Matching Clinical Diagnosis and Amyloid Biomarkers in Alzheimer’s Disease and Frontotemporal Dementia

 , , , ,
, , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Neuropsychological Assessment
2.3. PET Imaging Acquisition and Analysis
2.4. CSF Collection and Biomarkers Analysis
2.5. Apolipoprotein E ε4 Genotyping, FAD and FTD Genes Mutation Analysis
2.6. Statistical Analysis
2.7. Data Availability
3. Results
3.1. Description of the Sample
3.2. Aβ Biomarkers Positivity Prevalence
3.3. Concordance between CSF and Amyloid PET
3.4. Concordance between Aβ1–42 and Aβ42/40
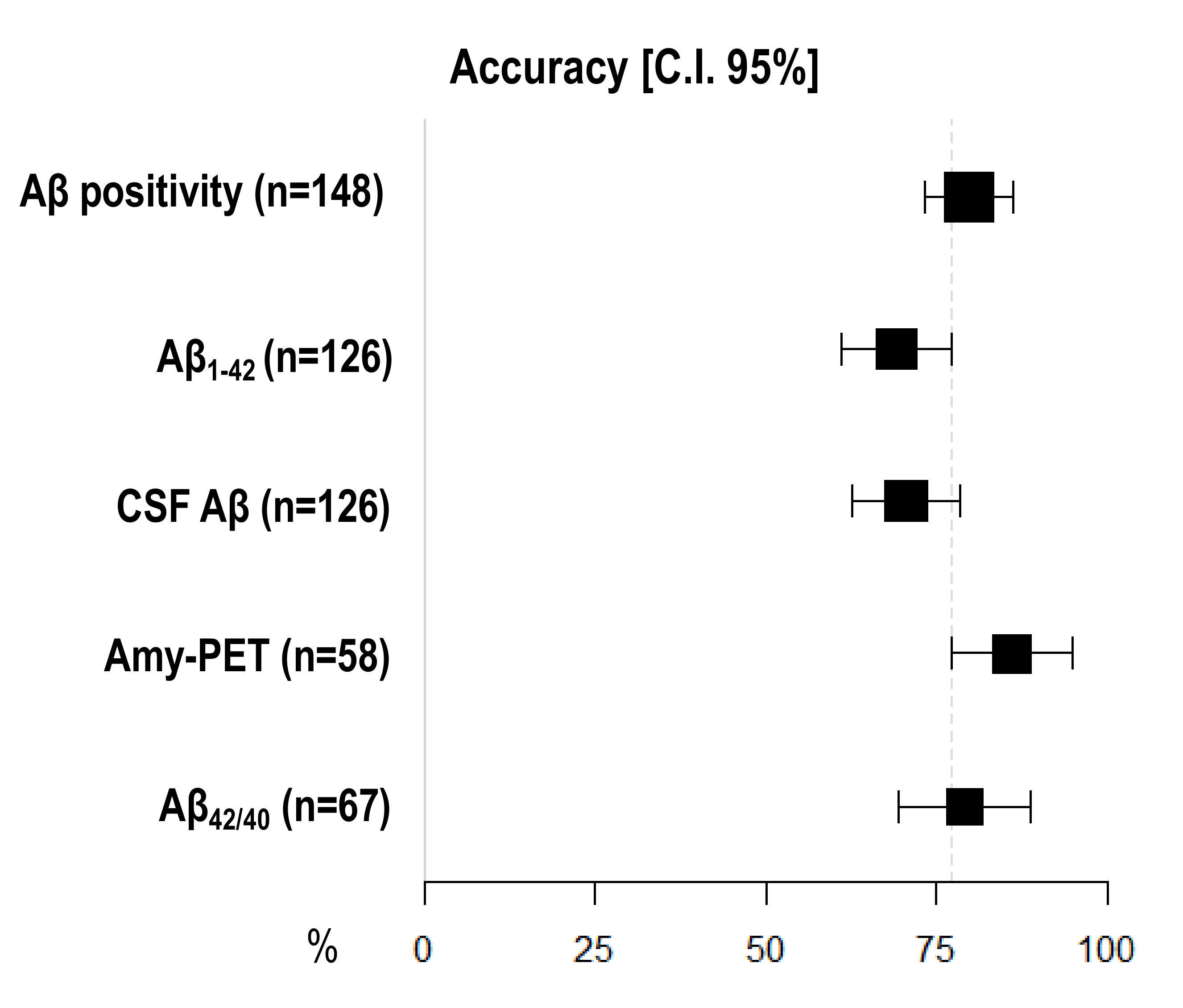
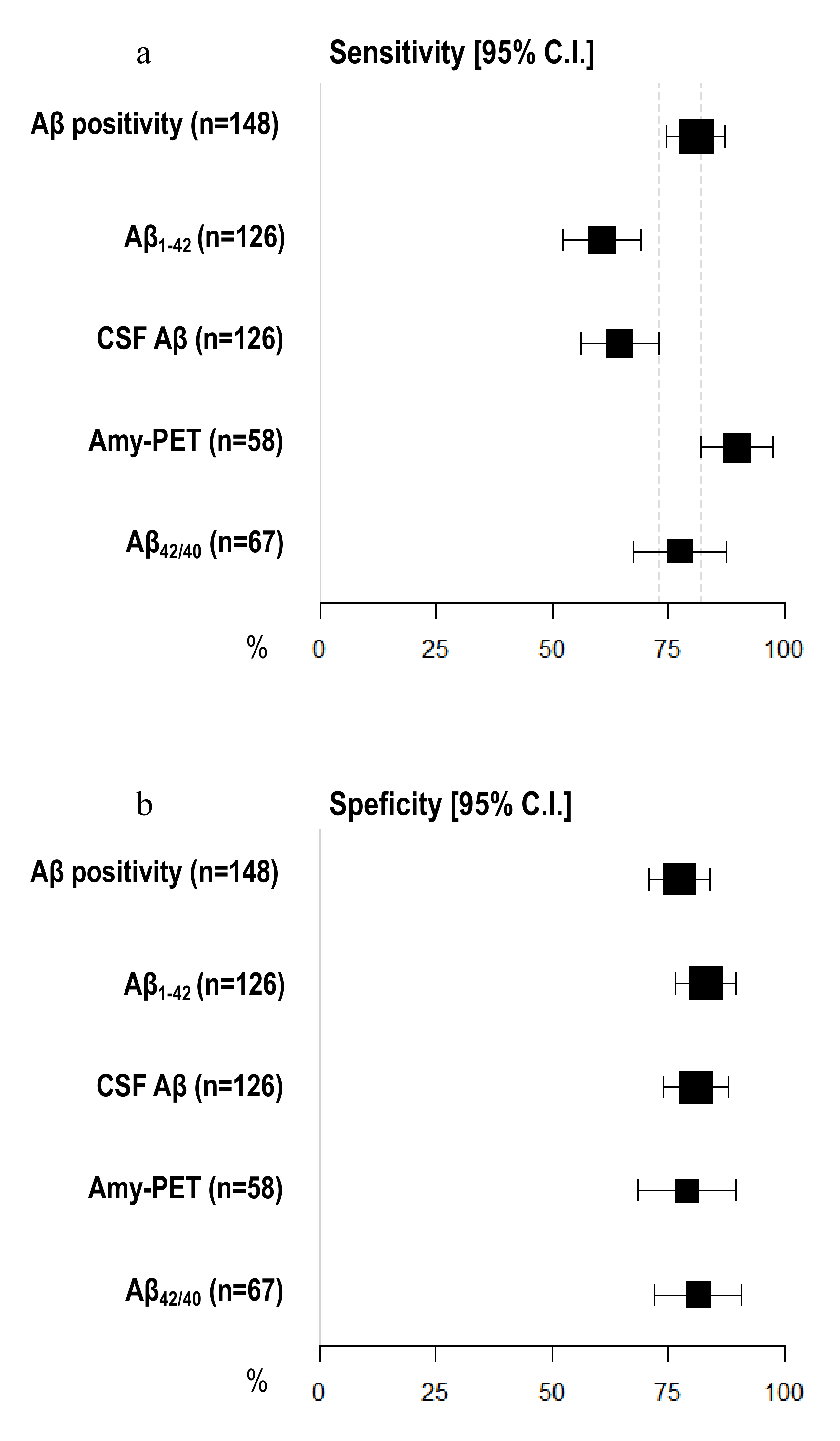
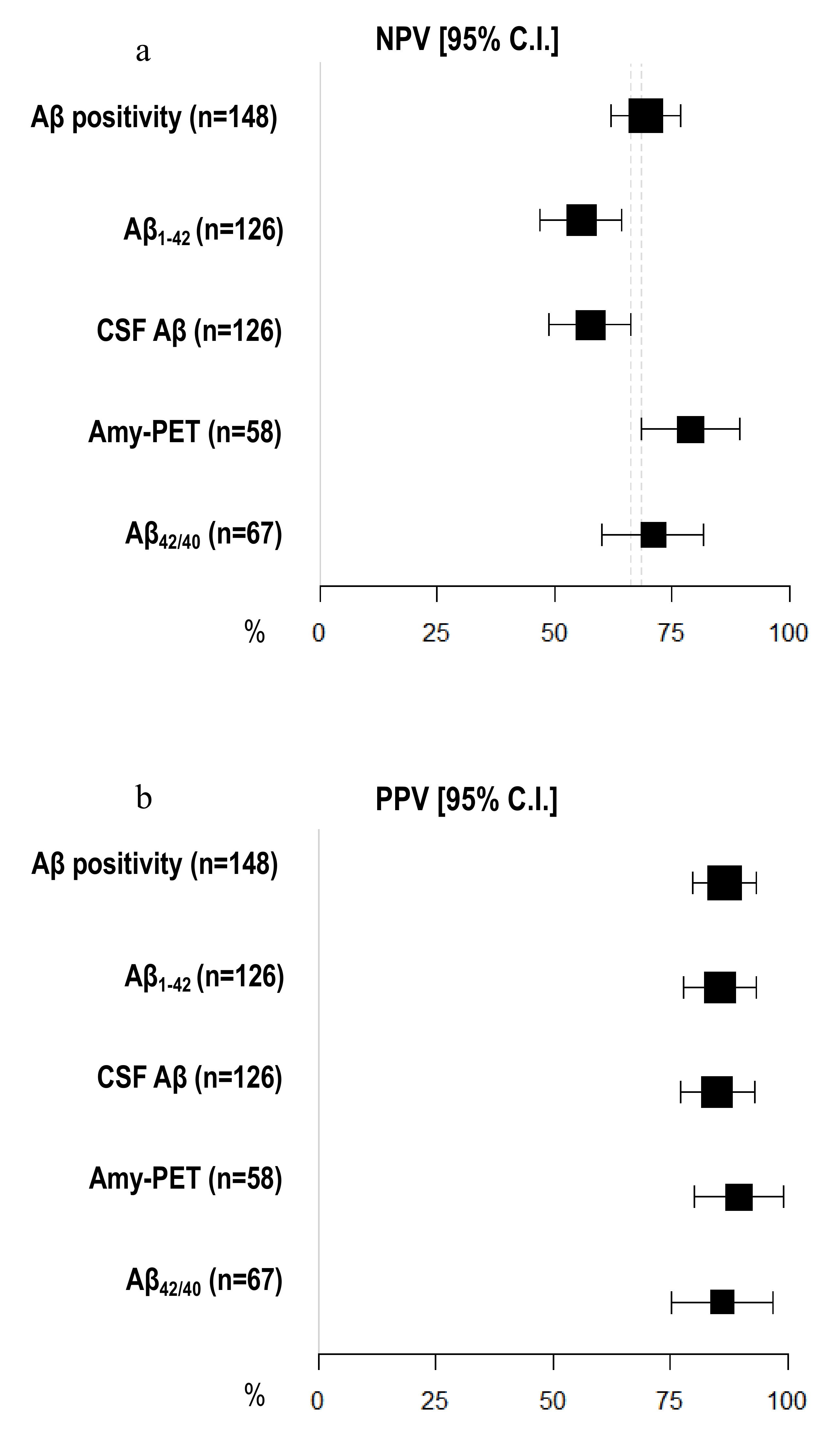
3.5. Diagnostic Accuracy of Amyloid Burden Biomarkers
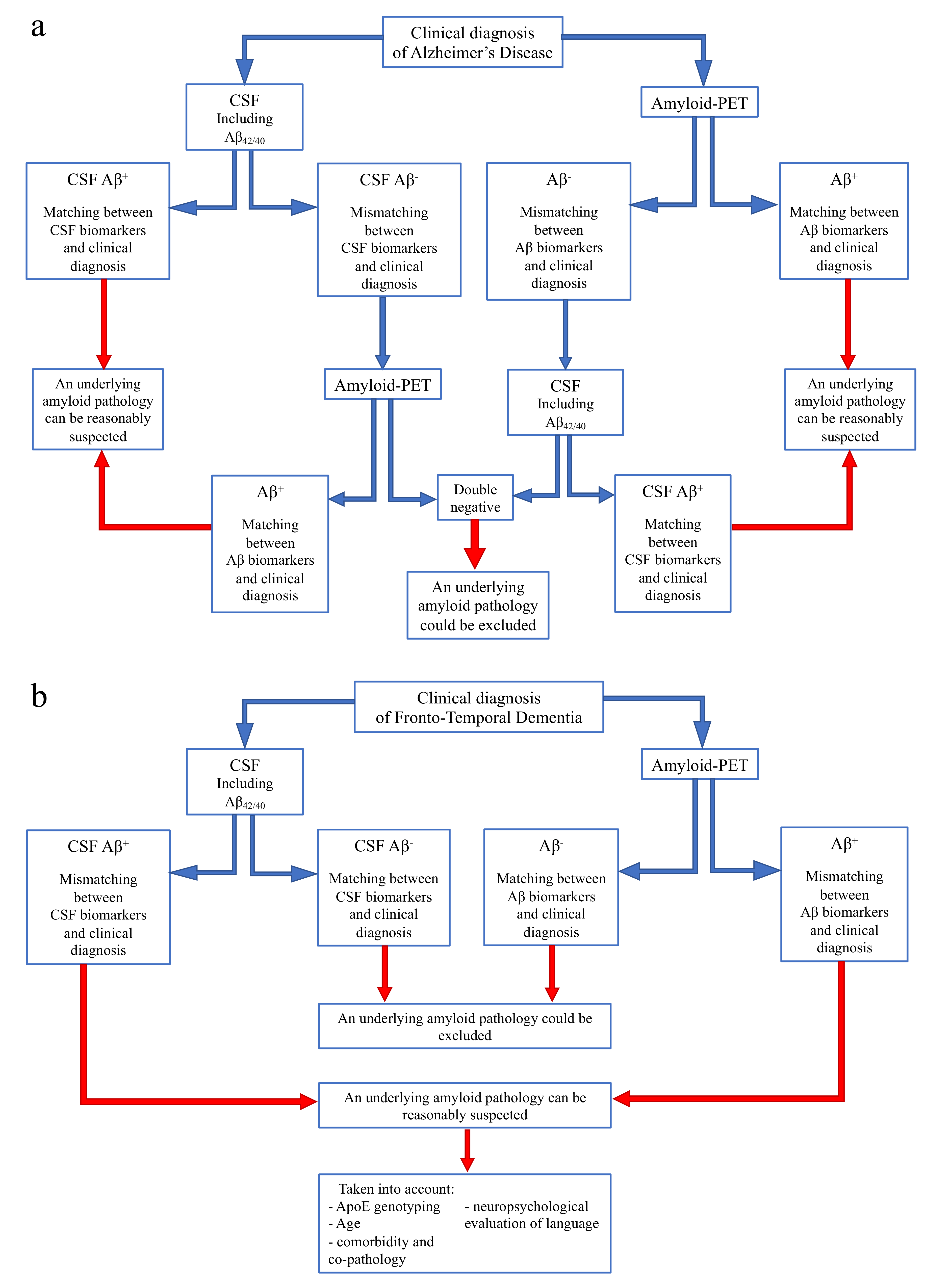
4. Discussion
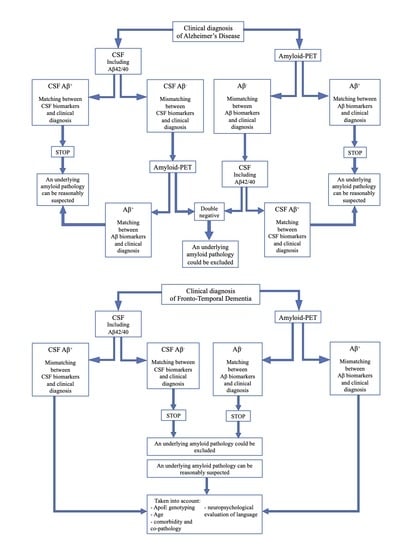
- If CSF analysis including Aβ42/40 ratio is positive, underlying Aβ pathology can be reasonably suspected and amyloid-PET might be avoided. Similarly, if amyloid-PET detects cortical amyloid deposition, CSF analysis cannot be performed. In conclusion, as also reported in a recent study [73], the matching between clinical diagnosis and a single amyloid biomarker could be sufficient, considering the high PPV of both CSF analysis and amyloid-PET.
