
RNA-Sequencing-Based Transcriptomic Score with Prognostic and Theranostic Values in Multiple Myeloma

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Expression Profiling
2.2. Statistical Analyses
2.3. Multiple Myeloma Cell Lines
2.4. Drug Response Analyses
2.5. EZH2 Inhibition in Primary MM Cells from Patients
3. Results
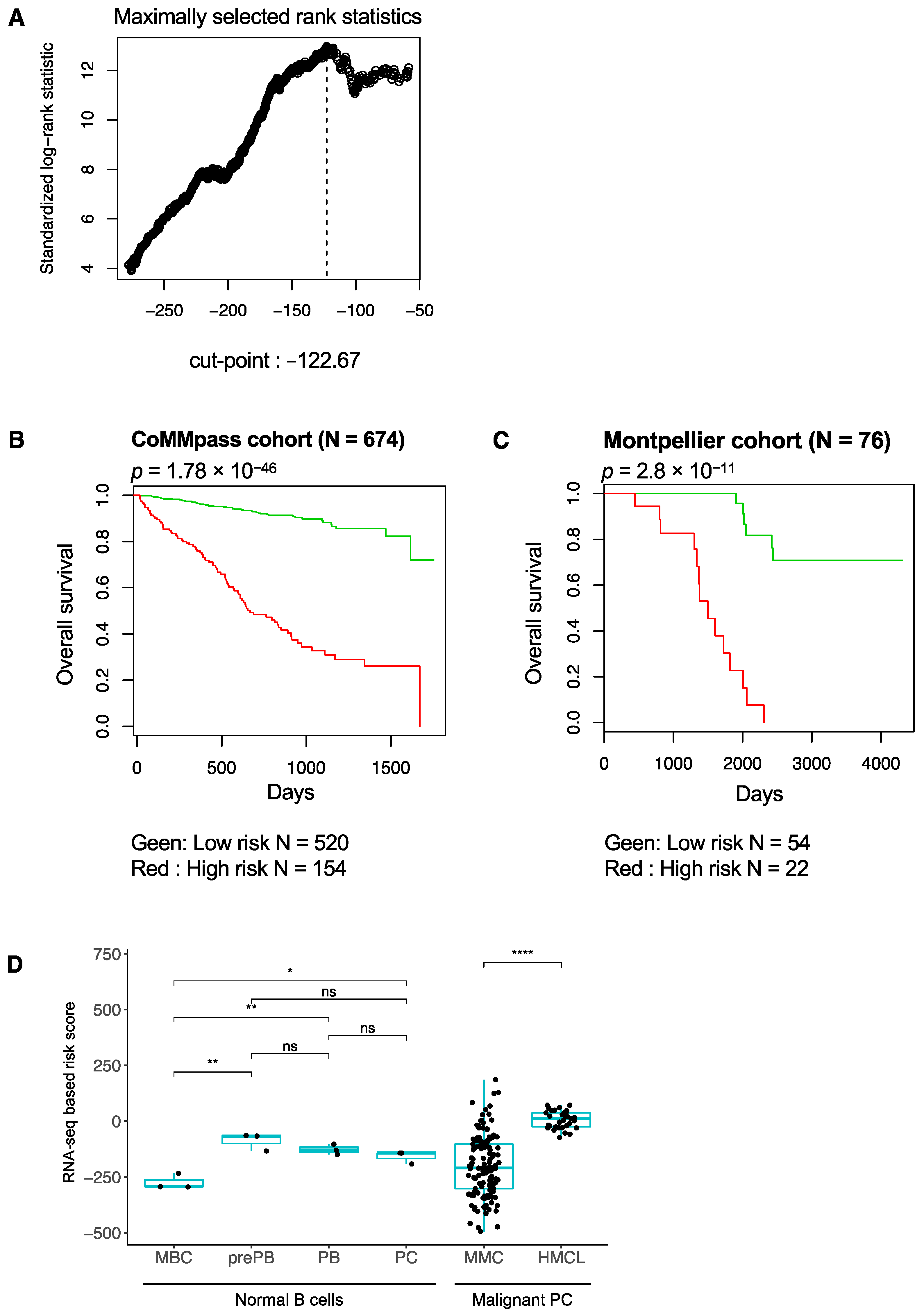
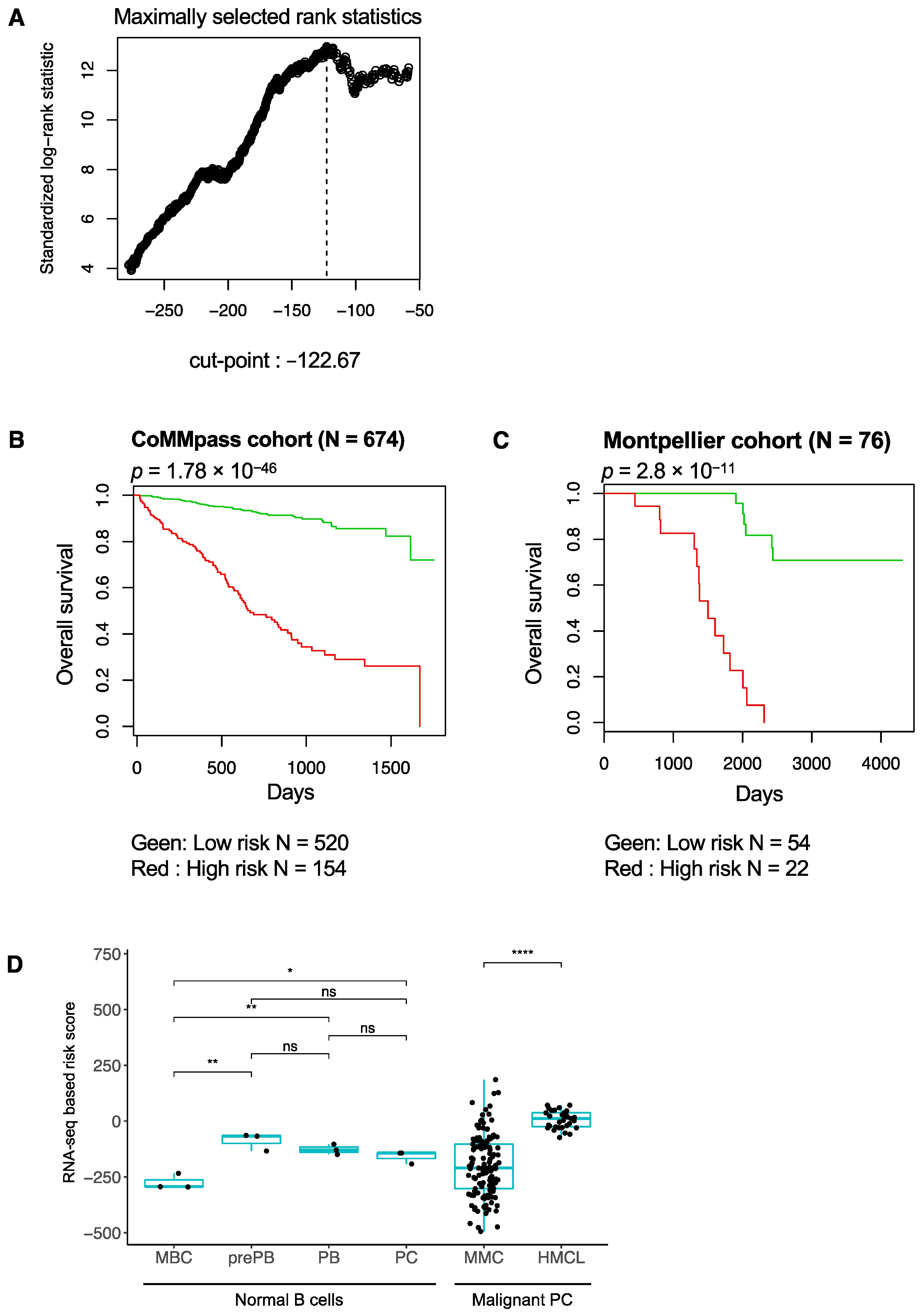
3.1. RNA-Seq-Based Gene Risk Score in Multiple Myeloma
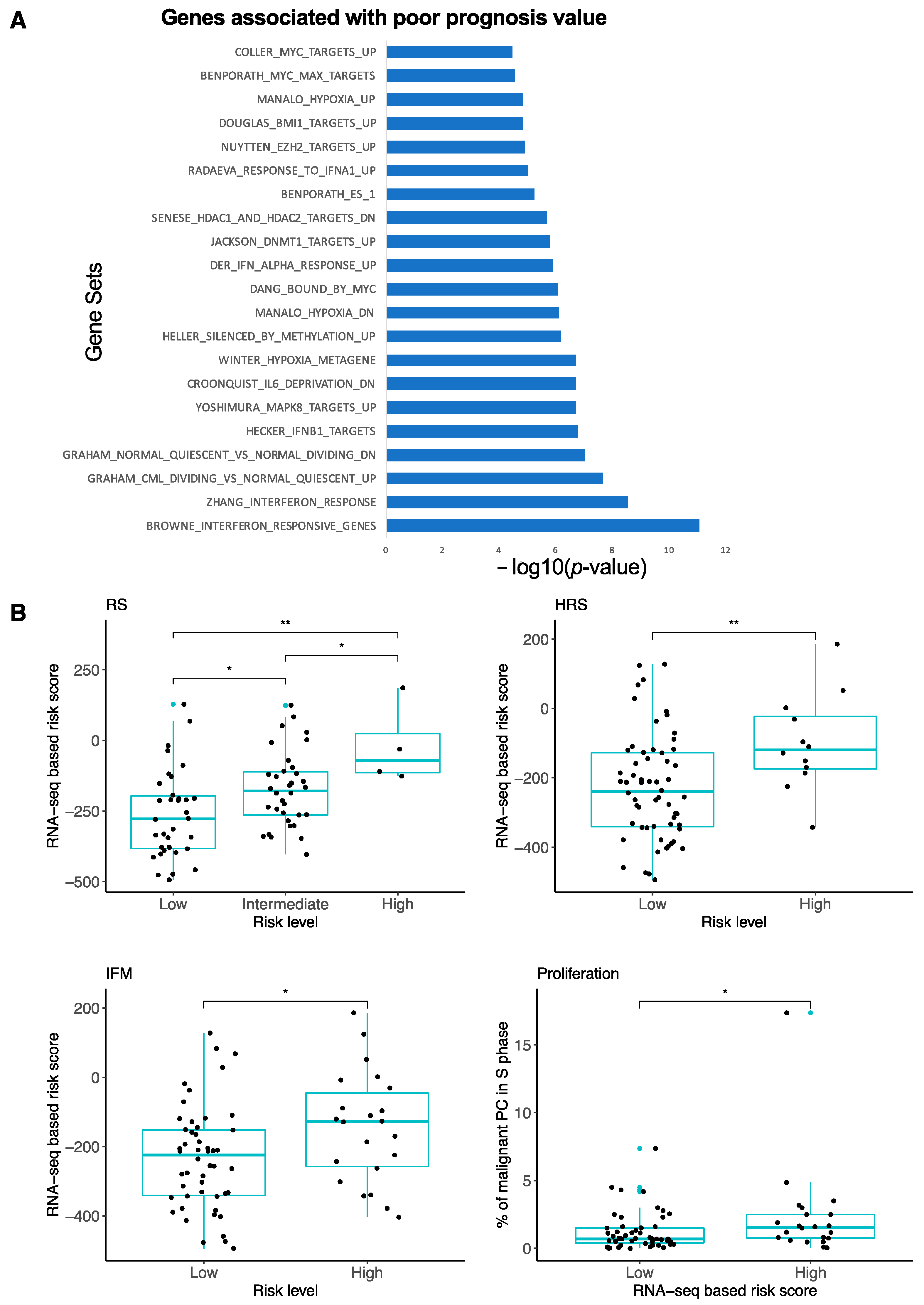
3.2. High-Risk MM Patients Identified with the RNA-Seq-Based Risk Score Are Characterized by Enrichment of Genes Related to Cell Proliferation, Growth Factor Signaling, MYC Pathway and Epigenetic Deregulation
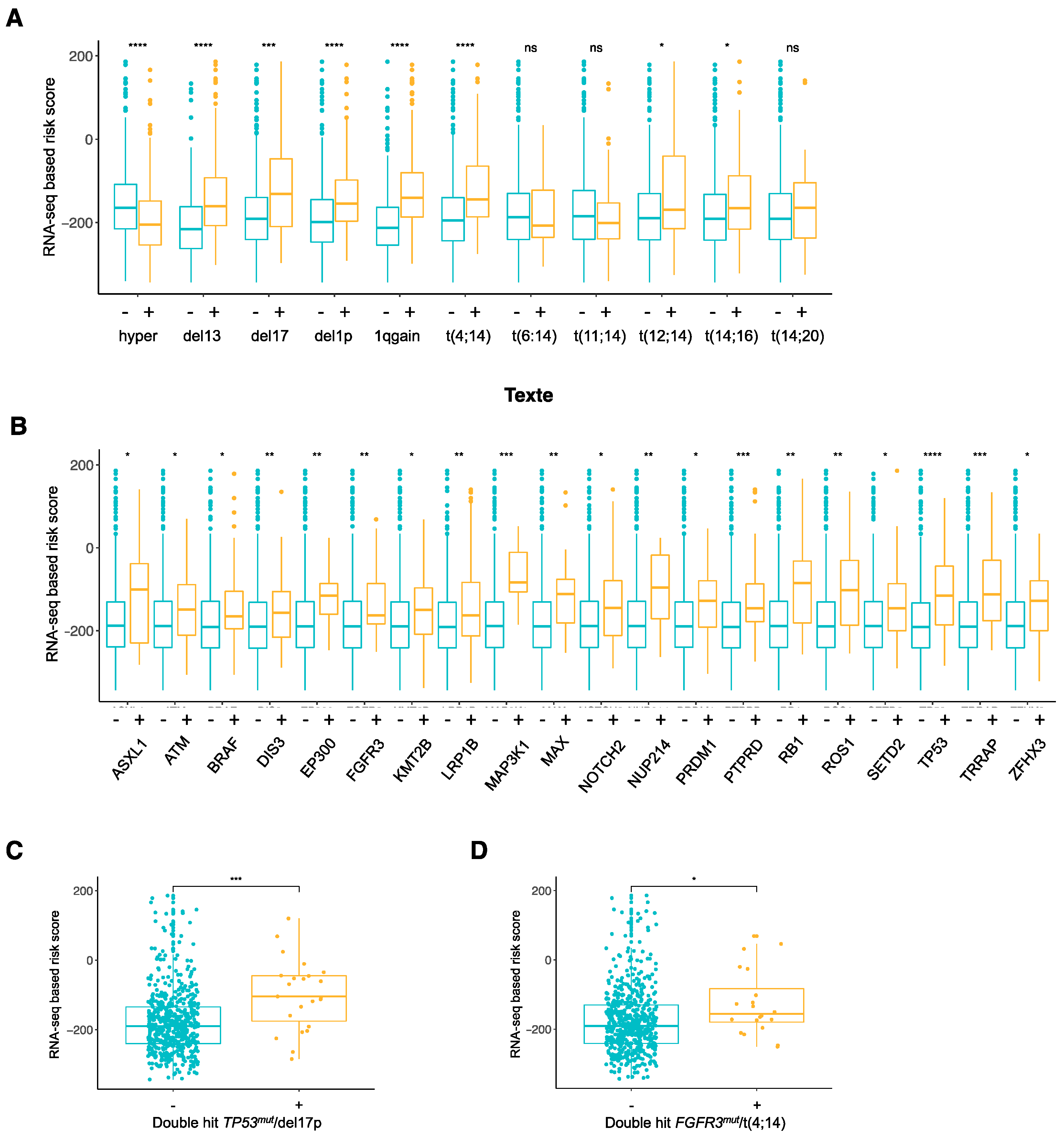
3.3. Association between RNA-Seq-Based Risk Score and Mutations in MM
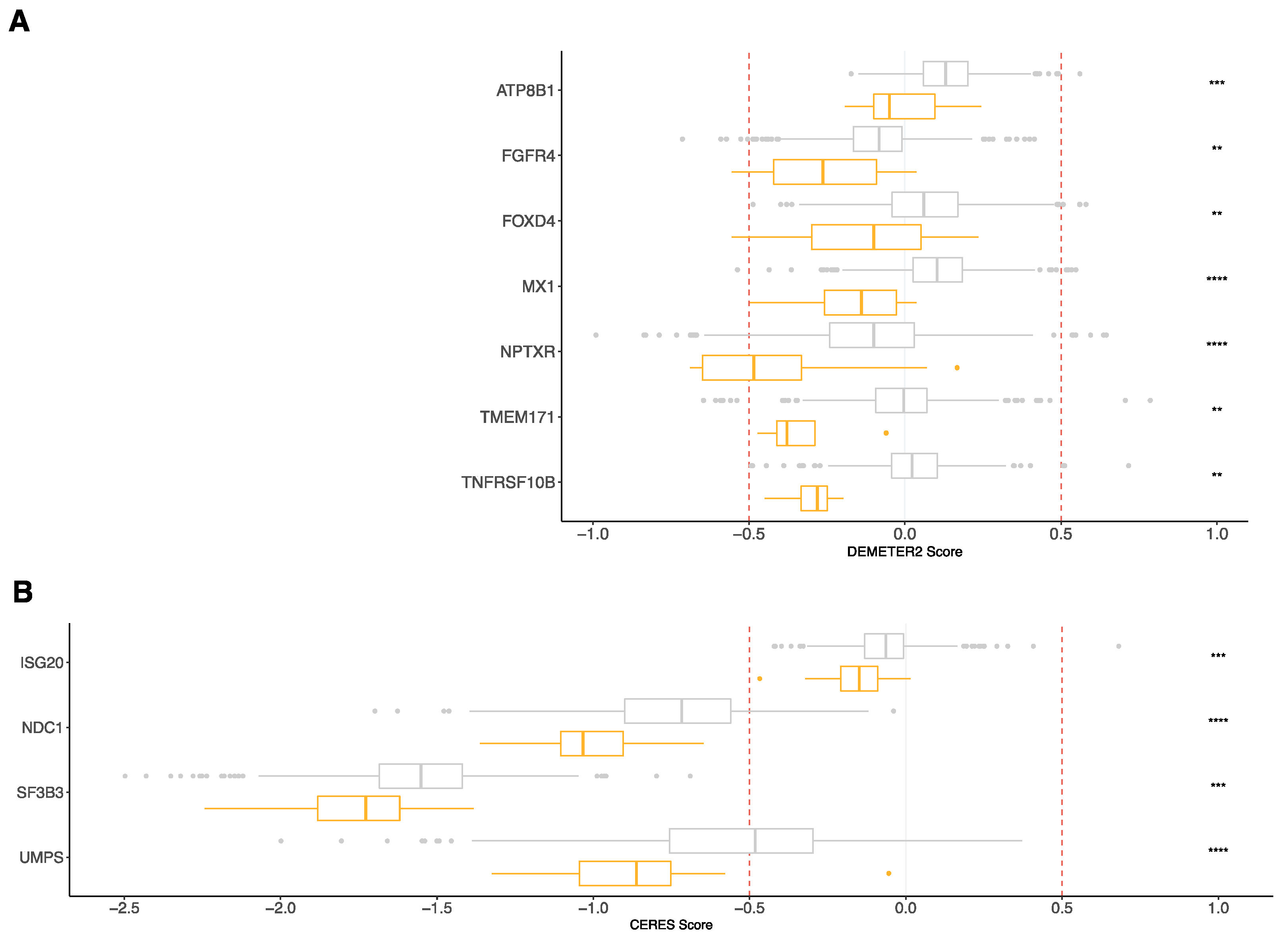
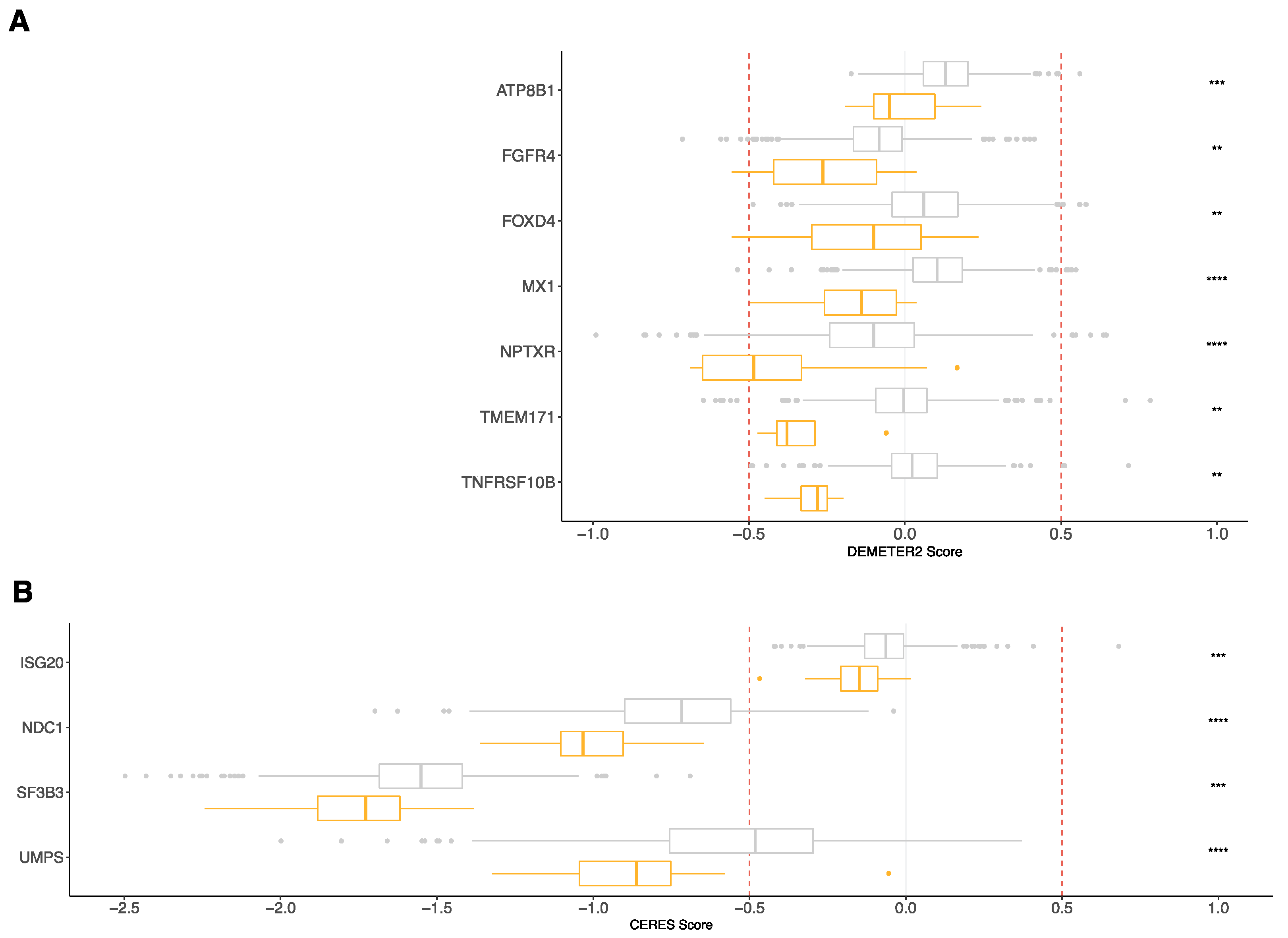
3.4. RNA-Seq Risk Score Revealed New Genes Significantly Associated with MM Pathophysiology
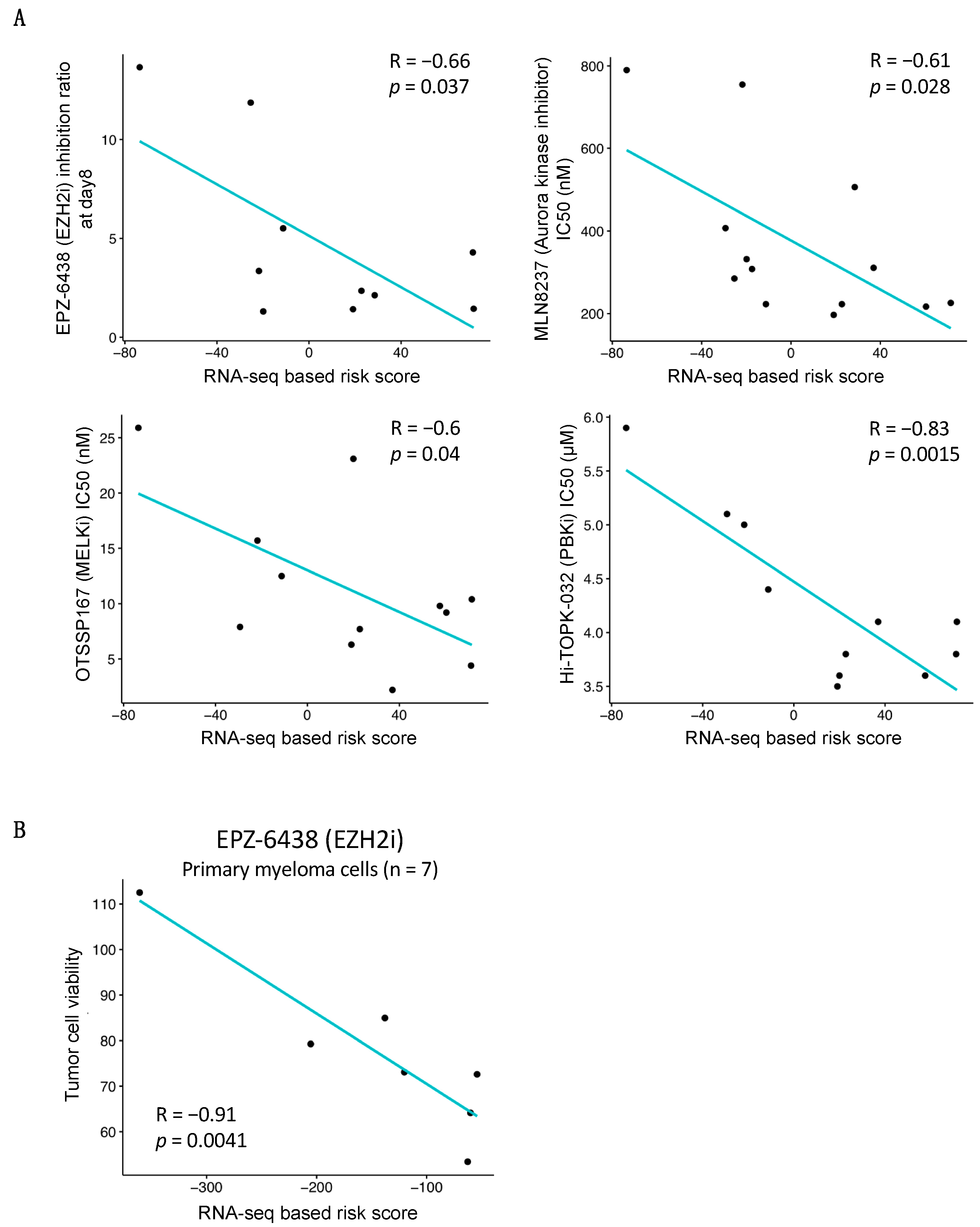
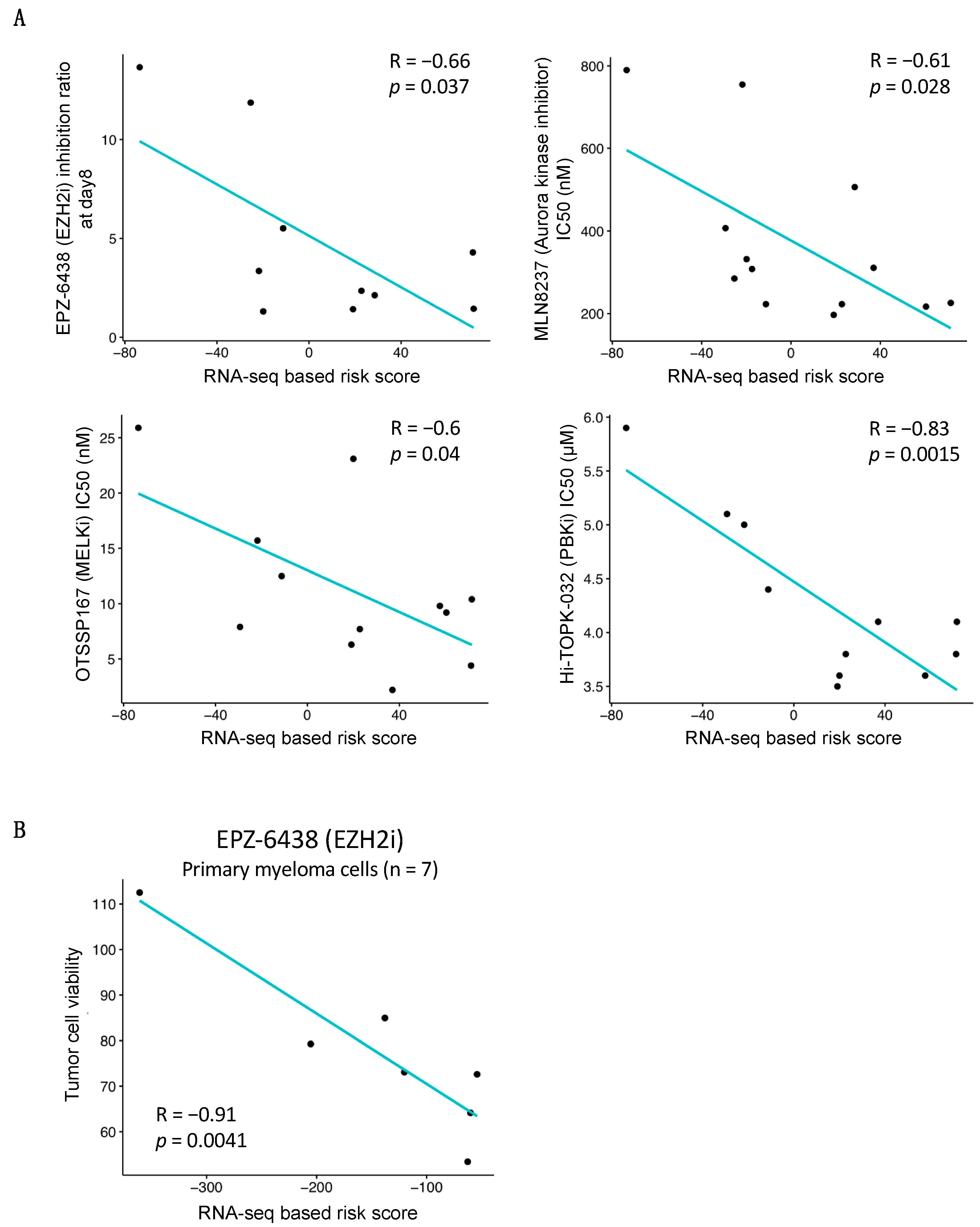
3.5. Association between RNA-Seq-Based Risk Score and Response to Treatment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Walker, B.A.; Leone, P.E.; Chiecchio, L.; Dickens, N.J.; Jenner, M.W.; Boyd, K.D.; Johnson, D.C.; Gonzalez, D.; Dagrada, G.P.; Protheroe, R.K.M.; et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood 2010, 116, e56–e65. [Google Scholar] [CrossRef]
- De Mel, S.; Lim, S.H.; Tung, M.L.; Chng, W.-J. Implications of Heterogeneity in Multiple Myeloma. BioMed Res. Int. 2014, 2014, 232546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergsagel, P.L.; Kuehl, W.M. Molecular Pathogenesis and a Consequent Classification of Multiple Myeloma. J. Clin. Oncol. 2005, 23, 6333–6338. [Google Scholar] [CrossRef] [PubMed]
- Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; Burington, B.; et al. The molecular classification of multiple myeloma. Blood 2006, 108, 2020–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaughnessy, J.D.; Zhan, F.; Burington, B.E.; Huang, Y.; Colla, S.; Hanamura, I.; Stewart, J.P.; Kordsmeier, B.; Randolph, C.; Williams, D.R.; et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome. Blood 2007, 109, 2276–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuiper, R.; Broyl, A.; De Knegt, Y.; Van Vliet, M.H.; Van Beers, E.H.; Van Der Holt, B.; El Jarari, L.; Mulligan, G.; Gregory, W.; Morgan, G.; et al. A gene expression signature for high-risk multiple myeloma. Leukemia 2012, 26, 2406–2413. [Google Scholar] [CrossRef] [Green Version]
- Decaux, O.; Lodé, L.; Magrangeas, F.; Charbonnel, C.; Gouraud, W.; Jézéquel, P.; Attal, M.; Harousseau, J.-L.; Moreau, P.; Bataille, R.; et al. Prediction of Survival in Multiple Myeloma Based on Gene Expression Profiles Reveals Cell Cycle and Chromosomal Instability Signatures in High-Risk Patients and Hyperdiploid Signatures in Low-Risk Patients: A Study of the Intergroupe Francophone du Myélome. J. Clin. Oncol. 2008, 26, 4798–4805. [Google Scholar] [CrossRef]
