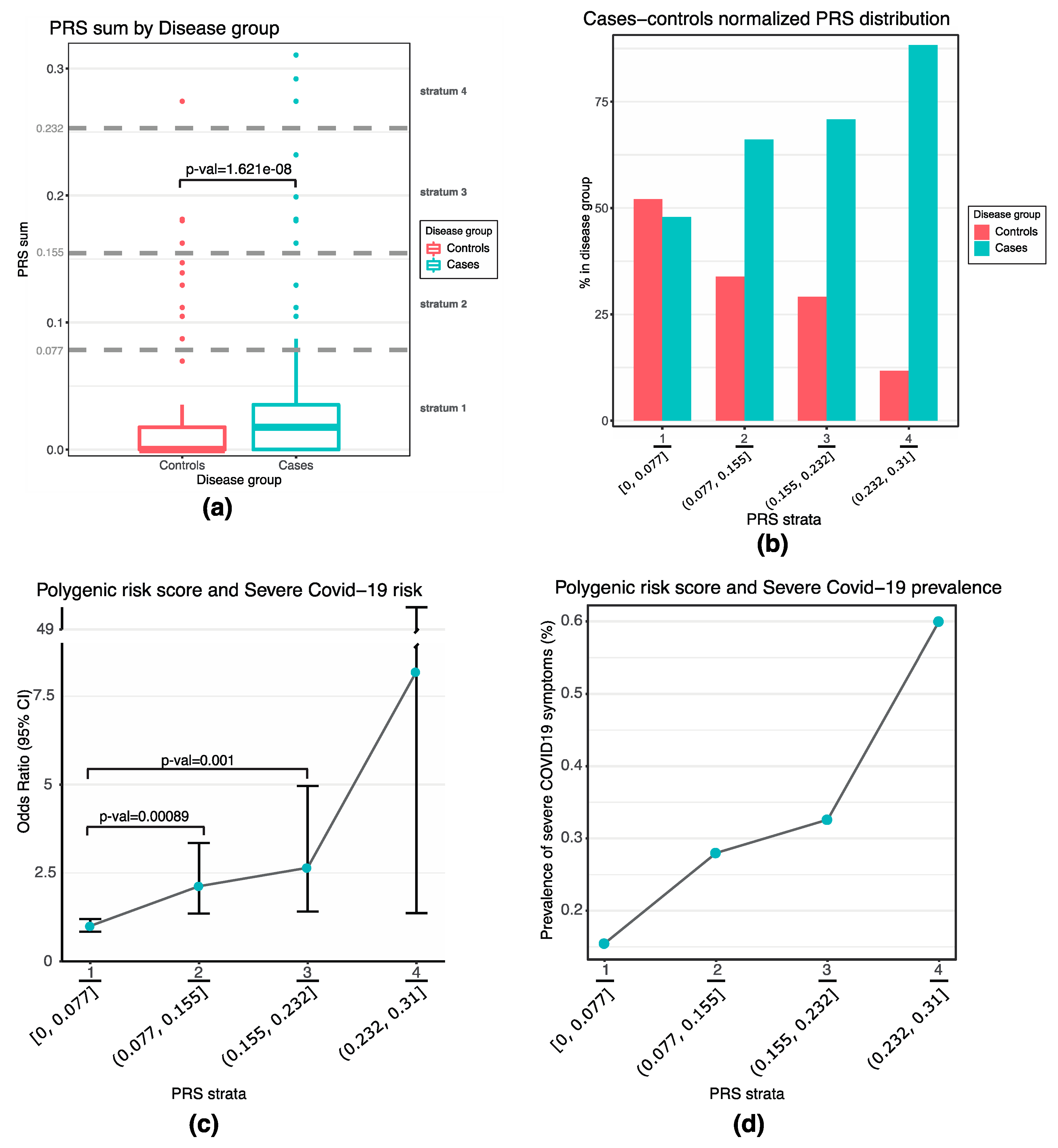
3.2. Set up of a Poligenic Risk Score Based on Hemostatic Gene Variants
3.3. Meta-Analysis Confirms the Involvement of MTHFR in Severe COVID-19 Predisposition
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johns Hopkins Coronavirus Resource Center. Available online: https://coronavirus.jhu.edu/map.html (accessed on 8 October 2021).
