
Protein Aggregation of NPAS3, Implicated in Mental Illness, Is Not Limited to the V304I Mutation

 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Brain Samples
2.2. Human Blood Samples
2.3. Insoluble Protein Purification from Brain
2.4. Serum Samples and Insoluble Protein Purification from Serum
2.5. Plasmids
2.6. Mammalian Cell Culture
2.7. Insoluble Protein Purification from Mammalian Cell Lysates
2.8. Recombinant Protein Expression in Bacteria
2.9. Western Blotting and Analysis
2.10. Immunocytochemistry and Microscopy
2.11. Statistical Analysis
3. Results
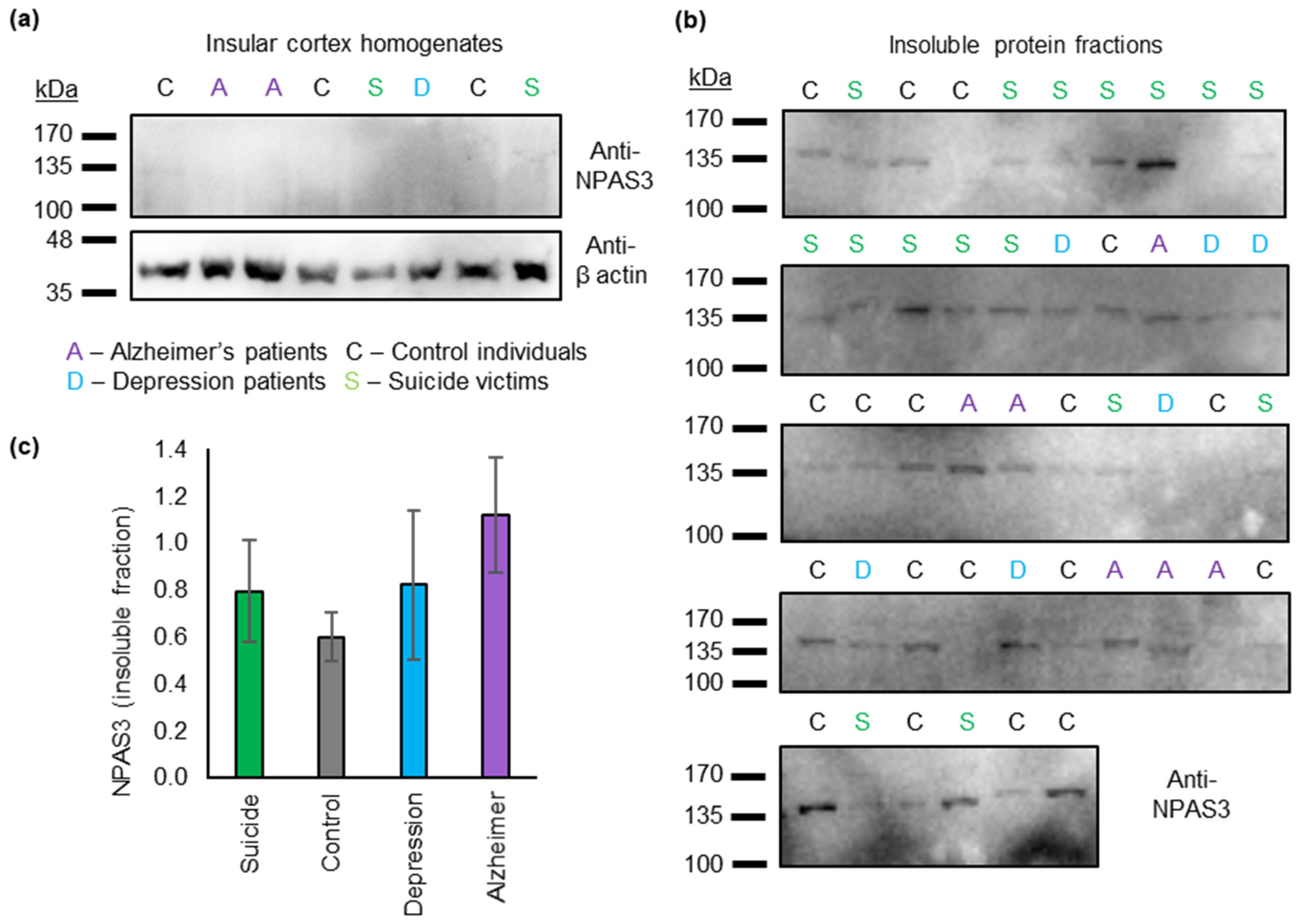
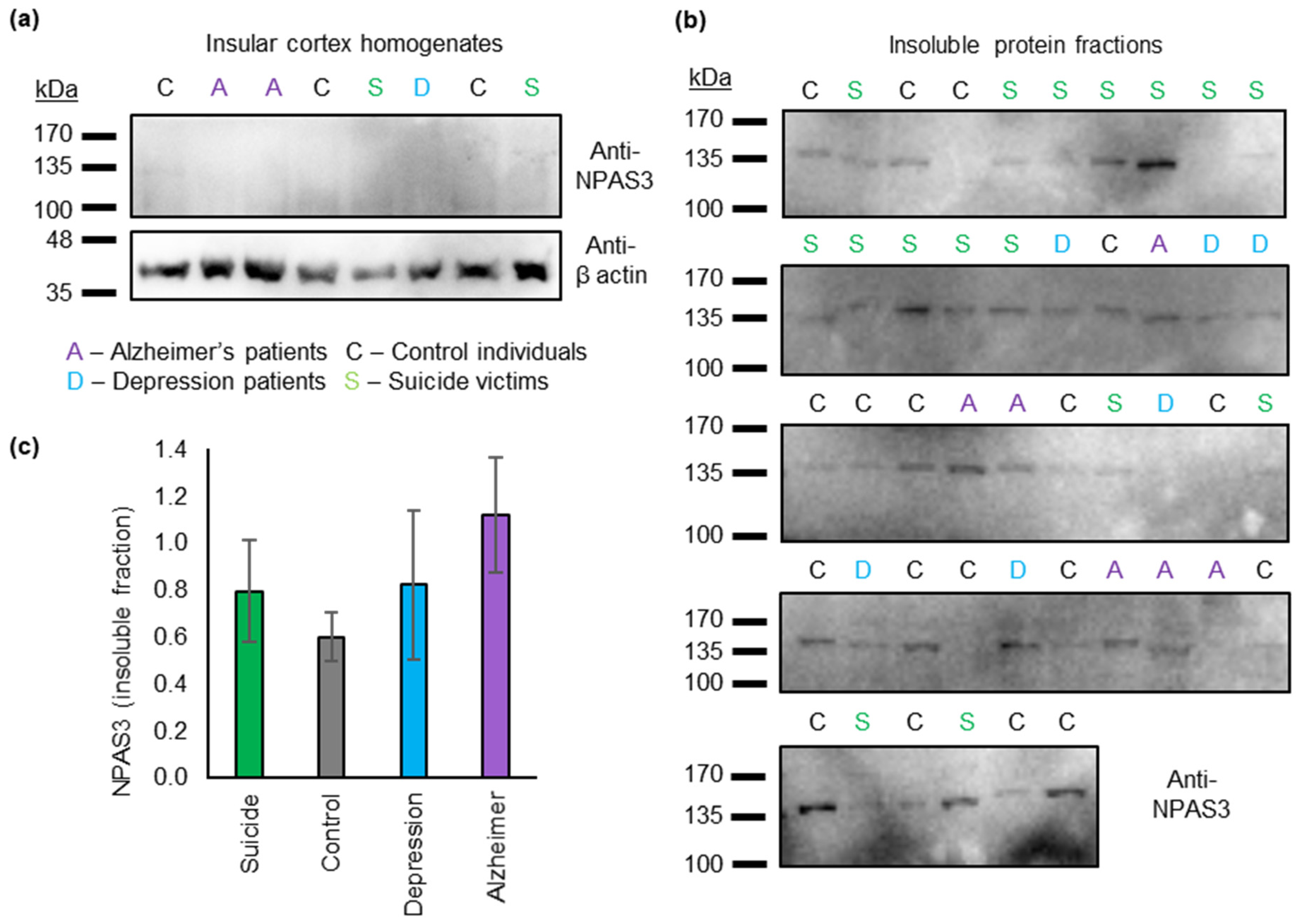
3.1. Insoluble NPAS3 Is Common in Human Post Mortem Insular Cortex
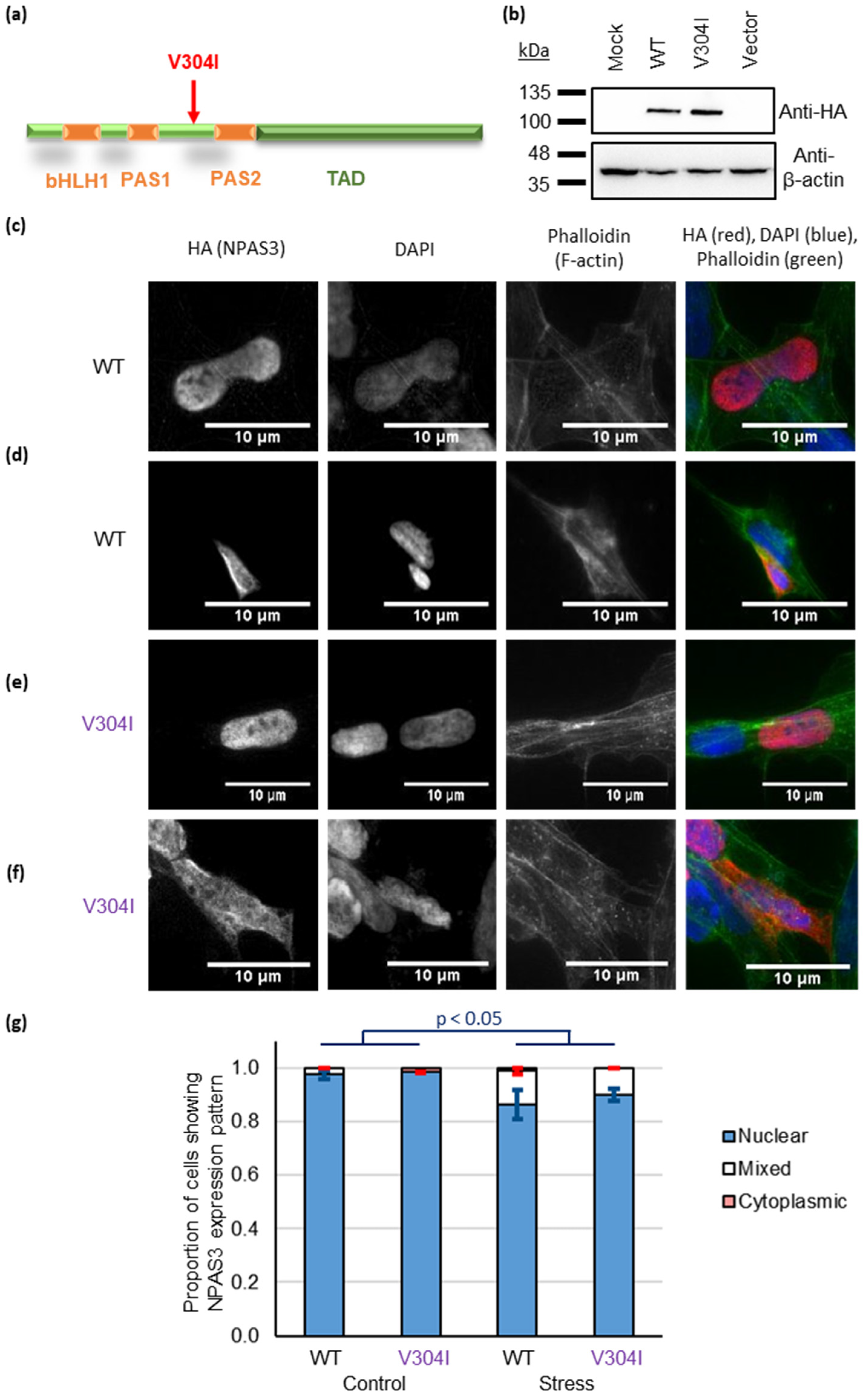
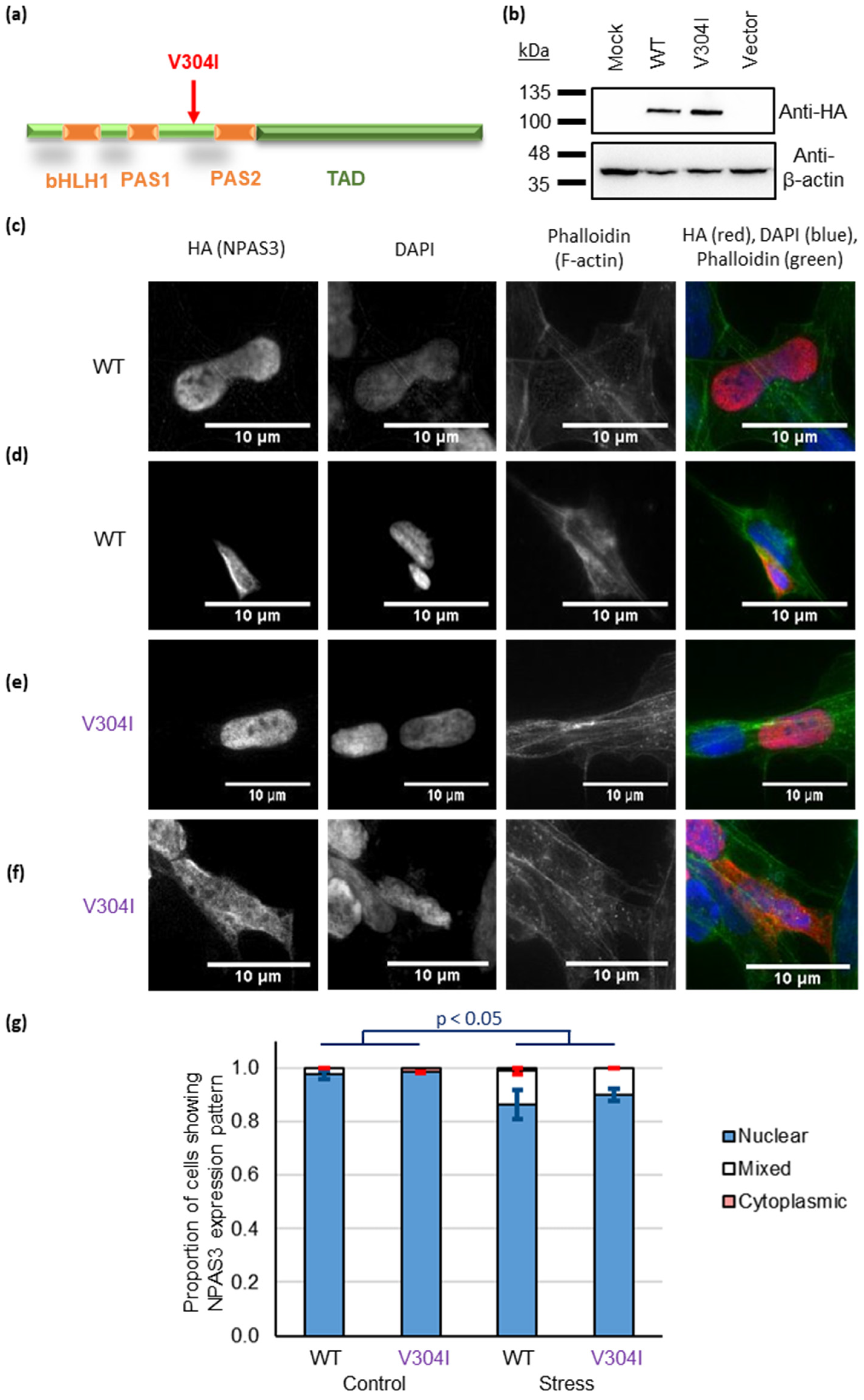
3.2. NPAS3 Aggregation in Cultured Cells Can Derive from Oxidative Stress and the V304I Mutation
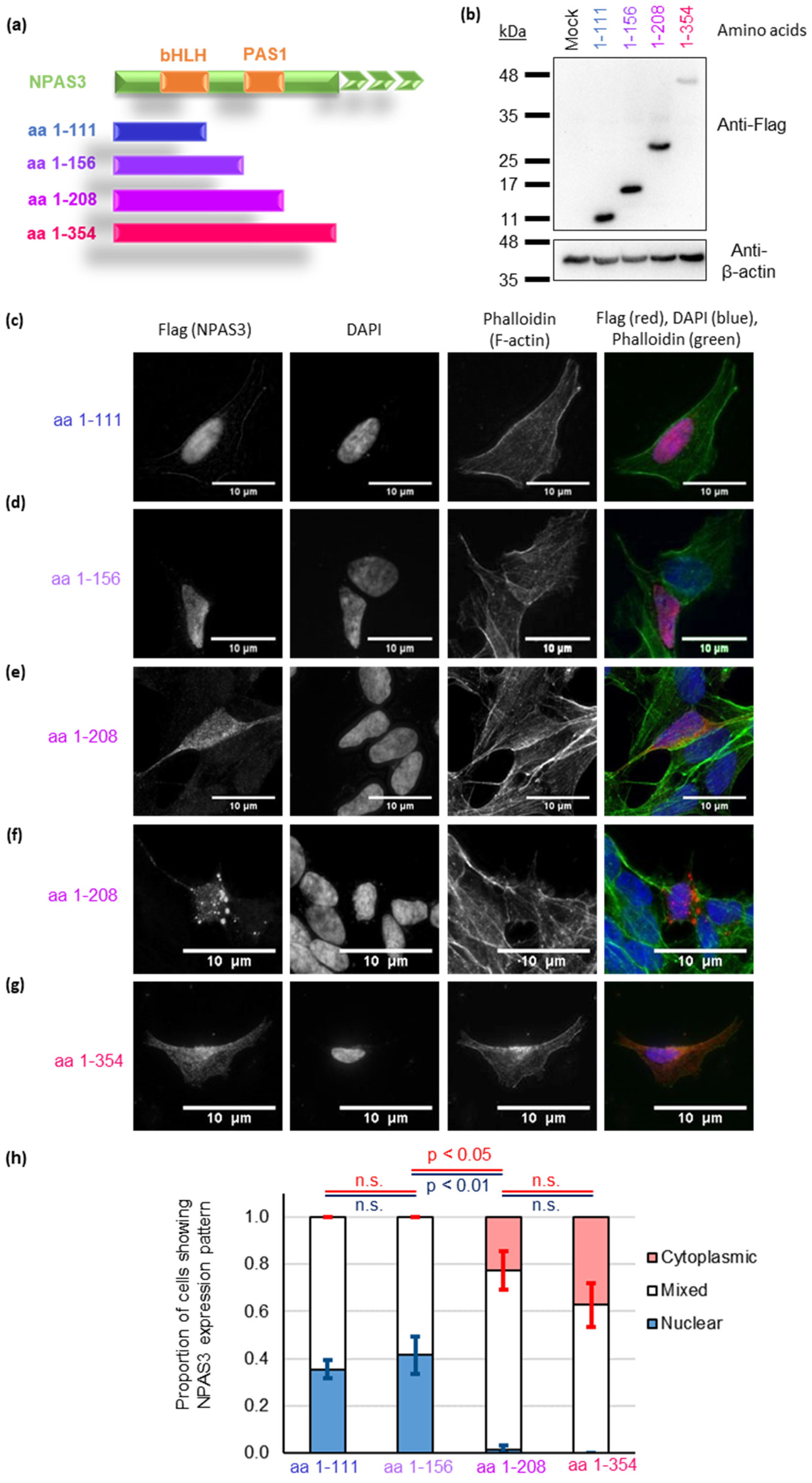
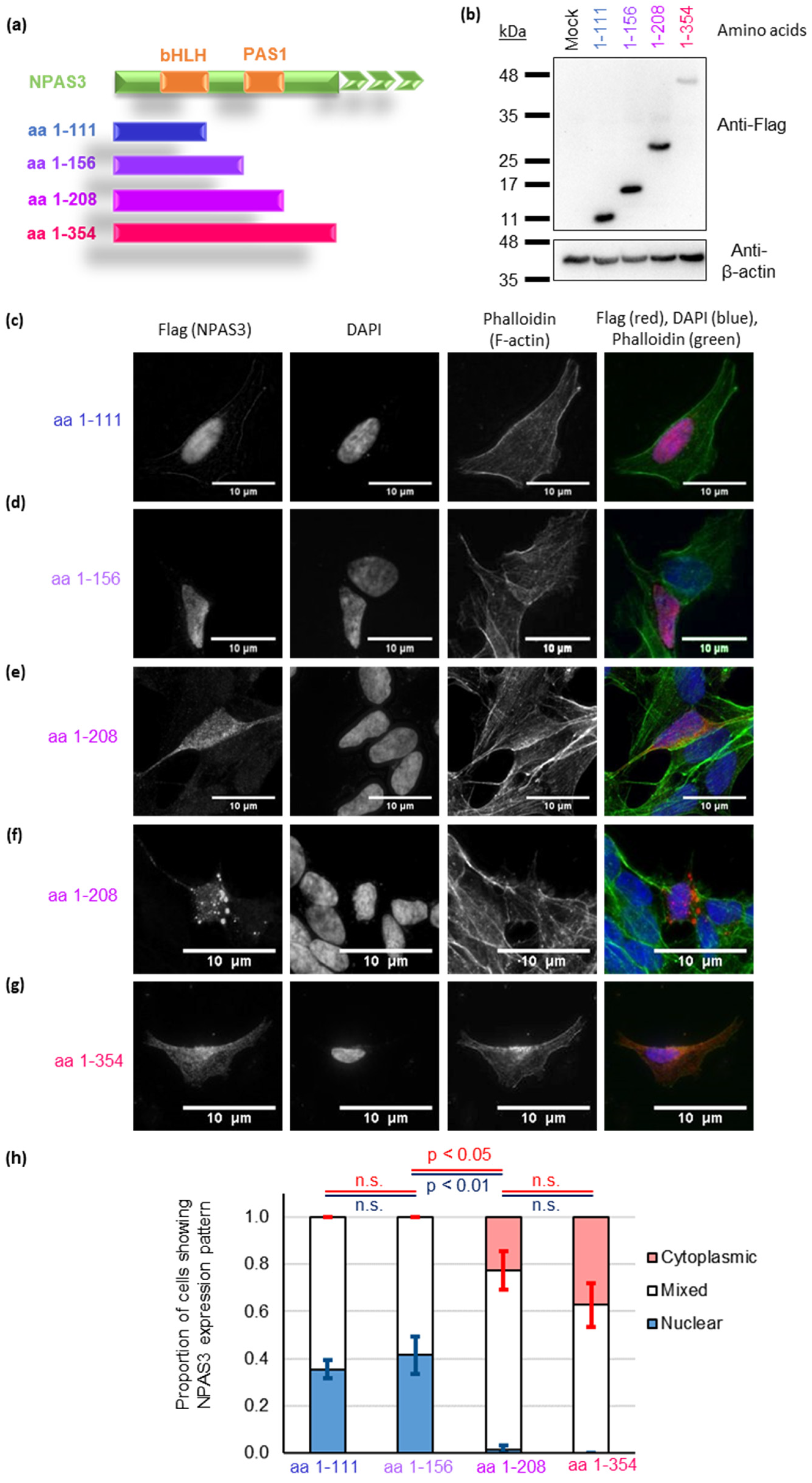
3.3. NPAS3 Aggregation in Cultured Cells Is Driven by Its PAS1 Domain
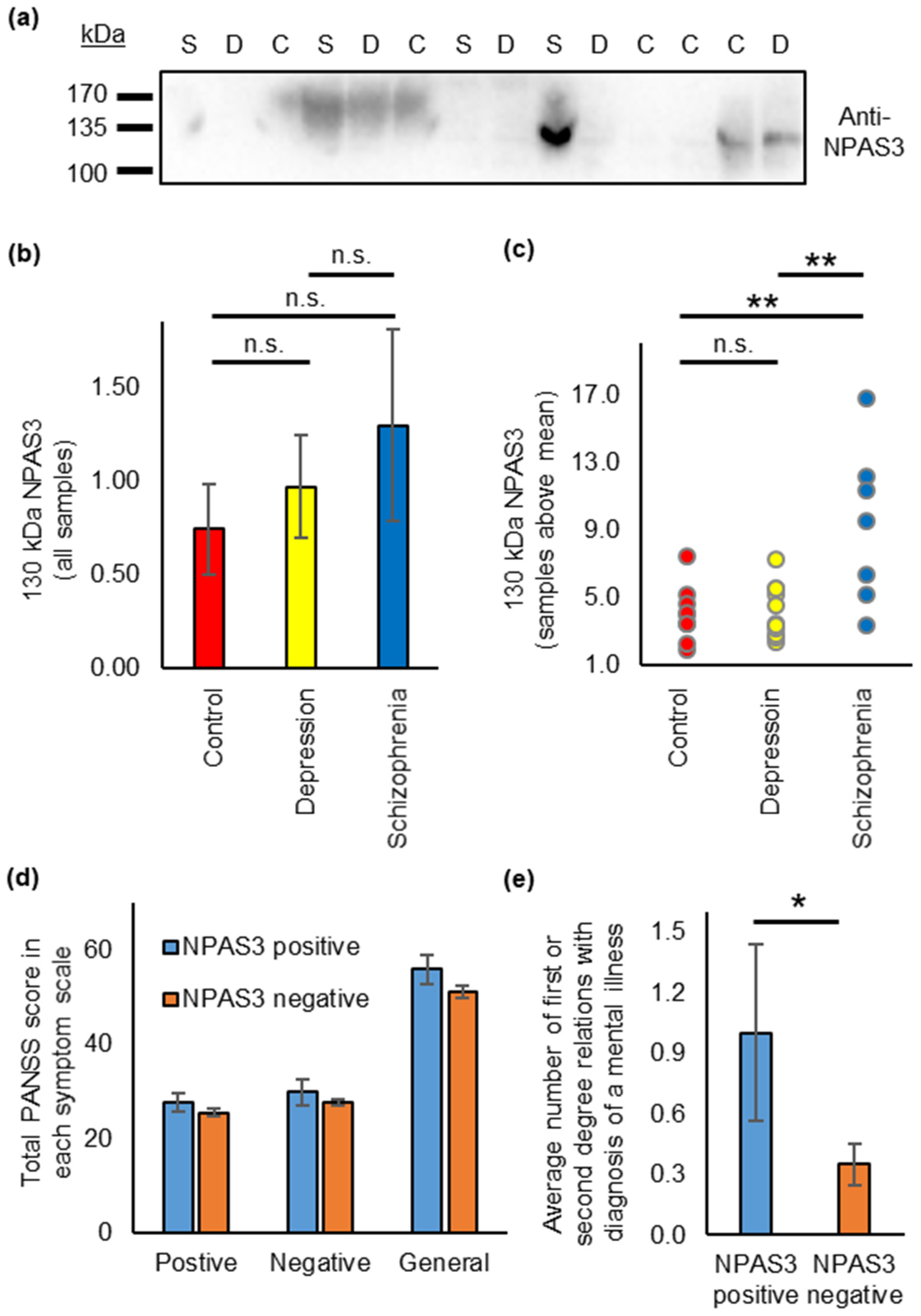
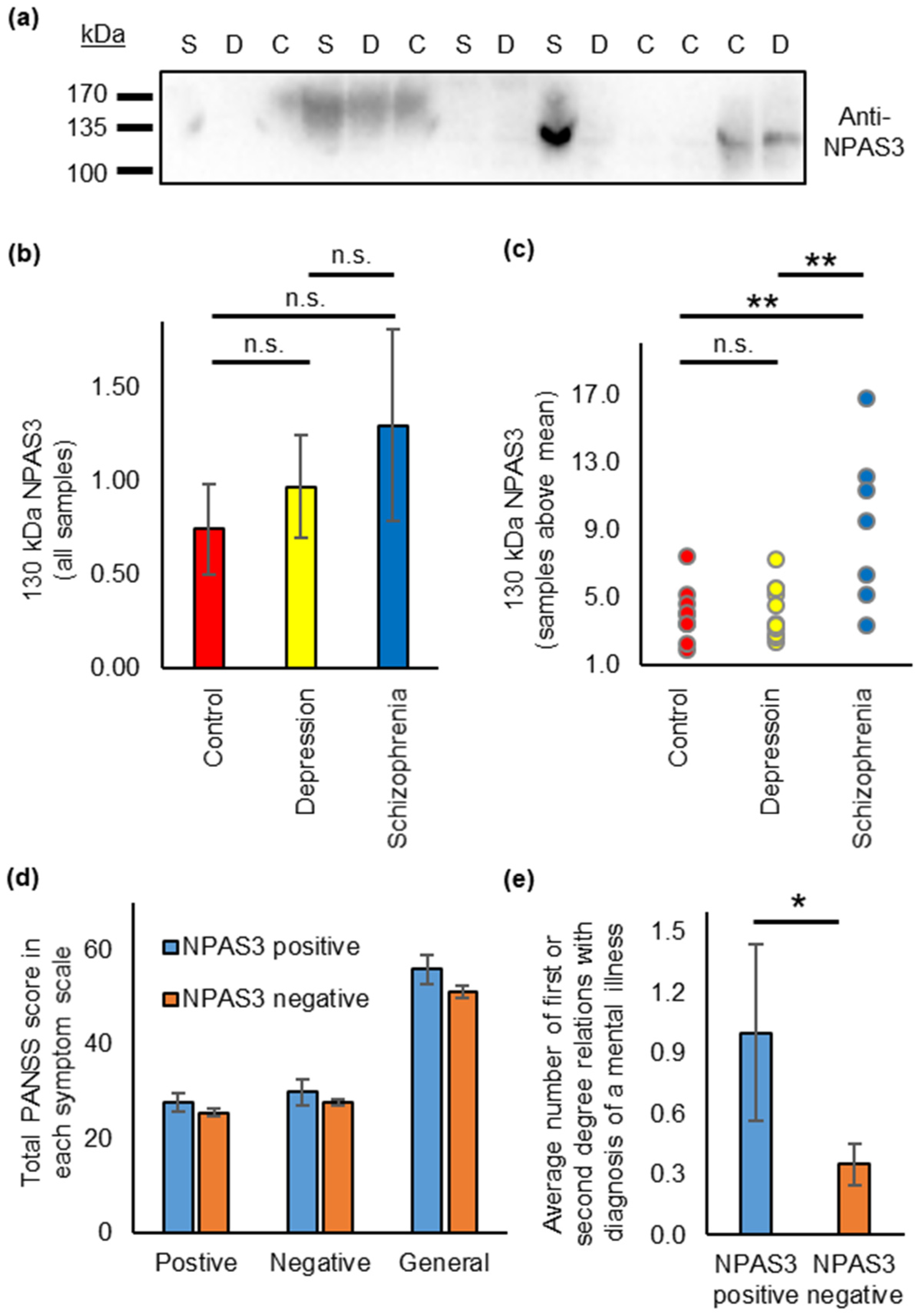