- Rème, T.; Hose, D.; Theillet, C.; Klein, B. Modeling risk stratification in human cancer. Bioinformatics 2013, 29, 1149–1157. [Google Scholar] [CrossRef] [Green Version]
- Dickens, N.; Walker, B.A.; Leone, P.E.; Johnson, D.C.; Brito, J.L.; Zeisig, A.; Jenner, M.; Boyd, K.; Gonzalez, D.; Gregory, W.M.; et al. Homozygous Deletion Mapping in Myeloma Samples Identifies Genes and an Expression Signature Relevant to Pathogenesis and Outcome. Clin. Cancer Res. 2010, 16, 1856–1864. [Google Scholar] [CrossRef] [Green Version]
- Hose, D.; Rème, T.; Hielscher, T.; Moreaux, J.; Messner, T.; Seckinger, A.; Benner, A.; Shaughnessy, J.D.; Barlogie, B.; Zhou, Y.; et al. Proliferation is a central independent prognostic factor and target for personalized and risk-adapted treatment in multiple myeloma. Haematologica 2011, 96, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Chng, W.J.; Braggio, E.; Mulligan, G.; Bryant, B.; Remstein, E.; Valdez, R.; Dogan, A.; Fonseca, R. The centrosome index is a powerful prognostic marker in myeloma and identifies a cohort of patients that might benefit from aurora kinase inhibition. Blood 2008, 111, 1603–1609. [Google Scholar] [CrossRef] [Green Version]
- Jourdan, M.; Reme, T.; Goldschmidt, H.; Fiol, G.; Pantesco, V.; DE Vos, J.; Rossi, J.-F.; Hose, D.; Klein, B. Gene expression of anti- and pro-apoptotic proteins in malignant and normal plasma cells. Br. J. Haematol. 2009, 145, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, J.D.; Qu, P.; Usmani, S.; Heuck, C.J.; Zhang, Q.; Zhou, Y.; Tian, E.; Hanamura, I.; Van Rhee, F.; Anaissie, E.; et al. Pharmacogenomics of bortezomib test-dosing identifies hyperexpression of proteasome genes, especially PSMD4, as novel high-risk feature in myeloma treated with Total Therapy. Blood 2011, 118, 3512–3524. [Google Scholar] [CrossRef] [PubMed]
- Stessman, H.; Baughn, L.B.; Sarver, A.; Xia, T.; Deshpande, R.; Mansoor, A.; Walsh, S.; Sunderland, J.; Dolloff, N.G.; Linden, M.; et al. Profiling Bortezomib Resistance Identifies Secondary Therapies in a Mouse Myeloma Model. Mol. Cancer Ther. 2013, 12, 1140–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, A.; Mukherjee, U.K.; Harding, T.; Jang, J.S.; Stessman, H.; Li, Y.; Abyzov, A.; Jen, J.; Kumar, S.; Rajkumar, V.; et al. Single-cell analysis of targeted transcriptome predicts drug sensitivity of single cells within human myeloma tumors. Leukemia 2015, 30, 1094–1102. [Google Scholar] [CrossRef]
- Mitra, A.; Harding, T.; Mukherjee, U.K.; Jang, J.S.; Li, Y.; Hongzheng, R.; Jen, J.; Sonneveld, P.; Kumar, S.; Kuehl, W.M.; et al. A gene expression signature distinguishes innate response and resistance to proteasome inhibitors in multiple myeloma. Blood Cancer J. 2017, 7, e581. [Google Scholar] [CrossRef]
- Gourzones, C.; Bellanger, C.; Lamure, S.; Gadacha, O.C.; De Paco, E.G.; Vincent, L.; Cartron, G.; Klein, B.; Moreaux, J. Antioxidant Defenses Confer Resistance to High Dose Melphalan in Multiple Myeloma Cells. Cancers 2019, 11, 439. [Google Scholar] [CrossRef] [Green Version]