- Yuki, K.; Fujiogi, M.; Koutsogiannaki, S. COVID-19 pathophysiology: A review. Clin. Immunol. 2020, 215, 108427. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, M.; Ribeiro Dos Santos, G.; Wang, L.; Cummings, D.A.T.; Azman, A.S.; Paireau, J.; Fontanet, A.; Cauchemez, S.; Salje, H. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature 2021, 590, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Asselta, R.; Paraboschi, E.M.; Mantovani, A.; Duga, S. ACE2 and TMPRSS2 variants and expression as candidates to sex and country differences in COVID-19 severity in Italy. Aging 2020, 12, 10087–10098. [Google Scholar] [CrossRef] [PubMed]
- Severe COVID-19 GWAS Group; Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernández, J.; Prati, D.; Baselli, G.; et al. Genomewide Association Study of Severe COVID-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.F.; Shastri, A.J.; Ye, C.; Weldon, C.H.; Filshtein-Sonmez, T.; Coker, D.; Symons, A.; Esparza-Gordillo, J.; 23andMe COVID-19 Team; Aslibekyan, S.; et al. Trans-ancestry analysis reveals genetic and nongenetic associations with COVID-19 susceptibility and severity. Nat. Genet. 2021, 53, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef]
- Kosmicki, J.A.; Horowitz, J.E.; Banerjee, N.; Lanche, R.; Marcketta, A.; Maxwell, E.; Bai, X.; Sun, D.; Backman, J.; Sharma, D.; et al. Genetic association analysis of SARS-CoV-2 infection in 455,838 UK Biobank participants. medRxiv 2020. [Google Scholar] [CrossRef]
- Horowitz, J.E.; Kosmicki, J.A.; Damask, A.; Sharma, D.; Roberts, G.H.L.; Justice, A.E.; Banerjee, N.; Coignet, M.V.; Yadav, A.; Leader, J.B.; et al. Common genetic variants identify targets for COVID-19 and individuals at high risk of severe disease. medRxiv 2021. [Google Scholar] [CrossRef]
- COVID-19 Host Genetics Initiative. Mapping the human genetic architecture of COVID-19. Nature 2021. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- Povysil, G.; Butler-Laporte, G.; Shang, N.; Wang, C.; Khan, A.; Alaamery, M.; Nakanishi, T.; Zhou, S.; Forgetta, V.; Eveleigh, R.J.M.; et al. Rare loss-of-function variants in type I IFN immunity genes are not associated with severe COVID-19. J. Clin. Investig. 2021, 147834. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Stefely, J.A.; Christensen, B.B.; Gogakos, T.; Cone Sullivan, J.K.; Montgomery, G.G.; Barranco, J.P.; Van Cott, E.M. Marked factor V activity elevation in severe COVID-19 is associated with venous thromboembolism. Am. J. Hematol. 2020, 95, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Kipshidze, N.; Dangas, G.; White, C.J.; Kipshidze, N.; Siddiqui, F.; Lattimer, C.R.; Carter, C.A.; Fareed, J. Viral Coagulopathy in Patients With COVID-19: Treatment and Care. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620936776. [Google Scholar] [CrossRef] [PubMed]
- McGonagle, D.; O’Donnell, J.S.; Sharif, K.; Emery, P.; Bridgewood, C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2020, 2, e437–e445. [Google Scholar] [CrossRef]
- Tsivgoulis, G.; Palaiodimou, L.; Zand, R.; Lioutas, V.A.; Krogias, C.; Katsanos, A.H.; Shoamanesh, A.; Sharma, V.K.; Shahjouei, S.; Baracchini, C.; et al. COVID-19 and cerebrovascular diseases: A comprehensive overview. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420978004. [Google Scholar] [CrossRef]
- Merrill, J.T.; Erkan, D.; Winakur, J.; James, J.A. Emerging evidence of a COVID-19 thrombotic syndrome has treatment implications. Nat. Rev. Rheumatol. 2020, 16, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Estes, S.K.; Ali, R.A.; Gandhi, A.A.; Yalavarthi, S.; Shi, H.; Sule, G.; Gockman, K.; Madison, J.A.; Zuo, M.; et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci Transl. Med. 2020, 12, eabd3876. [Google Scholar] [CrossRef]
- Myocardial Infarction Genetics Consortium; Kathiresan, S.; Voight, B.F.; Purcell, S.; Musunuru, K.; Ardissino, D.; Mannucci, P.M.; Anand, S.; Engert, J.C.; Samani, N.J.; et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat. Genet. 2009, 41, 334–341. [Google Scholar] [CrossRef] [Green Version]
- TOPMed Imputation Server. Available online: https://imputation.biodatacatalyst.nhlbi.nih.gov/index.html#! (accessed on 27 April 2021).