3.4. NPAS3 Is Also Found in Blood Serum, and Shows Expression Differences in Patients with Schizophrenia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Lam, M.; Chen, C.-Y.; Li, Z.; Martin, A.R.; Bryois, J.; Ma, X.; Gaspar, H.; Ikeda, M.; Benyamin, B.; Brown, B.C.; et al. Comparative genetic architectures of schizophrenia in East Asian and European populations. Nat. Genet. 2019, 51, 1670–1678. [Google Scholar] [CrossRef] [PubMed]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. Medrxiv 2020. [Google Scholar] [CrossRef]
- Wray, N.R.; Ripke, S.; Mattheisen, M.; Trzaskowski, M.; Byrne, E.M.; Abdellaoui, A.; Adams, M.J.; Agerbo, E.; Air, T.M.; Andlauer, T.M.F.; et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018, 50, 668–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradshaw, N.J.; Korth, C. Protein misassembly and aggregation as potential convergence points for non-genetic causes of chronic mental illness. Mol. Psychiatry 2019, 24, 936–951. [Google Scholar] [CrossRef]
- Leliveld, S.R.; Bader, V.; Hendriks, P.; Prikulis, I.; Sajnani, G.; Requena, J.R.; Korth, C. Insolubility of Disrupted-in-Schizophrenia 1 disrupts oligomer-dependent interactions with Nuclear Distribution Element 1 and is associated with sporadic mental disease. J. Neurosci. 2008, 28, 3839–3845. [Google Scholar] [CrossRef] [Green Version]