- Bhutani, M.; Zhang, Q.; Friend, R.; Voorhees, P.M.; Druhan, L.J.; Barlogie, B.; Sonneveld, P.; Morgan, G.; Symanowski, J.T.; Avalos, B.R.; et al. Investigation of a gene signature to predict response to immunomodulatory derivatives for patients with multiple myeloma: An exploratory, retrospective study using microarray datasets from prospective clinical trials. Lancet Haematol. 2017, 4, e443–e451. [Google Scholar] [CrossRef]
- Moreaux, J.; Reme, T.; Leonard, W.; Veyrune, J.-L.; Requirand, G.; Goldschmidt, H.; Hose, D.; Klein, B. Gene expression-based prediction of myeloma cell sensitivity to histone deacetylase inhibitors. Br. J. Cancer 2013, 109, 676–685. [Google Scholar] [CrossRef] [Green Version]
- Moreaux, J.; Rème, T.; Leonard, W.; Veyrune, J.-L.; Requirand, G.; Goldschmidt, H.; Hose, D.; Klein, B. Development of Gene Expression–Based Score to Predict Sensitivity of Multiple Myeloma Cells to DNA Methylation Inhibitors. Mol. Cancer Ther. 2012, 11, 2685–2692. [Google Scholar] [CrossRef] [Green Version]
- Herviou, L.; Kassambara, A.; Boireau, S.; Robert, N.; Requirand, G.; Müller-Tidow, C.; Vincent, L.; Seckinger, A.; Goldschmidt, H.; Cartron, G.; et al. PRC2 targeting is a therapeutic strategy for EZ score defined high-risk multiple myeloma patients and overcome resistance to IMiDs. Clin. Epigenetics 2018, 10, 121. [Google Scholar] [CrossRef] [PubMed]
- de Boussac, H.; Bruyer, A.; Jourdan, M.; Maes, A.; Robert, N.; Gourzones, C.; Vincent, L.; Seckinger, A.; Cartron, G.; Hose, D.; et al. Kinome expression profiling to target new therapeutic avenues in multiple myeloma. Haematologica 2020, 105, 784–795. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Moreaux, J.; Hose, D.; Kassambara, A.; Reme, T.; Moine, P.; Requirand, G.; Goldschmidt, H.; Klein, B. Osteoclast-gene expression profiling reveals osteoclast-derived CCR2 chemokines promoting myeloma cell migration. Blood 2011, 117, 1280–1290. [Google Scholar] [CrossRef] [Green Version]
- Moreaux, J.; Klein, B.; Bataille, R.; Descamps, G.; Maïga, S.; Hose, D.; Goldschmidt, H.; Jauch, A.; Rème, T.; Jourdan, M.; et al. A high-risk signature for patients with multiple myeloma established from the molecular classification of human myeloma cell lines. Haematologica 2010, 96, 574–582. [Google Scholar] [CrossRef] [Green Version]
- Vikova, V.; Jourdan, M.; Robert, N.; Requirand, G.; Boireau, S.; Bruyer, A.; Vincent, L.; Cartron, G.; Klein, B.; Elemento, O.; et al. Comprehensive characterization of the mutational landscape in multiple myeloma cell lines reveals potential drivers and pathways associated with tumor progression and drug resistance. Theranostics 2019, 9, 540–553. [Google Scholar] [CrossRef]
- Kassambara, A.; Hose, D.; Moreaux, J.; Walker, B.A.; Protopopov, A.; Reme, T.; Pellestor, F.; Pantesco, V.; Jauch, A.; Morgan, G.; et al. Genes with a spike expression are clustered in chromosome (sub)bands and spike (sub)bands have a powerful prognostic value in patients with multiple myeloma. Haematologica 2012, 97, 622–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Requirand, G.; Robert, N.; Boireau, S.; Vincent, L.; Seckinger, A.; Bouhya, S.; Ceballos, P.; Cartron, G.; Hose, D.; Klein, B.; et al. BrdU incorporation in multiparameter flow cytometry: A new cell cycle assessment approach in multiple myeloma. Cytom. Part B Clin. Cytom. 2019, 96, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [Green Version]