- Taliun, D.; Harris, D.N.; Kessler, M.D.; Carlson, J.; Szpiech, Z.A.; Torres, R.; Taliun, S.A.G.; Corvelo, A.; Gogarten, S.M.; Kang, H.M.; et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 2021, 590, 290–299. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Stouffer, S.A.; Suchman, E.A.; Devinney, L.C.; Star, S.A.; Williams, R.M. Adjustment during Army Life; Princeton University Press: Princeton, NJ, USA, 1949. [Google Scholar]
- Gao, F.; Chang, D.; Biddanda, A.; Ma, L.; Guo, Y.; Zhou, Z.; Keinan, A. XWAS: A Software Toolset for Genetic Data Analysis and Association Studies of the X Chromosome. J. Hered 2015, 106, 666–671. [Google Scholar] [CrossRef] [Green Version]
- Lander, E.; Kruglyak, L. Genetic dissection of complex traits: Guidelines for interpreting and reporting linkage results. Nat. Genet. 1995, 11, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.W.; Mak, T.S.H.; O’Reilly, P.F. Tutorial: A guide to performing polygenic risk score analyses. Nat. Protoc. 2020, 15, 2759–2772. [Google Scholar] [CrossRef]
- The Covid19 Host Genetics Initiative. Available online: https://www.covid19hg.org/ (accessed on 20 May 2021).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017; Available online: https://www.R-project.org/ (accessed on 10 January 2021).
- The Regeneron–Genetic Center Database. Available online: https://rgc-covid19.regeneron.com/home (accessed on 20 July 2021).
- Mantel, N.; Haenszel, W. Statistical aspects of the analysis of data from retrospective studies of disease. J. Natl. Cancer Inst. 1959, 22, 719–748. [Google Scholar]
- Mosteller, F.; Bush, R.R. Selected quantitative techniques. In Handbook of Social Psychology; Lindzey, G., Ed.; Addison-Wesley: Cambridge, MA, USA, 1954; Volume 1, pp. 289–334. [Google Scholar]
- Liptak, T. On the combination of independent tests. Magyar. Tud Akad Mat. Kutato Int. Kozl. 1958, 3, 171–197. [Google Scholar]
- Rosendaal, F.R.; Reitsma, P.H. Genetics of venous thrombosis. J. Thromb. Haemost. 2009, 7 (Suppl. S1), 301–304. [Google Scholar] [CrossRef]
- Nyholt, D.R. A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am. J. Hum. Genet. 2004, 74, 765–769. [Google Scholar] [CrossRef] [Green Version]
- Nicodemus, K.K.; Liu, W.; Chase, G.A.; Tsai, Y.Y.; Fallin, M.D. Comparison of type I error for multiple test corrections in large single-nucleotide polymorphism studies using principal components versus haplotype blocking algorithms. BMC Genet. 2005, 6 (Suppl. S1), S78. [Google Scholar] [CrossRef] [Green Version]
- Genotype-Tissue Expression-GTEx Portal. Available online: https://gtexportal.org/home/locusBrowserPage/FGA (accessed on 1 September 2021).
- Atlas of GWAS Summary Statistics. Available online: https://atlas.ctglab.nl/ (accessed on 1 September 2021).
- Zhu, Z.; Wang, X.; Li, X.; Lin, Y.; Shen, S.; Liu, C.L.; Hobbs, B.D.; Hasegawa, K.; Liang, L.; International COPD Genetics Consortium; et al. Genetic overlap of chronic obstructive pulmonary disease and cardiovascular disease-related traits: A large-scale genome-wide cross-trait analysis. Respir Res. 2019, 20, 64. [Google Scholar] [CrossRef]
- Watanabe, K.; Stringer, S.; Frei, O.; Umićević Mirkov, M.; de Leeuw, C.; Polderman, T.J.C.; van der Sluis, S.; Andreassen, O.A.; Neale, B.M.; Posthuma, D. A global overview of pleiotropy and genetic architecture in complex traits. Nat. Genet. 2019, 51, 1339–1348. [Google Scholar] [CrossRef]