- Kamnasaran, D.; Muir, W.J.; Ferguson-Smith, M.A.; Cox, D.W. Disruption of the neuronal PAS3 gene in a family affected with schizophrenia. J. Med. Genet. 2003, 40, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Pickard, B.S.; Malloy, M.P.; Porteous, D.J.; Blackwood, D.H.R.; Muir, W.J. Disruption of a brain transcription factor, NPAS3, is associated with schizophrenia and learning disability. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 136B, 26–32. [Google Scholar] [CrossRef]
- Macintyre, G.; Alford, T.; Xiong, L.; Rouleau, G.A.; Tibbo, P.G.; Cox, D.W. Association of NPAS3 exonic variation with schizophrenia. Schizophr. Res. 2010, 120, 143–149. [Google Scholar] [CrossRef]
- Pickard, B.S.; Christoforou, A.; Thomson, P.A.; Fawkes, A.; Evans, K.L.; Morris, S.W.; Porteous, D.J.; Blackwood, D.H.; Muir, W.J. Interacting haplotypes at the NPAS3 locus alter risk of schizophrenia and bipolar disorder. Mol. Psychiatry 2009, 14, 874–884. [Google Scholar] [CrossRef]
- Huang, J.; Perlis, R.H.; Lee, P.H.; Rush, A.J.; Fava, M.; Sachs, G.S.; Lieberman, J.; Hamilton, S.P.; Sullivan, P.; Sklar, P.; et al. Cross-disorder genomewide analysis of schizophrenia, bipolar disorder, and depression. Am. J. Psychiatry 2010, 167, 1254–1263. [Google Scholar] [CrossRef] [Green Version]
- Lavedan, C.; Licamele, L.; Volpi, S.; Hamilton, J.; Heaton, C.; Mack, K.; Lannan, R.; Thompson, A.; Wolfgang, C.D.; Polymeropoulos, M.H. Association of the NPAS3 gene and five other loci with response to the antipsychotic