- Iii, E.R.M.; De Weck, A.; Schlabach, M.R.; Billy, E.; Mavrakis, K.J.; Hoffman, G.; Belur, D.; Castelletti, D.; Frias, E.; Gampa, K.; et al. Project DRIVE: A compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale, deep RNAi screening. Cell 2017, 170, 577–592.e10. [Google Scholar] [CrossRef] [Green Version]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFarland, J.; Ho, Z.V.; Kugener, G.; Dempster, J.M.; Montgomery, P.G.; Bryan, J.; Krill-Burger, J.M.; Green, T.M.; Vazquez, F.; Boehm, J.S.; et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun. 2018, 9, 4610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreaux, J.; Bruyer, A.; Veyrune, J.-L.; Goldschmidt, H.; Hose, D.; Klein, B. DNA methylation score is predictive of myeloma cell sensitivity to 5-azacitidine. Br. J. Haematol. 2013, 164, 613–616. [Google Scholar] [CrossRef] [Green Version]
- Bergsagel, P.L.; Kuehl, W.M.; Zhan, F.; Sawyer, J.; Barlogie, B.; Shaughnessy, J.J. Cyclin D dysregulation: An early and unifying pathogenic event in multiple myeloma. Blood 2005, 106, 296–303. [Google Scholar] [CrossRef] [Green Version]
- Broyl, A.; Hose, D.; Lokhorst, H.; de Knegt, Y.; Peeters, J.; Jauch, A.; Bertsch, U.; Buijs, A.; Stevens-Kroef, M.; Beverloo, H.B.; et al. Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood 2010, 116, 2543–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chng, W.J.; Dispenzieri, A.; Chim, C.-S.; Fonseca, R.; Goldschmidt, H.; Lentzsch, S.; Munshi, N.; Palumbo, A.; Miguel, J.S.; Sonneveld, P.; et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia 2014, 28, 269–277. [Google Scholar] [CrossRef]
- Kortüm, K.M.; Langer, C.; Monge, J.; Bruins, L.; Egan, J.B.; Zhu, Y.X.; Shi, C.X.; Jedlowski, P.; Schmidt, J.; Ojha, J.; et al. Targeted sequencing using a 47 gene multiple myeloma mutation panel (M(3) P) in -17p high risk disease. Br. J. Haematol. 2014, 168, 507–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez, C.; Jara-Acevedo, M.; Sánchez, L.A.C.; Castillo, D.; Ordóñez, G.R.; Sarasquete, M.E.; Puig, N.; Martínez-López, J.; Prieto-Conde, M.I.; García-Álvarez, M.; et al. A Next-Generation Sequencing Strategy for Evaluating the Most Common Genetic Abnormalities in Multiple Myeloma. J. Mol. Diagn. 2017, 19, 99–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryland, G.L.; Jones, K.; Chin, M.; Markham, J.; Aydogan, E.; Kankanige, Y.; Caruso, M.; Guinto, J.; Dickinson, M.; Prince, H.M.; et al. Novel genomic findings in multiple myeloma identified through routine diagnostic sequencing. J. Clin. Pathol. 2018, 71, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Kortüm, K.M.; Mai, E.K.; Hanafiah, N.H.; Shi, C.-X.; Zhu, Y.