- Wu, Y.; Byrne, E.M.; Zheng, Z.; Kemper, K.E.; Yengo, L.; Mallett, A.J.; Yang, J.; Visscher, P.M.; Wray, N.R. Genome-wide association study of medication-use and associated disease in the UK Biobank. Nat. Commun. 2019, 10, 1891. [Google Scholar] [CrossRef] [Green Version]
- Astle, W.J.; Elding, H.; Jiang, T.; Allen, D.; Ruklisa, D.; Mann, A.L.; Mead, D.; Bouman, H.; Riveros-Mckay, F.; Kostadima, M.A.; et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell 2016, 167, 1415–1429.e19. [Google Scholar] [CrossRef] [Green Version]
- International Consortium for Blood Pressure Genome-Wide Association Studies. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 2011, 478, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Guo, R.; Kim, S.H.; Shah, H.; Zhang, S.; Liang, J.H.; Fang, Y.; Gentili, M.; Leary, C.N.O.; Elledge, S.J.; et al. SARS-CoV-2 hijacks folate and one-carbon metabolism for viral replication. Nat. Commun. 2021, 12, 1676. [Google Scholar] [CrossRef]
- Sharma, P.; Senthilkumar, R.D.; Brahmachari, V.; Sundaramoorthy, E.; Mahajan, A.; Sharma, A.; Sengupta, S. Mining literature for a comprehensive pathway analysis: A case study for retrieval of homocysteine related genes for genetic and epigenetic studies. Lipids Health Dis. 2006, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Karst, M.; Hollenhorst, J.; Achenbach, J. Life-threatening course in coronavirus disease 2019 (COVID-19): Is there a link to methylenetetrahydrofolic acid reductase (MTHFR) polymorphism and hyperhomocysteinemia? Med. Hypotheses 2020, 144, 110234. [Google Scholar] [CrossRef]
- Ponti, G.; Pastorino, L.; Manfredini, M.; Ozben, T.; Oliva, G.; Kaleci, S.; Iannella, R.; Tomasi, A. COVID-19 spreading across world correlates with C677T allele of the methylenetetrahydrofolate reductase (MTHFR) gene prevalence. J. Clin. Lab. Anal. 2021, 35, e23798. [Google Scholar] [CrossRef] [PubMed]
- Ponti, G.; Roli, L.; Oliva, G.; Manfredini, M.; Trenti, T.; Kaleci, S.; Iannella, R.; Balzano, B.; Coppola, A.; Fiorentino, G.; et al. Homocysteine (Hcy) assessment to predict outcomes of ho-spitalized COVID-19 patients: A multicenter study on 313 Covid-19 patients. Clin. Chem. Lab. Med. 2021, 59, e354–e357. [Google Scholar] [CrossRef]
- Durand, P.; Lussier-Cacan, S.; Blache, D. Acute methionine load-induced hyperhomocysteinemia enhances platelet aggregation, thromboxane biosynthesis, and macrophage-derived tissue factor activity in rats. FASEB J. 1997, 11, 1157–1168. [Google Scholar] [CrossRef] [PubMed]
- Fryer, R.H.; Wilson, B.D.; Gubler, D.B.; Fitzgerald, L.A.; Rodgers, G.M. Homocysteine, a risk factor for premature vascular disease and thrombosis, induces tissue factor activity in endothelial cells. Arterioscler. Thromb. 1993, 13, 1327–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Cao, W.; Jiang, W.; Xiao, M.; Li, Y.; Tang, N.; Liu, Z.; Yan, X.; Zhao, Y.; Li, T.; et al. Profile of natural anticoagulant, coagulant factor and anti-phospholipid antibody in critically ill COVID-19 patients. J. Thromb. Thrombolysis 2020, 50, 580–586. [Google Scholar] [CrossRef]
- Stanne, T.M.; Pedersen, A.; Gisslén, M.; Jern, C. Low admission protein C levels are a risk factor for disease worsening and mortality in hospitalized patients with COVID-19. Thromb. Res. 2021, 204, 13–15. [Google Scholar] [CrossRef]