iloperidone identified in a whole genome association study. Mol. Psychiatry 2009, 14, 804–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erbel-Sieler, C.; Dudley, C.; Zhou, Y.; Wu, X.; Estill, S.J.; Han, T.; Diaz-Arrastia, R.; Brunskill, E.W.; Potter, S.S.; McKnight, S.L. Behavioral and regulatory abnormalities in mice deficient in the NPAS1 and NPAS3 transcription factors. Proc. Natl. Acad. Sci. USA 2004, 101, 13648–13653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Arbez, N.; Nucifora, L.G.; Sell, G.L.; DeLisi, L.E.; Ross, C.A.; Margolis, R.L.; Nucifora, F.C. A mutation in NPAS3 segregates with mental illness in a small family. Mol. Psychiatry 2014, 19, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Nucifora, L.G.; Wu, Y.C.; Lee, B.J.; Sha, L.; Margolis, R.L.; Ross, C.A.; Sawa, A.; Nucifora, F.C., Jr. A mutation in NPAS3 that segregates with schizophrenia in a small family leads to protein aggregation. Mol. Neuropsychiatry 2016, 2, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucl. Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottis, P.; Bader, V.; Trossbach, S.V.; Kretzschmar, H.; Michel, M.; Leliveld, S.R.; Korth, C. Convergence of two independent mental disease genes on the protein level: Recruitment of dysbindin to cell-invasive Disrupted-in-Schizophrenia 1 aggresomes. Biol. Psychiatry 2011, 70, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Bader, V.; Tomppo, L.; Trossbach, S.V.; Bradshaw, N.J.; Prikulis, I.; Leliveld, S.R.; Lin, C.