- In case of a mismatch between clinical diagnosis and one amyloid biomarker, we suggest performing the other analysis, due to the low NPV of both CSF and amyloid-PET. In particular:
- ○
- If CSF shows Aβ-, amyloid-PET should be performed, also considering previous results highlighting an advantage of amyloid-PET when used as a second biomarker [74].
- ○
- If amyloid-PET is negative for cortical deposition, CSF analysis including Aβ42/40 ratio is suggested.
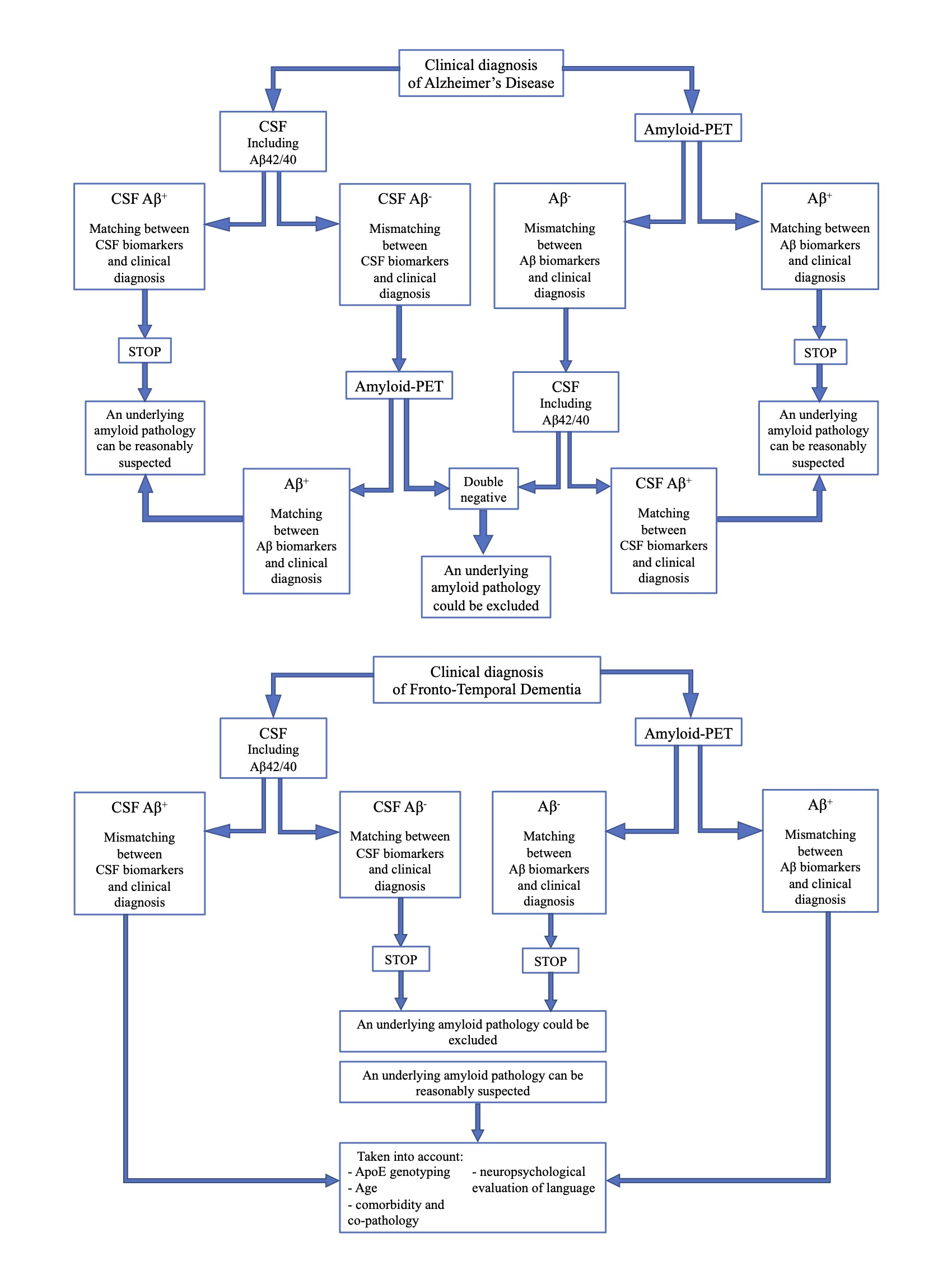
In conclusion, if both the analyses show Aβ-, an underlying amyloid pathology could most probably be excluded, and a revision of clinical diagnosis should be considered. On the contrary, if the second analysis detects Aβ+, and matching between clinical diagnosis and biomarkers is achieved, amyloid pathology can be reasonably suspected.- If CSF analysis or amyloid-PET shows Aβ-, an underlying amyloid pathology could most probably be excluded. Thus, the performance of the other biomarker is not required, due to the high concordance between these two analyses in FTD cases.
- If CSF or amyloid PET detects Aβ+, some factors should be taken into account, in particular ApoE genotyping, age, and a deep neuropsychological evaluation of language to exclude a diagnosis of PPA. Moreover, the presence of comorbidity and co-pathology should be considered, as also suggested by other recent reports [73].