- Corrêa, T.D.; Cordioli, R.L.; Campos Guerra, J.C.; Caldin da Silva, B.; Dos Reis Rodrigues, R.; de Souza, G.M.; Midega, T.D.; Campos, N.S.; Carneiro, B.V.; Campos, F.N.D.; et al. Coagulation profile of COVID-19 patients admitted to the ICU: An exploratory study. PLoS ONE 2020, 15, e0243604. [Google Scholar] [CrossRef]
- Panigada, M.; Bottino, N.; Tagliabue, P.; Grasselli, G.; Novembrino, C.; Chantarangkul, V.; Pesenti, A.; Peyvandi, F.; Tripodi, A. Hypercoagulability of COVID-19 patients in intensive care unit: A report of thromboelastography findings and other parameters of hemostasis. J. Thromb. Haemost. 2020, 18, 1738–1742. [Google Scholar] [CrossRef]
- Hardy, M.; Michaux, I.; Lessire, S.; Douxfils, J.; Dogné, J.M.; Bareille, M.; Horlait, G.; Bulpa, P.; Chapelle, C.; Laporte, S.; et al. Prothrombotic hemostasis disturbances in patients with severe COVID-19: Individual daily data. Data Brief. 2020, 33, 106519. [Google Scholar] [CrossRef]
- Cao, W.J.; Niiya, M.; Zheng, X.W.; Shang, D.Z.; Zheng, X.L. Inflammatory cytokines inhibit ADAMTS13 synthesis in hepatic stellate cells and endothelial cells. J. Thromb. Haemost. 2008, 6, 1233–1235. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, A.; Ball, C.; Nolasco, L.; Moake, J.F.; Dong, J.F. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood 2004, 104, 100–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Ward, S.E.; Curley, G.F.; Lavin, M.; Fogarty, H.; Karampini, E.; McEvoy, N.L.; Clarke, J.; Boylan, M.; Alalqam, R.; Worrall, A.P.; et al. Von Willebrand factor propeptide in severe coronavirus disease 2019 (COVID-19): Evidence of acute and sustained endothelial cell activation. Br. J. Haematol. 2021, 192, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Fagot Gandet, F.; et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020, 46, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, F.; Artoni, A.; Novembrino, C.; Aliberti, S.; Panigada, M.; Boscarino, M.; Gualtierotti, R.; Rossi, F.; Palla, R.; Martinelli, I.; et al. Hemostatic alterations in COVID-19. Haematologica 2021, 106, 1472–1475. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.E.; Fogarty, H.; Karampini, E.; Lavin, M.; Schneppenheim, S.; Dittmer, R.; Morrin, H.; Glavey, S.; Ni Cheallaigh, C.; Bergin, C.; et al. ADAMTS13 regulation of VWF multimer distribution in severe COVID-19. J. Thromb. Haemost. 2021, 19, 1914–1921. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, N.; Montagnana, M.; Pizzolo, F.; Friso, S.; Salvagno, G.L.; Forni, G.L.; Gianesin, B.; Morandi, M.; Lunardi, C.; Lippi, G.; et al. A relative ADAMTS13 deficiency supports the presence of a secondary microangiopathy in COVID 19. Thromb. Res. 2020, 193, 170–172. [Google Scholar] [CrossRef]
- Philippe, A.; Gendron, N.; Bory, O.; Beauvais, A.; Mirault, T.; Planquette, B.; Sanchez, O.; Diehl, J.L.; Chocron, R.; Smadja, D.M. Von Willebrand factor collagen-binding capacity predicts in-hospital mortality in COVID-19 patients: Insight from VWF/ADAMTS13 ratio imbalance. Angiogenesis 2021, 24, 407–411. [Google Scholar] [CrossRef]
- Marco, A.; Marco, P. Von Willebrand factor and ADAMTS13 activity as clinical severity markers in patients with COVID-19. J. Thromb. Thrombolysis 2021, 1–7. [Google Scholar] [CrossRef]