-X.; Bruins, L.; Barrio, S.; Jedlowski, P.; Merz, M.; Xu, J.; et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood 2016, 128, 1226–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corre, J.; Cleynen, A.; Du Pont, S.R.; Buisson, L.; Bolli, N.; Attal, M.; Munshi, N.; Avet-Loiseau, H. Multiple myeloma clonal evolution in homogeneously treated patients. Leukemia 2018, 32, 2636–2647. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.S.; Li, Y.; Mitra, A.; Bi, L.; Abyzov, A.; Van Wijnen, A.J.; Baughn, L.B.; Van Ness, B.; Rajkumar, V.; Kumar, S.; et al. Molecular signatures of multiple myeloma progression through single cell RNA-Seq. Blood Cancer J. 2019, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Jovanović, K.K.; Roche-Lestienne, C.; Ghobrial, I.M.; Facon, T.; Quesnel, B.; Manier, S. Targeting MYC in multiple myeloma. Leukemia 2017, 32, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Van Valckenborgh, E.; Menu, E.; De Bruyne, E.; Vanderkerken, K. Understanding the hypoxic niche of multiple myeloma: Therapeutic implications and contributions of mouse models. Dis. Model. Mech. 2012, 5, 763–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viziteu, E.; Grandmougin, C.; Goldschmidt, H.; Seckinger, A.; Hose, D.; Klein, B.; Moreaux, J. Chetomin, targeting HIF-1α/p300 complex, exhibits antitumour activity in multiple myeloma. Br. J. Cancer 2016, 114, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Sprynski, A.C.; Hose, D.; Caillot, L.; Réme, T.; Shaughnessy, J.J.D.; Barlogie, B.; Seckinger, A.; Moreaux, J.; Hundemer, M.; Jourdan, M.; et al. The role of IGF-1 as a major growth factor for myeloma cell lines and the prognostic relevance of the expression of its receptor. Blood 2009, 113, 4614–4626. [Google Scholar] [CrossRef]
- Matthes, T.; Manfroi, B.; Huard, B. Revisiting IL-6 antagonism in multiple myeloma. Crit. Rev. Oncol. 2016, 105, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Arora, T.; Jelinek, D.F. Differential myeloma cell responsiveness to interferon-alpha correlates with differential induction of p19(INK4d) and cyclin D2 expression. J. Biol. Chem. 1998, 273, 11799–11805. [Google Scholar] [CrossRef] [Green Version]
- Ferlin-Bezombes, M.; Jourdan, M.; Liautard, J.; Brochier, J.; Rossi, J.F.; Klein, B. IFN-alpha is a survival factor for human myeloma cells and reduces dexamethasone-induced apoptosis. J. Immunol. 1998, 161, 2692–2699. [Google Scholar] [PubMed]
- Barlogie, B.; Kyle, R.A.; Anderson, K.C.; Greipp, P.R.; Lazarus, H.M.; Hurd, D.D.; McCoy, J.; Jr, D.F.M.; Dakhil, S.R.; Lanier, K.S.; et al. Standard Chemotherapy Compared With High-Dose Chemoradiotherapy for Multiple Myeloma: Final Results of Phase III US Intergroup Trial S9321. J. Clin. Oncol. 2006, 24, 929–936. [Google Scholar] [CrossRef]
- Cunningham, D.; Powles, R.; Malpas, J.; Raje, N.; Milan, S.; Viner, C.; Montes, A.; Hickish, T.; Nicolson, M.; Johnson, P.; et al. A randomized trial of maintenance interferon following high-dose chemotherapy in multiple myeloma: Long-term follow-up results. Br. J. Haematol. 1998, 102, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Kassambara, A.; Hose, D.; Moreaux, J.; Rème, T.; Torrent, J.; Rossi, J.F.; Goldschmidt, H.; Klein, B. Identification of Pluripotent and Adult Stem Cell Genes Unrelated to Cell Cycle and Associated with Poor Prognosis in Multiple Myeloma. PLoS ONE 2012, 7, e42161. [Google Scholar] [CrossRef]