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- Strozyk, D.; Blennow, K.; White, L.R.; Launer, L.J. CSF Abeta 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology 2003, 60, 652–656. [Google Scholar] [CrossRef]
- Lewczuk, P.; Esselmann, H.; Otto, M.; Maler, J.M.; Henkel, A.W.; Henkel, M.K.; Eikenberg, O.; Antz, C.; Krause, W.R.; Reulbach, U.; et al. Neurochemical diagnosis of Alzheimer’s dementia by CSF Abeta42, Abeta42/Abeta40 ratio and total tau. Neurobiol. Aging 2004, 25, 273–281. [Google Scholar] [CrossRef]
- Tapiola, T.; Overmyer, M.; Lehtovirta, M.; Helisalmi, S.; Ramberg, J.; Alafuzoff, I.; Riekkinen, P.S.; Soininen, H. The level of cerebrospinal fluid tau correlates with neurofibrillary tangles in Alzheimer’s disease. Neuroreport 1997, 8, 3961–3963. [Google Scholar] [CrossRef] [PubMed]
- Buerger, K.; Ewers, M.; Pirttila, T.; Zinkowski, R.; Alafuzoff, I.; Teipel, S.J.; DeBernardis, J.; Kerkman, D.; McCulloch, C.; Hampel, H.; et al. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain 2006, 129, 3035–3041. [Google Scholar] [CrossRef] [PubMed]
- Tapiola, T.; Alafuzoff, I.; Herukka, S.K.; Parkkinen, L.; Hartikainen, P.; Soininen, H.; Pirttilä, T. Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch. Neurol. 2009, 66, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Ikonomovic, M.D.; Klunk, W.E.; Abrahamson, E.E.; Mathis, C.A.; Price, J.C.; Tsopelas, N.D.; Lopresti, B.J.; Ziolko, S.; Bi, W.; Paljug, W.R.; et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain 2008, 131, 1630–1645. [Google Scholar] [CrossRef] [PubMed]
- Sabri, O.; Sabbagh, M.N.; Seibyl, J.; Barthel, H.; Akatsu, H.; Ouchi, Y.; Senda, K.; Murayama, S.; Ishii, K.; Takao, M.; et al. Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer’s disease: Phase 3 study. Alzheimers Dement. 2015, 11, 964–974. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Gamez, J.E.; Singh, U.; Sadowsky, C.H.; Villena, T.; Sabbagh, M.N.; Beach, T.G.; Duara, R.; Fleisher, A.S.; Frey, K.A.; et al. Phase 3 trial of flutemetamol labeled with radioactive fluorine 18 imaging and neuritic plaque density. JAMA Neurol. 2015, 72, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.M.; Pontecorvo, M.J.; Beach, T.G.; Bedell, B.J.; Coleman, R.E.; Doraiswamy, P.M.; Fleisher, A.S.; Reiman, E.M.; Sabbagh, M.N.; Sadowsky, C.H.; et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-β plaques: A prospective cohort study. Lancet Neurol. 2012, 11, 669–678. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA research framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Fagan, A.M. What does it mean to be “amyloid-positive”? Brain 2015, 138, 514–516. [Google Scholar] [CrossRef]
- Fagan, A.M.; Mintun, M.A.; Shah, A.R.; Aldea, P.; Roe, C.M.; Mach, R.H.; Marcus, D.; Morris, J.C.; Holtzman, D.M. Cerebrospinal fluid tau and ptau181 increase with cortical amyloid deposition in cognitively normal individuals: Implications for future clinical trials of Alzheimer’s disease. EMBO Mol. Med. 2009, 1, 371–380. [Google Scholar] [CrossRef]
- Zwan, M.D.; Rinne, J.O.; Hasselbalch, S.G.; Nordberg, A.; Lleó, A.; Herukka, S.K.; Soininen, H.; Law, I.; Bahl, J.M.; Carter, S.F.; et al. Use of amyloid-PET to determine cutpoints for CSF markers: A multicenter study. Neurol. Int. 2016, 86, 50–58. [Google Scholar] [CrossRef]
- Blennow, K.; Mattsson, N.; Schöll, M.; Hansson, O.; Zetterberg, H. Amyloid biomarkers in Alzheimer’s disease. Trends Pharmacol. Sci. 2015, 36, 297–309. [Google Scholar] [CrossRef]
- Rohrer, J.D.; Lashley, T.; Schott, J.M.; Warren, J.E.; Mead, S.; Isaacs, A.M.; Beck, J.; Hardy, J.; de Silva, R.; Warrington, E.; et al. Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain 2011, 134, 2565–2581. [Google Scholar] [CrossRef] [PubMed]
- Knibb, J.A.; Xuereb, J.H.; Patterson, K.; Hodges, J.R. Clinical and pathological characterization of progressive aphasia. Ann. Neurol. 2006, 59, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Alladi, S.; Xuereb, J.; Bak, T.; Nestor, P.; Knibb, J.; Patterson, K.; Hodges, J.R. Focal cortical presentations of Alzheimer’s disease. Brain 2007, 130, 2636–2645. [Google Scholar] [CrossRef]
- Blennow, K. A review of fluid biomarkers for Alzheimer’s Disease: Moving from CSF to blood. Neurol. Ther. 2017, 6, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Paterson, R.W.; Slattery, C.F.; Poole, T.; Nicholas, J.M.; Magdalinou, N.K.; Toombs, J.; Chapman, M.D.; Lunn, M.P.; Heslegrave, A.J.; Foiani, M.S.; et al. Cerebrospinal fluid in the differential diagnosis of Alzheimer’s disease: Clinical utility of an extended panel of biomarkers in a specialist cognitive clinic. Alzheimers Res. Ther. 2018, 10, 32. [Google Scholar] [CrossRef]