- Sweeney, J.M.; Barouqa, M.; Krause, G.J.; Gonzalez-Lugo, J.D.; Rahman, S.; Gil, M.R. Low ADAMTS13 Activity Correlates with Increased Mortality in COVID-19 Patients. TH Open 2021, 5, e89–e103. [Google Scholar] [CrossRef] [PubMed]
- Pascreau, T.; Zia-Chahabi, S.; Zuber, B.; Tcherakian, C.; Farfour, E.; Vasse, M. ADAMTS 13 deficiency is associated with abnormal distribution of von Willebrand factor multimers in patients with COVID-19. Thromb. Res. 2021, 204, 138–140. [Google Scholar] [CrossRef] [PubMed]
- De Bruijn, S.; Maes, M.B.; De Waele, L.; Vanhoorelbeke, K.; Gadisseur, A. First report of a de novo iTTP episode associated with an mRNA-based anti-COVID-19 vaccination. J. Thromb. Haemost. 2021, 19, 2014–2018. [Google Scholar] [CrossRef] [PubMed]
- Yocum, A.; Simon, E.L. Thrombotic Thrombocytopenic Purpura after Ad26.COV2-S Vaccination. Am. J. Emerg Med. 2021, 49, 441.e3–441.e4. [Google Scholar] [CrossRef]
- Albánez, S.; Ogiwara, K.; Michels, A.; Hopman, W.; Grabell, J.; James, P.; Lillicrap, D. Aging and ABO blood type influence von Willebrand factor and factor VIII levels through interrelated mechanisms. J. Thromb. Haemost. 2016, 14, 953–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Chr | Gene | Protein | Gene Size (kb) | Analyzed Region (kb) | Coordinates of the Analyzed Region (hg38) | Analyzed SNPs (n) |
|---|---|---|---|---|---|---|
| 1 | MTHFR | MTHR | 20.3 | 520.3 | 1:11535729-12056103 | 1821 |
| 1 | F5 | FA5 (FV) | 74.5 | 574.5 | 1:169261953-169836531 | 2276 |
| 1 | F13B | F13B (FXIII) | 28.0 | 528 | 1:196789190-197317267 | 956 |
| 1 | MTR | METH | 108.7 | 608.7 | 1:236545280-237153981 | 2270 |
| 2 | TFPI | TFPI | 90.2 | 590.2 | 2:187214230-187804492 | 1655 |
| 2 | PROC | PROC | 10.8 | 510.8 | 2:127168419-127679246 | 1492 |
| 3 | PROS1 | PROS | 101.0 | 601 | 3:93623036-94224090 | 592 |
| 4 | FGB | FIBB | 49.7 | 549.7 | 4:154312979-154862750 | 1606 |
| 4 | FGA | |||||
| 4 | FGG | |||||
| 4 | F11 | FA11 (FXI) | 23.7 | 523.7 | 4:186015963-186539681 | 2251 |
| 5 | ITGA2 | ITA2 (GPIa) | 105.4 | 605.4 | 5:52739325-53344779 | 2469 |
| 5 | THBS4 | TSP4 | 47.9 | 547.9 | 5:79785348-80333284 | 2047 |
| 5 | F12 | FA12 (FXII) | 7.4 | 507.4 | 5:177152137-177659576 | 890 |
| 6 | F13A1 | F13A (FXIII) | 176.6 | 676.6 | 6:5894077-6570691 | 2895 |
| 6 | THBS2 | TSP2 | 38.3 | 538.3 | 6:168965779-169504114 | 2080 |
| 7 | CD36 | CD36 (GPIIIb) | 304.8 | 804.8 | 7:80119575-80924418 | 2110 |
| 7 | SERPINE1 | PAI-1 | 12.1 | 512.1 | 7:100877088-101389266 | 1929 |
| 8 | PLAT | TPA | 32.9 | 532.9 | 8:41924717-42457676 | 927 |
| 9 | ADAMTS13 | ATS13 (ADAMTS13) | 37.4 | 537.4 | 9:133171999-133709403 | 2195 |
| 11 | F2 | THRB | 20.3 | 520.3 | 11:46469192-46989506 | 697 |
| 12 | VWF | VWF | 175.7 | 675.7 | 12:5698873-6374670 | 2122 |
| 13 | CPB2 | CBPB2 (TAFI) | 51.8 | 551.8 | 13:45803186-46355076 | 1811 |
| 13 | F7 | FA7 (FVII) | 14.8 | 514.8 | 13:112855787-113370681 | 1411 |
| 13 | F10 | FA10 (FX) | 26.7 | 526.7 | 13:112872798-113399529 | 1540 |
| 15 | THBS1 | TSP1 | 17.8 | 517.8 | 15:39331078-39848921 | 1443 |
| 17 | GPIBA | GPIBA | 2.7 | 502.7 | 17:4682274-5185030 | 1502 |
| 17 | ITGB3 | ITB3 (GPIIIa) | 58.8 | 558.8 | 17:47003841-47562711 | 1695 |
| 18 | LMAN1 | LMAN1 | 31.4 | 531.4 | 18:59077823-59609276 | 2327 |
| 20 | THBD | TRBM | 4.0 | 504 | 20:22795632-23299664 | 1488 |