-Y.; Ishizuka, K.; Sawa, A.; Ramos, A.; et al. Proteomic, genomic and translational approaches identify CRMP1 for a role in schizophrenia and its underlying traits. Hum. Mol. Genet. 2012, 21, 4406–4418. [Google Scholar] [CrossRef]
- Bradshaw, N.J.; Bader, V.; Prikulis, I.; Lueking, A.; Müllner, S.; Korth, C. Aggregation of the protein TRIOBP-1 and its potential relevance to schizophrenia. PLoS ONE 2014, 9, e111196. [Google Scholar] [CrossRef] [Green Version]
- Bradley, W.G.; Andrew, A.S.; Traynor, B.J.; Chiò, A.; Butt, T.H.; Stommel, E.W. Gene-Environment-Time Interactions in Neurodegenerative Diseases: Hypotheses and Research Approaches. Ann. Neurosci. 2018, 25, 261–267. [Google Scholar] [CrossRef]
- Ishigaki, S.; Sobue, G. Importance of Functional Loss of FUS in FTLD/ALS. Front. Mol. Biosci. 2018, 5, 44. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Med. 2019, 12, 25. [Google Scholar] [CrossRef]
- Palkovits, M. Microdissection of individual brain nuclei an areas. In General Neurochemical Techniques; Boulton, A.A., Baker, G.B., Eds.; Neuromethods; Humana Press: Clifton, NJ, USA, 1985; pp. 1–17. [Google Scholar]