- Neary, D.; Snowden, J.S.; Gustafson, L.; Passant, U.; Stuss, D.; Black, S.; Freedman, M.; Kertesz, A.; Robert, P.H.; Albert, M.; et al. Frontotemporal lobar degeneration: A consensus on clinical diagnostic criteria. Neurology 1998, 51, 1546–1554. [Google Scholar] [CrossRef]
- Bracco, L.; Amaducci, L.; Pedone, D.; Bino, G.; Lazzaro, M.P.; Carella, F.; D’Antona, R.; Gallato, R.; Denes, G. Italian Multicentre Study on Dementia (SMID): A neuropsychological test battery for assessing Alzheimer’s disease. J. Psychiatr. Res. 1990, 24, 213–226. [Google Scholar] [CrossRef]
- Caffarra, P.; Vezzadini, G.; Dieci, F.; Zonato, F.; Venneri, A. Rey-Osterrieth complex figure: Normative values in an Italian population sample. Neurol. Sci. 2002, 22, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Baddeley, A.; Della Sala, S.; Papagno, C.; Spinnler, H. Dual-task performance in dysexecutive and nondysexecutive patients with a frontal lesion. Neuropsychology 1997, 11, 187–194. [Google Scholar] [CrossRef]
- Spinnler, H.; Tognoni, G. Standardizzazione e taratura italiana di test neuropsicologici: Gruppo italiano per lo studio neuropsicologico dell’invecchiamento. Ital. J. Neurol. Sci. 1987, 6, 21–120. [Google Scholar]
- Giovagnoli, A.R.; Del Pesce, M.; Mascheroni, S.; Simoncelli, M.; Laiacona, M.; Capitani, E. Trailmaking test: Normative values from 287 normaladultcontrols. Ital. J. Neurol. Sci. 1996, 17, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Brazzelli, M.; Della Sala, S.; Laiacona, M. Calibration of the Italian version of the Rivermead Behavioural Memory Test: A test for the ecological evaluation of memory. Boll. Psicol. Appl. 1993, 206, 33–42. [Google Scholar]
- Colombo, L.; Sartori, G.; Brivio, C. Stima del quoziente intellettivo tramite l’applicazione del TIB (Test Breve di Intelligenza). Giomale Ital. Psicol. 2002, 3, 613–663. [Google Scholar]
- Nelson, H. National Adult Reading Test (NART): For the Assessment of Premorbid Intelligence in Patients with Dementia: Test Manual; NFER-Nelson: Windsor, UK, 1982. [Google Scholar]
- Hamilton, M. A rating scale for depression. J. Neurol. Neurosurg. Psychiatry 1960, 23, 56–62. [Google Scholar] [CrossRef]
- Minoshima, S.; Drzezga, A.E.; Barthel, H.; Bohnen, N.; Djekidel, M.; Lewis, D.H.; Mathis, C.A.; McConathy, J.; Nordberg, A.; Sabri, O.; et al. SNMMI Procedure Standard/EANM Practice Guideline for Amyloid PET Imaging of the Brain 1.0. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2016, 57, 1316–1322. [Google Scholar] [CrossRef]
- Molinuevo, J.L.; Gispert, J.D.; Dubois, B.; Heneka, M.T.; Lleo, A.; Engelborghs, S.; Pujol, J.; de Souza, L.C.; Alcolea, D.; Jessen, F.; et al. The AD-CSF-index discriminates Alzheimer’s disease patients from healthy controls: A validation study. J. Alzheimers Dis. JAD 2013, 36, 67–77. [Google Scholar] [CrossRef]
- Alcolea, D.; Pegueroles, J.; Muñoz, L.; Camacho, V.; López-Mora, D.; Fernández-León, A.; Le Bastard, N.; Huyck, E.; Nadal, A.; Olmedo, V.; et al. Agreement of amyloid PET and CSF biomarkers for Alzheimer’s disease on Lumipulse. Ann. Clin. Transl. Neurol. 2019, 6, 1815–1824. [Google Scholar] [CrossRef]
- Sorbi, S.; Nacmias, B.; Forleo, P.; Latorraca, S.; Gobbini, I.; Bracco, L.; Piacentini, S.; Amaducci, L. ApoE allele frequencies in Italian sporadic and familial Alzheimer’s disease. Neurosci. Lett. 1994, 177, 100–102. [Google Scholar] [CrossRef]
- Primer3 (v.0.4.0) Pick primers from a DNA sequence. Available online: http://bioinfo.ut.ee/primer3-0.4.0/primer3/ (accessed on 8 September 2020).
- Bessi, V.; Piaceri, I.; Padiglioni, S.; Bagnoli, S.; Berti, V.; Sorbi, S.; Nacmias, B. Crossed aphasia in nonfluent variant of primary progressive aphasia carrying a GRN mutation. J. Neurol. Sci. 2018, 15, 34–37. [Google Scholar] [CrossRef]
- Mazzeo, S.; Polito, C.; Padiglioni, S.; Berti, V.; Bagnoli, S.; Lombardi, G.; Piaceri, I.; Carraro, M.; De Cristofaro, M.T.; Passeri, A.; et al. Linguistic profiles, brain metabolic patterns and rates of amyloid-β biomarker positivity in patients with mixed primary progressive aphasia. Neurobiol. Aging 2020, 96, 155–164. [Google Scholar] [CrossRef]
- Varma, A.R.; Snowden, J.S.; Lloyd, J.J.; Talbot, P.R.; Mann, D.M.; Neary, D. Evaluation of the NINCDS-ADRDA criteria in the differentiation of Alzheimer’s disease and frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 1999, 66, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Rabinovici, G.D.; Rosen, H.J.; Alkalay, A.; Kornak, J.; Furst, A.J.; Agarwal, N.; Mormino, E.C.; O’Neil, J.P.; Janabi, M.; Karydas, A.; et al. Amyloid vs FDG-PET in the differential diagnosis of AD and FTLD. Neurology 2001, 77, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Landau, S.M.; Lu, M.; Joshi, A.D.; Pontecorvo, M.; Mintun, M.A.; Trojanowski, J.Q.; Shaw, L.M.; Jagust, W.J.; Alzheimer’s Disease Neuroimaging Initiative. Comparing positron emission tomography imaging and cerebrospinal fluid measurements of β-amyloid. Ann. Neurol. 2013, 74, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Jagust, W.J.; Landau, S.M.; Shaw, L.M.; Trojanowski, J.Q.; Koeppe, R.A.; Reiman, E.M.; Foster, N.L.; Petersen, R.C.; Weiner, M.W.; Price, J.C.; et al. Relationships between biomarkers in aging and dementia. Neurology 2009, 73, 1193–1199. [Google Scholar] [CrossRef]