| X | F8 | FA8 (FVIII) | 186.9 | 686.9 | 23:154585788-155272723 | 570 |
| X | F9 | FA9 (FIX) | 32.7 | 532.7 | 23:139280735-139813458 | 778 |
| Total | 32 genes | - | 1893 | 16,893 | - | 49,845 |
| (a) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Uncorrected Analyses | Corrected Analyses | ||||||||
| Chr | Gene | SNP | MAF Cases | MAF Controls | p Value | OR (95%CI) | p Value | OR (95%CI) | |
| 2 | PROC | chr2:127192625:G:A | 0.065 | 0.037 | 8.51 × 10−4 | 1.82 (1.28–2.61) | 8.77 × 10−5 | 2.23 (1.50–3.34) | |
| 1 | MTHFR | chr1:11753033:G:A | 0.247 | 0.183 | 1.33 × 10−4 | 1.47 (1.20–1.79) | 1.08 × 10−4 | 1.56 (1.25–1.95) | |
| 1 | MTR | chr1:237145686:A:G | 0.023 | 0.010 | 4.53 × 10−3 | 2.39 (1.29–4.43) | 3.14 × 10−4 | 3.49 (1.77–6.88) | |
| 9 | ADAMTS13 | chr9:133179750:G:C | 0.030 | 0.011 | 1.58 × 10−4 | 2.77 (1.60–4.80) | 4.26 × 10−4 | 2.90 (1.60–5.24) | |
| 6 | THBS2 | chr6:169195156:A:T | 0.039 | 0.021 | 6.25 × 10−3 | 1.87 (1.19–2.96) | 7.84 × 10−4 | 2.35 (1.43–3.87) | |
| 17 | ITGB3 | chr17:47019591:G:C | 0.321 | 0.270 | 7.05 × 10−3 | 1.28 (1.07–1.53) | 1.41 × 10−3 | 1.39 (1.14–1.71) | |
| 7 | SERPINE1 | chr7:101070945:A:G | 0.324 | 0.267 | 2.55 × 10−3 | 1.32 (1.10–1.58) | 1.73 × 10−3 | 1.38 (1.13–1.69) | |
| 6 | F13A1 | chr6:6163858:G:C | 0.108 | 0.070 | 6.99 × 10−4 | 1.61 (1.22–2.13) | 1.80 × 10−3 | 1.67 (1.21–2.31) | |
| 18 | LMAN1 | chr18:59208206:A:G | 0.401 | 0.337 | 1.57 × 10−3 | 1.32 (1.11–1.56) | 1.81 × 10−3 | 1.36 (1.12–1.65) | |
| 4 | F11 | chr4:186056516:A:G | 0.065 | 0.106 | 1.31 × 10−3 | 0.59 (0.42–0.82) | 1.84 × 10−3 | 0.56 (0.39–0.81) | |
| 7 | CD36 | chr7:80591832:AAATCAGC:A | 0.039 | 0.021 | 6.25 × 10−3 | 1.87 (1.19–2.96) | 1.98 × 10−3 | 2.15 (1.33–3.50) | |
| 15 | THBS1 | chr15:39455553:G:C | 0.178 | 0.133 | 2.17 × 10−3 | 1.42 (1.13–1.77) | 2.50 × 10−3 | 1.46 (1.14–1.86) | |
| 12 | VWF | chr12:6068637:T:C | 0.054 | 0.031 | 3.71 × 10−3 | 1.76 (1.20–2.60) | 2.50 × 10−3 | 1.95 (1.27–3.01) | |
| 13 | F7–F10 | chr13:113257337:C:T | 0.288 | 0.234 | 3.50 × 10−3 | 1.32 (1.10–1.59) | 3.13 × 10−3 | 1.37 (1.11–1.69) | |
| 20 | THBD | chr20:22949512:A:G | 0.048 | 0.025 | 1.51 × 10−3 | 1.94 (1.28–2.93) | 3.18 × 10−3 | 2.01 (1.26–3.19) | |
| 4 | FG_genes | chr4:154774926:G:A | 0.054 | 0.033 | 8.81 × 10−3 | 1.67 (1.13–2.45) | 4.45 × 10−3 | 1.88 (1.22–2.91) | |
| 1 | F13B | chr1:196913749:A:AT | 0.036 | 0.017 | 1.84 × 10−3 | 2.12 (1.31–3.44) | 5.04 × 10−3 | 2.12 (1.25–3.59) | |
| 13 | CPB2 | chr13:45931697:A:G | 0.008 | 0.026 | 3.59 × 10−3 | 0.28 (0.12–0.70) | 5.29 × 10−3 | 0.18 (0.056–0.61) | |
| 8 | PLAT | chr8:42278531:C:T | 0.017 | 0.007 | 1.79 × 10−2 | 2.33 (1.13–4.77) | 5.69 × 10−3 | 2.88 (1.36–6.08) | |
| 5 | ITGA2 | chr5:53162220:CAGAG:C | 0.029 | 0.013 | 4.56 × 10−3 | 2.15 (1.25–3.71) | 7.60 × 10−3 | 2.30 (1.25–4.23) | |
| 2 | TFPI | chr2:187303697:A:G | 0.030 | 0.013 | 1.13 × 10−3 | 2.38 (1.39–4.07) | 8.74 × 10−3 | 2.25 (1.23–4.14) | |
| 1 | F5 | chr1:169677905:G:A | 0.116 | 0.151 | 1.90 × 10−2 | 0.74 (0.57–0.95) | 9.74 × 10−3 | 0.68 (0.51–0.91) | |
| 5 | THBS4 | chr5:80174425:G:T | 0.203 | 0.163 | 1.23 × 10−2 | 1.31 (1.06–1.61) | 9.83 × 10−3 | 1.37 (1.08–1.73) | |
| 3 | PROS1 | chr3:94043799:A:C | 0.032 | 0.014 | 1.84 × 10−3 | 2.24 (1.33–3.76) | 1.19 × 10−2 | 2.10 (1.18–3.74) | |
| 11 | F2 | chr11:46517560:T:C | 0.331 | 0.382 | 1.39 × 10−2 | 0.80 (0.63–0.96) | 1.19 × 10−2 | 0.77 (0.63–0.95) | |