- Palkovits, M. Isolated removal of hypothalamic or other brain nuclei of the rat. Brain Res. 1973, 59, 449–450. [Google Scholar] [CrossRef]
- The ICD-10 Classification of Mental and Behavioural Disorders: Diagnostic Criteria for Research; World Health Orgnaization: Geneva, Switzerland, 1993.
- Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013.
- Leliveld, S.R.; Hendriks, P.; Michel, M.; Sajnani, G.; Bader, V.; Trossbach, S.; Prikulis, I.; Hartmann, R.; Jonas, E.; Willbold, D.; et al. Oligomer assembly of the C-terminal DISC1 domain (640-854) is controlled by self-association motifs and disease-associated polymorphism S704C. Biochemistry 2009, 48, 7746–7755. [Google Scholar] [CrossRef]
- Luoma, L.M.; Berry, F.B. Molecular analysis of NPAS3 functional domains and variants. BMC Molecular Biol. 2018, 19, 14. [Google Scholar] [CrossRef]
- Bradshaw, N.J.; Yerabham, A.S.K.; Marreiros, R.; Zhang, T.; Nagel-Steger, L.; Korth, C. An unpredicted aggregation-critical region of the actin-polymerizing protein TRIOBP-1/Tara, determined by elucidation of its domain structure. J. Biol. Chem. 2017, 292, 9583–9598. [Google Scholar] [CrossRef] [Green Version]
- Wiemann, S.; Pennacchio, C.; Hu, Y.; Hunter, P.; Harbers, M.; Amiet, A.; Bethel, G.; Busse, M.; Carninci, P.; Diekhans, M.; et al. The ORFeome Collaboration: A genome-scale human ORF-clone resource. Nat. Methods 2016, 13, 191–192. [Google Scholar] [CrossRef]