- Mattsson, N.; Insel, P.S.; Landau, S.; Jagust, W.; Donohue, M.; Shaw, L.M.; Trojanowski, J.Q.; Zetterberg, H.; Blennow, K.; Weiner, M. Alzheimer’s Disease Neuroimaging Initiativea. Diagnostic accuracy of CSF Ab42 and florbetapir PET for Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2014, 1, 534–543. [Google Scholar] [CrossRef]
- Spies, P.E.; Slats, D.; Sjogren, J.M.; Kremer, B.P.; Verhey, F.R.; Rikkert, M.G.; Verbeek, M.M. The cerebrospinal fluid amyloid beta42/40 ratio in the differentiation of Alzheimer’s disease from non-Alzheimer’s dementia. Curr. Alzheimer Res. 2010, 7, 470–476. [Google Scholar] [CrossRef]
- Slaets, S.; Le Bastard, N.; Martin, J.J.; Sleegers, K.; Van Broeckhoven, C.; De Deyn, P.P.; Engelborghs, S. Cerebrospinal fluid Abeta1-40 improves differential dementia diagnosis in patients with intermediate P-tau181P levels. J. Alzheimers Dis. 2013, 36, 759–767. [Google Scholar] [CrossRef]
- Portelius, E.; Westman-Brinkmalm, A.; Zetterberg, H.; Blennow, K. Determination of beta-amyloid peptide signatures in cerebrospinal fluid using immunoprecipitation-mass spectrometry. J. Proteome Res. 2006, 5, 1010–1016. [Google Scholar] [CrossRef]
- Hansson, O.; Lehmann, S.; Otto, M.; Zetterberg, H.; Lewczuk, P. Advantages and disadvantages of the use of the CSF Amyloid β (Aβ) 42/40 ratio in the diagnosis of Alzheimer’s Disease. Alzheimers Res. Ther. 2019, 11, 34. [Google Scholar] [CrossRef]
- Leuzy, A.; Carter, S.F.; Chiotis, K.; Almkvist, O.; Wall, A.; Nordberg, A. Concordance and Diagnostic Accuracy of [11C]PIB PET and Cerebrospinal Fluid Biomarkers in a Sample of Patients with Mild Cognitive Impairment and Alzheimer’s Disease. J. Alzheimers Dis. 2015, 45, 1077–1088. [Google Scholar] [CrossRef]
- Jung, N.Y.; Kim, E.S.; Kim, H.S.; Jeon, S.; Lee, M.J.; Pak, K.; Lee, J.H.; Lee, Y.M.; Lee, K.; Shin, J.H.; et al. Comparison of Diagnostic Performances Between Cerebrospinal Fluid Biomarkers and Amyloid PET in a Clinical Setting. J. Alzheimers Dis. 2020, 74, 473–490. [Google Scholar] [CrossRef] [PubMed]
- De Wilde, A.; Reimand, J.; Teunissen, C.E.; Zwan, M.; Windhorst, A.D.; Boellaard, R.; van der Flier, W.M.; Scheltens, P.; van Berckel, B.N.M.; Bouwman, F.; et al. Discordant amyloid-β PET and CSF biomarkers and its clinical consequences. Alzheimers Res. Ther. 2019, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Reimand, J.; de Wilde, A.; Teunissen, C.E.; Zwan, M.; Windhorst, A.D.; Boellaard, R.; Barkhof, F.; van der Flier, W.M.; Scheltens, P.; van Berckel, B.N.M.; et al. PET and CSF amyloid-β status are differently predicted by patient features: Information from discordant cases. Alzheimers Res. Ther. 2019, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Leuzy, A.; Chiotis, K.; Hasselbalch, S.G.; Rinne, J.O.; de Mendonça, A.; Otto, M.; Lleó, A.; Castelo-Branco, M.; Santana, I.; Johansson, J.; et al. Pittsburgh compound B imaging and cerebrospinal fluid amyloid-β in a multicentre European memory clinic study. Brain 2016, 139, 2540–2553. [Google Scholar] [CrossRef]
- Palmqvist, S.; Zetterberg, H.; Mattsson, N.; Johansson, P.; Alzheimer’s Disease Neuroimaging Initiative; Minthon, L.; Blennow, K.; Olsson, M.; Hansson, O.; Swedish BioFINDER Study Group. Detailed comparison of amyloid PET and CSF biomarkers for identifying early Alzheimer disease. Neurology 2015, 85, 1240–1249. [Google Scholar] [CrossRef]
- Mattsson, N.; Insel, P.S.; Donohue, M.; Landau, S.; Jagust, W.J.; Shaw, L.M.; Trojanowski, J.Q.; Zetterberg, H.; Blennow, K.; Weiner, M.W.; et al. Independent information from cerebrospinal fluid amyloid-β and florbetapir imaging in Alzheimer’s disease. Brain 2015, 138, 772–783. [Google Scholar] [CrossRef]
- Palmqvist, S.; Mattsson, N.; Hansson, O.; Initiative, A. Cerebrospinal fluid analysis detects cerebral amyloid-β accumulation earlier than positron emission tomography. Brain 2016, 139, 1226–1236. [Google Scholar] [CrossRef]
- Morris, E.; Chalkidou, A.; Hammers, A.; Peacock, J.; Summers, J.; Keevil, S. Diagnostic accuracy of (18)F amyloid PET tracers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 374–385. [Google Scholar] [CrossRef]
- Mattsson, N.; Rajendran, L.; Zetterberg, H.; Gustavsson, M.; Andreasson, U.; Olsson, M.; Brinkmalm, G.; Lundkvist, J.; Jacobson, L.H.; Perrot, L.; et al. BACE1 Inhibition induces a specific cerebrospinal fluid beta-amyloid pattern that identifies drug effects in the central nervous system. PLoS ONE 2012, 7, e31084. [Google Scholar] [CrossRef]