- Bruyer, A.; Maes, K.; Herviou, L.; Kassambara, A.; Seckinger, A.; Cartron, G.; Rème, T.; Robert, N.; Requirand, G.; Boireau, S.; et al. DNMTi/HDACi combined epigenetic targeted treatment induces reprogramming of myeloma cells in the direction of normal plasma cells. Br. J. Cancer 2018, 118, 1062–1073. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, A.; Kumar, S.; Hofmeister, C.; Laubach, J.; Vij, R.; Dueck, A.; Gano, K.; Stewart, A.K. A Phase Ib Study of the combination of Aurora Kinase Inhibitor alisertib (MLN8237) and bortezomib in Relapsed or Refractory Multiple Myeloma. Br. J. Haematol. 2016, 174, 323–325. [Google Scholar] [CrossRef] [Green Version]
- Bolomsky, A.; Heusschen, R.; Schlangen, K.; Stangelberger, K.; Muller, J.; Schreiner, W.; Zojer, N.; Caers, J.; Ludwig, H. Maternal embryonic leucine zipper kinase is a novel target for proliferation-associated high-risk myeloma. Haematologica 2018, 103, 325–335. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread Genetic Heterogeneity in Multiple Myeloma: Implications for Targeted Therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.; Brunet, J.-P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nat. Cell Biol. 2011, 471, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Dao, D.D.; Sawyer, J.R.; Epstein, J.; Hoover, R.G.; Barlogie, B.; Tricot, G. Deletion of the retinoblastoma gene in multiple myeloma. Leukemia 1994, 8, 1280–1284. [Google Scholar] [PubMed]
- Weißbach, S.; Langer, C.; Puppe, B.; Nedeva, T.; Bach, E.; Kull, M.; Bargou, R.; Einsele, H.; Rosenwald, A.; Knop, S.; et al. The molecular spectrum and clinical impact ofDIS3mutations in multiple myeloma. Br. J. Haematol. 2014, 169, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Price-Troska, T.; Gonzalez-Paz, N.; Van Wier, S.; Jacobus, S.; Blood, E.; Henderson, K.; Oken, M.; Van Ness, B.; Greipp, P.; et al. Clinical significance of TP53 mutation in myeloma. Leukemia 2007, 21, 582–584. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.; Boyle, E.M.; Wardell, C.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef]
- Chavan, S.S.; He, J.; Tytarenko, R.; Deshpande, S.; Patel, P.; Bailey, M.; Stein, C.K.; Stephens, O.; Weinhold, N.; Petty, N.; et al. Bi-allelic inactivation is more prevalent at relapse in multiple myeloma, identifying RB1 as an independent prognostic marker. Blood Cancer J. 2017, 7, e535. [Google Scholar] [CrossRef] [PubMed]
- Benard, B.; Christofferson, A.; Legendre, C.; Jessica, A.; Sara, N.; Jennifer, Y.; Daniel, A.; Winnie, L.; Sagar, L.; Jonathan, J.K. FGFR3 Mutations Are an Adverse Prognostic Factor in Patients with t(4;14)(p16;q32) Multiple Myeloma: An Mmrf Commpass Analysis. Blood 2017, 130, 3027. [Google Scholar]