- Seiler, C.Y.; Park, J.G.; Sharma, A.; Hunter, P.; Surapaneni, P.; Sedillo, C.; Field, J.; Algar, R.; Price, A.; Steel, J.; et al. DNASU plasmid and PSI:Biology-Materials repositories: Resources to accelerate biological research. Nucleic Acids Res. 2013, 42, D1253–D1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campeau, E.; Ruhl, V.E.; Rodier, F.; Smith, C.L.; Rahmberg, B.L.; Fuss, J.O.; Campisi, J.; Yaswen, P.; Cooper, P.K.; Kaufman, P.D. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE 2009, 4, e6529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrotis, A.; Pengo, N.; Burden, J.J.; Ketteler, R. Redundancy of human ATG4 protease isoforms in autophagy and LC3/GABARAP processing revealed in cells. Autophagy 2019, 15, 976–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottis, P.; Topic, B.; Loos, M.; Li, K.W.; de Souza Silva, M.A.; Schulz, D.; Smit, A.B.; Huston, J.P.; Korth, C. Aging-induced proteostatic changes in the rat hippocampus identify ARP3, NEB2 and BRAG2 as a molecular circuitry for cognitive impairment. PLoS ONE 2013, 8, e75112. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- JASP Team JASP, Version 0.14.1; 2020. Available online: https://jasp-stats.org/2020/12/17/jasp-0-14-1-minor-updates/ (accessed on 1 October 2021).