| 17 | GP1BA | chr17:4921551:T:C | 0.020 | 0.010 | 2.59 × 10−2 | 2.06 (1.08–3.95) | 1.31 × 10−2 | 2.50 (1.21–5.15) | |
| 5 | F12 | chr5:177464930:A:G | 0.026 | 0.013 | 1.71 × 10−2 | 1.97 (1.12–3.46) | 1.38 × 10−2 | 2.17 (1.17–4.03) | |
| (b) | |||||||||
| Chr | Gene | SNP | pValue_M | OR (95%CI)_M | pValue_F | OR (95%CI)_F | P_Comb_Stouffer | ||
| X | F9 | chrX:139407485:G:C | 8.5 × 10−4 | 1.27 (0.96–1.68) | 0.49 | 0.91 (0.63–1.31) | 0.0046 | ||
| Corrected Analyses | ||||||
|---|---|---|---|---|---|---|
| Chr | Gene | SNP | SNP ID | Minor Allele | p Value | OR (95%CI) |
| 1 | F5 | chr1:169549811:C:T | rs6025 | T | 0.92 | 0.96 (0.48–1.94) |
| 6 | F13A1 | chr6:6318562:C:A | rs5985 | A | 0.91 | 0.99 (0.78–1.24) |
| 11 | F2 | chr11:46739505:G:A | rs1799963 | A | 0.015 | 0.23 (0.070–0.75) |
| Italians (Our Cohort) | Regeneron Cohort | Meta-Analysis | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | SNP | Analysed Individuals (n) | p Value | OR (95%CI) | Analysed Individuals (n) | p Value | OR (95%CI) | Pooled OR (95%CI) | p Value (M-H) | p Value (F Weighted) |
| PROC | chr2:127192625:G:A | 2000 | 8.51 × 10−4 | 1.82 (1.28–2.61) | 429151 | 0.026 | 0.71 (0.53–0.96) | 0.98 (0.79–1.22) | 0.87 | 0.12 |
| MTHFR | chr1:11753033:G:A | 2000 | 1.33 × 10−4 | 1.47 (1.20–1.79) | 654056 | 0.030 | 1.13 (1.01–1.28) | 1.21 (1.09–1.33) | 2.55 × 10−4 | 4.34 × 10−14 |
| MTR | chr1:237145686:A:G | 2000 | 4.53 × 10−3 | 2.39 (1.29–4.43) | 898324 | 0.98 | 1.00 (0.88–1.14) | 1.03 (0.91–1.18) | 0.61 | 0.0027 |
| ADAMTS13 | chr9:133179750:G:C | 2000 | 1.58 × 10−4 | 2.77 (1.60–4.80) | 656078 | 0.030 | 0.75 (0.58–0.97) | 0.89 (0.72–1.11) | 0.33 | 0.056 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cappadona, C.; Paraboschi, E.M.; Ziliotto, N.; Bottaro, S.; Rimoldi, V.; Gerussi, A.; Azimonti, A.; Brenna, D.; Brunati, A.; Cameroni, C.; et al. MEDTEC Students against Coronavirus: Investigating the Role of Hemostatic Genes in the Predisposition to COVID-19 Severity. J. Pers. Med. 2021, 11, 1166. https://doi.org/10.3390/jpm11111166
Cappadona C, Paraboschi EM, Ziliotto N, Bottaro S, Rimoldi V, Gerussi A, Azimonti A, Brenna D, Brunati A, Cameroni C, et al. MEDTEC Students against Coronavirus: Investigating the Role of Hemostatic Genes in the Predisposition to COVID-19 Severity. Journal of Personalized Medicine. 2021; 11(11):1166. https://doi.org/10.3390/jpm11111166
Chicago/Turabian StyleCappadona, Claudio, Elvezia Maria Paraboschi, Nicole Ziliotto, Sandro Bottaro, Valeria Rimoldi, Alessio Gerussi, Andrea Azimonti, Daniele Brenna, Andrea Brunati, Charlotte Cameroni, and et al. 2021. "MEDTEC Students against Coronavirus: Investigating the Role of Hemostatic Genes in the Predisposition to COVID-19 Severity" Journal of Personalized Medicine 11, no. 11: 1166. https://doi.org/10.3390/jpm11111166
APA StyleCappadona, C., Paraboschi, E. M., Ziliotto, N., Bottaro, S., Rimoldi, V., Gerussi, A., Azimonti, A., Brenna, D., Brunati, A., Cameroni, C., Campanaro, G., Carloni, F., Cavadini, G., Ciravegna, M., Composto, A., Converso, G., Corbella, P., D’Eugenio, D., Dal Rì, G., ... Asselta, R. (2021). MEDTEC Students against Coronavirus: Investigating the Role of Hemostatic Genes in the Predisposition to COVID-19 Severity. Journal of Personalized Medicine, 11(11), 1166. https://doi.org/10.3390/jpm11111166