- Weinhold, N.; Ashby, C.; Rasche, L.; Chavan, S.S.; Stein, C.; Stephens, O.W.; Tytarenko, R.; Bauer, M.; Meißner, T.; Deshpande, S.; et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood 2016, 128, 1735–1744. [Google Scholar] [CrossRef]
- Xiao, H.; Chen, K.; Xu, H. MP85-05 Alternative splicing of EZH2 pre-mRNA by SF3B3 contributes to the tumorigenic potential of renal cancer. J. Urol. 2016, 195, 3428–3441. [Google Scholar] [CrossRef]
- Pawlyn, C.; Bright, M.D.; Buros, A.F.; Stein, C.K.; Walters, Z.; Aronson, L.I.; Mirabella, F.; Jones, J.R.; Kaiser, M.F.; Walker, B.A.; et al. Overexpression of EZH2 in multiple myeloma is associated with poor prognosis and dysregulation of cell cycle control. Blood Cancer J. 2017, 7, e549. [Google Scholar] [CrossRef]
- Ronca, R.; Ghedini, G.C.; Maccarinelli, F.; Sacco, A.; Locatelli, S.L.; Foglio, E.; Taranto, S.; Grillo, E.; Matarazzo, S.; Castelli, R.; et al. FGF Trapping Inhibits Multiple Myeloma Growth through c-Myc Degradation–Induced Mitochondrial Oxidative Stress. Cancer Res. 2020, 80, 2340–2354. [Google Scholar] [CrossRef] [Green Version]
- Alijaj, N.; Moutel, S.; Gouveia, Z.L.; Gray, M.; Roveri, M.; Dzhumashev, D.; Weber, F.; Meier, G.; Luciani, P.; Rössler, J.K.; et al. Novel FGFR4-Targeting Single-Domain Antibodies for Multiple Targeted Therapies against Rhabdomyosarcoma. Cancers 2020, 12, 3313. [Google Scholar] [CrossRef]
- Kimberley, F.C.; Screaton, G.R. Following a TRAIL: Update on a ligand and its five receptors. Cell Res. 2004, 14, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Menoret, E.; Bougie, P.G.; Geffroy-Luseau, A.; Daniels, S.; Moreau, P.; le Gouill, S.; Harousseau, J.-L.; Bataille, R.; Amiot, M.; Deceunynck, C. Mcl-1L cleavage is involved in TRAIL-R1– and TRAIL-R2–mediated apoptosis induced by HGS-ETR1 and HGS-ETR2 human mAbs in myeloma cells. Blood 2006, 108, 1346–1352. [Google Scholar] [CrossRef] [PubMed]
- Surget, S.; Chiron, D.; Gomez-Bougie, P.; Descamps, G.; Ménoret, E.; Bataille, R.; Moreau, P.; Le Gouill, S.; Amiot, M.; Pellat-Deceunynck, C. Cell Death via DR5, but not DR4, Is Regulated by p53 in Myeloma Cells. Cancer Res. 2012, 72, 4562–4573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardeleben, C.; Sharma, S.; Reeve, J.R.; Bassilian, S.; Frost, P.; Hoang, B.; Shi, Y.; Lichtenstein, A. Metabolomics Identifies Pyrimidine Starvation as the Mechanism of 5-Aminoimidazole-4-Carboxamide-1-β-Riboside-Induced Apoptosis in Multiple Myeloma Cells. Mol. Cancer Ther. 2013, 12, 1310–1321. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-J.; Wei, M.; Zhang, C.; Maxeiner, S.; Pak, C.; Botelho, S.C.; Trotter, J.; Sterky, F.H.; Südhof, T.C. Presynaptic Neuronal Pentraxin Receptor Organizes Excitatory and Inhibitory Synapses. J. Neurosci. 2017, 37, 1062–1080. [Google Scholar] [CrossRef] [PubMed]
- Kanda, M.; Shimizu, D.; Sawaki, K.; Nakamura, S.; Umeda, S.; Miwa, T.; Tanaka, H.; Tanaka, C.; Hayashi, M.; Iguchi, Y.; et al. Therapeutic monoclonal antibody targeting of neuronal pentraxin receptor to control metastasis in gastric cancer. Mol. Cancer 2020, 19, 131. [Google Scholar] [CrossRef]