- Bader, V.; Ottis, P.; Pum, M.; Huston, J.P.; Korth, C. Generation, purification, and characterization of cell-invasive DISC1 protein species. J. Vis. Exp. 2012, 66, e4132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.-Y.; Shen, Y.; Xu, Q. Propagation of dysbindin-1B aggregates: Exosome-mediated transmission of neurotoxic deposits. Neuroscience 2015, 291, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Trossbach, S.V.; Bader, V.; Hecher, L.; Pum, M.E.; Masoud, S.T.; Prikulis, I.; Schäble, S.; de Souza Silva, M.A.; Su, P.; Boulat, B.; et al. Misassembly of full-length Disrupted-in-Schizophrenia 1 protein is linked to altered dopamine homeostasis and behavioral deficits. Mol. Psychiatry 2016, 21, 1561–1572. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.; Duncan, C.E.; Beveridge, N.J.; Webster, M.J.; Cairns, M.J.; Weickert, C.S. Expression of NPAS3 in the Human Cortex and Evidence of Its Posttranscriptional Regulation by miR-17 During Development, With Implications for Schizophrenia. Schizophr. Bull. 2013, 39, 396–406. [Google Scholar] [CrossRef]
- Chou, C.-C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Duong, D.M.; et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Barmada, S.J.; Skibinski, G.; Korb, E.; Rao, E.J.; Wu, J.Y.; Finkbeiner, S. Cytoplasmic Mislocalization of TDP-43 Is Toxic to Neurons and Enhanced by a Mutation Associated with Familial Amyotrophic Lateral Sclerosis. J. Neurosci. 2010, 30, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.-J.; Vanderwyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M.; et al. Tar DNA Binding Protein-43 (TDP-43) associates with stress granules: Analysis of culture cells and pathological brain tissue. PLoS ONE 2010, 5, e13250. [Google Scholar] [CrossRef] [Green Version]
- Brunskill, E.W.; Witte, D.P.; Shreiner, A.B.; Potter, S.S. Characterization of Npas3, a novel basic helix-loop-helix PAS gene expressed in the developing mouse nervous system. Mech. Dev. 1999, 88, 237–241. [Google Scholar] [CrossRef]
- Kay, S.R.; Fiszbein, A.; Opter, L.A. The Postive and Negative Syndrome Scale (PANSS) for Schizophrenia. Schizophr. Bull. 1987, 13, 261–276. [Google Scholar] [CrossRef]
- Gogolla, N. The insular cortex. Curr. Biol. 2017, 27, R573–R591. [Google Scholar] [CrossRef] [Green Version]
- Fallini, C.; Khalil, B.; Smith, C.L.; Rossoll, W. Traffic jam at the nuclear pore: All roads lead to nucleocytoplasmic transport defects in ALS/FTD. Neurobiol. Dis. 2020, 140, 104836. [Google Scholar] [CrossRef]
- Rossi, J.J.; Rosenfeld, J.A.; Chan, K.M.; Streff, H.; Nankivell, V.; Peet, D.J.; Whitelaw, M.L.; Bersten, D.C. Molecular characterisation of rare loss-of-function NPAS3 and NPAS4 variants identified in individuals with neurodevelopmental disorders. Sci. Rep. 2021, 11, 6602. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Ba, A.N.; Pogouste, A.; Provart, N.; Moses, A.M. NLStradamus: A simple Hidden Markov Model for nuclear localization signal prediction. BMC Bioinform. 2009, 10, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonsson, I.P.C.; Whitelaw, M.L.; Poellinger, L. Role of the PAS Domain in Regulation of Dimerization and DNA Binding Specificity of the Dioxin Receptor. Mol. Cell. Biol. 1998, 18, 4079–4088. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samardžija, B.; Pavešić Radonja, A.; Zaharija, B.; Bergman, M.; Renner, É.; Palkovits, M.; Rubeša, G.; Bradshaw, N.J. Protein Aggregation of NPAS3, Implicated in Mental Illness, Is Not Limited to the V304I Mutation. J. Pers. Med. 2021, 11, 1070. https://doi.org/10.3390/jpm11111070
Samardžija B, Pavešić Radonja A, Zaharija B, Bergman M, Renner É, Palkovits M, Rubeša G, Bradshaw NJ. Protein Aggregation of NPAS3, Implicated in Mental Illness, Is Not Limited to the V304I Mutation. Journal of Personalized Medicine. 2021; 11(11):1070. https://doi.org/10.3390/jpm11111070
Chicago/Turabian StyleSamardžija, Bobana, Aristea Pavešić Radonja, Beti Zaharija, Mihaela Bergman, Éva Renner, Miklós Palkovits, Gordana Rubeša, and Nicholas J. Bradshaw. 2021. "Protein Aggregation of NPAS3, Implicated in Mental Illness, Is Not Limited to the V304I Mutation" Journal of Personalized Medicine 11, no. 11: 1070. https://doi.org/10.3390/jpm11111070
APA StyleSamardžija, B., Pavešić Radonja, A., Zaharija, B., Bergman, M., Renner, É., Palkovits, M., Rubeša, G., & Bradshaw, N. J. (2021). Protein Aggregation of NPAS3, Implicated in Mental Illness, Is Not Limited to the V304I Mutation. Journal of Personalized Medicine, 11(11), 1070. https://doi.org/10.3390/jpm11111070