- Xu, C.; Tian, G.; Jiang, C.; Xue, H.; Kuerbanjiang, M.; Sun, L.; Gu, L.; Zhou, H.; Liu, Y.; Zhang, Z.; et al. NPTX2 promotes colorectal cancer growth and liver metastasis by the activation of the canonical Wnt/β-catenin pathway via FZD6. Cell Death Dis. 2019, 10, 217. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Univariate COX Analyses | ||
|---|---|---|
| Prognostic Variable | Proportional HR | p-Value |
| RNA-seq-based risk score | 8.787 | <0.0001 |
| del17p | 1.609 | NS |
| 1qgain | 0.958 | NS |
| t(12;14) | 1.890 | NS |
| ATRmut | 1.780 | NS |
| CREBBPmut | 1.419 | NS |
| MAP3K1mut | 0.991 | NS |
| PMS1mut | 5.013 | 0.006 |
| TP53mut | 1.162 | NS |
| Multivariate COX Analysis (Cytogenetic Abnormalities) | ||
|---|---|---|
| Prognostic Variable | Proportional HR | p-Value |
| RNA-seq-based risk score | 8.082 | <0.0001 |
| del17p | 1.576 | NS |
| 1qgain | 0.954 | NS |
| t(12;14) | 1.600 | NS |
| Multivariate COX Analysis (Mutations) | ||
|---|---|---|
| Prognostic Variable | Proportional HR | p-Value |
| RNA-seq-based risk score | 8.741 | <0.0001 |
| ATRmut | 1.805 | NS |
| CREBBPmut | 1.436 | NS |
| MAP3K1mut | 1.050 | NS |
| PMS1mut | 5.099 | 0.006 |
| TP53mut | 1.161 | NS |
| Gene | Dataset | T.Statistic | p-Value |
|---|---|---|---|
| ATP8B1 | RNAi (Broad) | −4.4246306 | 1.18 × 10−5 |
| FGFR4 | Combined RNAi (Broad, Novartis, Marcotte) | −4.6107503 | 4.74 × 10−6 |
| FOXD4 | Combined RNAi (Broad, Novartis, Marcotte) | −4.8020113 | 1.91 × 10−6 |
| ISG20 | CRISPR (Avana) Public 20Q1 | −3.949974 | 8.56 × 10−5 |
| MX1 | Combined RNAi (Broad, Novartis, Marcotte) | −7.990899 | 5.30 × 10−15 |
| NDC1 | CRISPR (Avana) Public 20Q1 | −5.029908 | 6.15 × 10−7 |
| NPTXR | Combined RNAi (Broad, Novartis, Marcotte) | −5.638059 | 2.47 × 10−8 |
| SF3B3 | CRISPR (Avana) Public 20Q1 | −4.0305542 | 6.13 × 10−5 |
| TMEM171 | RNAi (Novartis) | −3.6151729 | 3.39 × 10−3 |
| TNFRSF10B | RNAi (Novartis) | −3.996171 | 7.67 × 10−5 |
| UMPS | CRISPR (Avana) Public 20Q1 | −4.6553134 | 3.83 × 10−6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alaterre, E.; Vikova, V.; Kassambara, A.; Bruyer, A.; Robert, N.; Requirand, G.; Bret, C.; Herbaux, C.; Vincent, L.; Cartron, G.; et al. RNA-Sequencing-Based Transcriptomic Score with Prognostic and Theranostic Values in Multiple Myeloma. J. Pers. Med. 2021, 11, 988. https://doi.org/10.3390/jpm11100988
Alaterre E, Vikova V, Kassambara A, Bruyer A, Robert N, Requirand G, Bret C, Herbaux C, Vincent L, Cartron G, et al. RNA-Sequencing-Based Transcriptomic Score with Prognostic and Theranostic Values in Multiple Myeloma. Journal of Personalized Medicine. 2021; 11(10):988. https://doi.org/10.3390/jpm11100988
Chicago/Turabian StyleAlaterre, Elina, Veronika Vikova, Alboukadel Kassambara, Angélique Bruyer, Nicolas Robert, Guilhem Requirand, Caroline Bret, Charles Herbaux, Laure Vincent, Guillaume Cartron, and et al. 2021. "RNA-Sequencing-Based Transcriptomic Score with Prognostic and Theranostic Values in Multiple Myeloma" Journal of Personalized Medicine 11, no. 10: 988. https://doi.org/10.3390/jpm11100988