- Reiman, E.M.; Quiroz, Y.T.; Fleisher, A.S.; Chen, K.; Velez-Pardo, C.; Jimenez-Del-Rio, M.; Fagan, A.M.; Shah, A.R.; Alvarez, S.; Arbelaez, A.; et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: A case-control study. Lancet Neurol. 2012, 11, 1048–1056. [Google Scholar] [CrossRef]
- Potter, R.; Patterson, B.W.; Elbert, D.L.; Ovod, V.; Kasten, T.; Sigurdson, W.; Mawuenyega, K.; Blazey, T.; Goate, A.; Chott, R.; et al. Increased in vivo amyloid-b42 production, exchange, and loss in presenilin mutation carriers. Sci. Transl. Med. 2013, 5, 189ra77. [Google Scholar] [CrossRef] [PubMed]
- Scholl, M.; Wall, A.; Thordardottir, S.; Ferreira, D.; Bogdanovic, N.; Långström, B.; Almkvist, O.; Graff, C.; Nordberg, A. Low PiB PET retention in presence of pathologic CSF biomarkers in Arctic APP mutation carriers. Neurology 2012, 79, 229–236. [Google Scholar] [CrossRef]
- Mattsson, N.; Axelsson, M.; Haghighi, S.; Malmestrom, C.; Wu, G.; Anckarsäter, R.; Sankaranarayanan, S.; Andreasson, U.; Fredrikson, S.; Gundersen, A.; et al. Reduced cerebrospinal fluid BACE1 activity in multiple sclerosis. Mult. Scler. 2009, 15, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Selnes, P.; Blennow, K.; Zetterberg, H.; Grambaite, R.; Rosengren, L.; Johnsen, L.; Stenset, V.; Fladby, T. Effects of cerebrovascular disease on amyloid precursor protein metabolites in cerebrospinal fluid. Cereb. Fluid Res. 2010, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Mikulskis, A.; Fagan, A.M.; Teunissen, C.; Zetterberg, H.; Vanderstichele, H.; Molinuevo, J.L.; Shaw, L.M.; Vandijck, M.; Verbeek, M.M.; et al. The impact of preanalytical variables on measuring cerebrospinal fluid biomarkers for Alzheimer’s disease diagnosis: A review. Alzheimers Dement. 2018, 14, 1313–1333. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, R.; Jansen, W.J.; Rabinovici, G.D.; Knol, D.L.; van der Flier, W.M.; van Berckel, B.N.; Scheltens, P.; Visser, P.J.; Amyloid PET Study Group; Verfaillie, S.C.; et al. Prevalence of amyloid PET positivity in dementia syndromes: A meta-analysis. JAMA 2015, 313, 1939–1949. [Google Scholar] [CrossRef]
- Clark, C.M.; Schneider, J.A.; Bedell, B.J.; Beach, T.G.; Bilker, W.B.; Mintun, M.A.; Pontecorvo, M.J.; Hefti, F.; Carpenter, A.P.; Flitter, M.L.; et al. Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA 2011, 305, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Weston, P.S.; Paterson, R.W.; Dickson, J.; Barnes, A.; Bomanji, J.B.; Kayani, I.; Lunn, M.P.; Mummery, C.J.; Warren, J.D.; Rossor, M.N.; et al. Diagnosing Dementia in the Clinical Setting: Can Amyloid PET Provide Additional Value Over Cerebrospinal Fluid? J. Alzheimers Dis. 2016, 54, 1297–1302. [Google Scholar] [CrossRef]
- Robinson, J.L.; Lee, E.B.; Xie, S.X.; Rennert, L.; Suh, E.; Bredenberg, C.; Caswell, C.; Van Deerlin, V.M.; Yan, N.; Yousef, A.; et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 2018, 141, 2181–2193. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, D.; Gorno-Tempini, M.L.; Rabinovici, G.D.; Santos-Santos, M.A.; Seeley, W.; Miller, B.L.; Pijnenburg, Y.; Keulen, M.A.; Groot, C.; van Berckel, B.N.M.; et al. Prevalence of amyloid-b pathology in distinct variants of primary progressive aphasia. Ann. Neurol. 2018, 84, 729–740. [Google Scholar] [CrossRef]
- Chételat, G.; Arbizu, J.; Barthel, H.; Garibotto, V.; Law, I.; Morbelli, S.; van de Giessen, E.; Agosta, F.; Barkhof, F.; Brooks, D.J.; et al. Amyloid-PET and 18F-FDG-PET in the diagnostic investigation of Alzheimer’s disease and other dementias. Lancet Neurol. 2020, 19, 951–962. [Google Scholar] [CrossRef]
- Ramusino, M.C.; Garibotto, V.; Bacchin, R.; Altomare, D.; Dodich, A.; Assal, F.; Mendes, A.; Costa, A.; Tinazzi, M.; Morbelli, S.D.; et al. Incremental value of amyloid-PET versus CSF in the diagnosis of Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Fink, H.A.; Linskens, E.J.; Silverman, P.C.; McCarten, J.R.; Hemmy, L.S.; Ouellette, J.M.; Greer, N.L.; Wilt, T.J.; Butler, M. Accuracy of Biomarker Testing for Neuropathologically Defined Alzheimer Disease in Older Adults With Dementia. Ann. Intern. Med. 2020, 172, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Gorno-Tempini, M.L.; Hillis, A.E.; Weintraub, S.; Kertesz, A.; Mendez, M.; Cappa, S.F.; Ogar, J.M.; Rohrer, J.D.; Black, S.; Boeve, B.F.; et al. Classification of primary progressive aphasia and its variants. Neurology 2011, 76, 1006–1014. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demographic | AD (n = 95) | FTD (n = 53) | p |
|---|---|---|---|
| Age at baseline (±SD) | 66.69 (±7.98) | 67.04 (±8.17) | 0.650 |
| Age at onset (±SD) | 63.49 (±8.88) | 64.24 (±8.27) | 0.661 |
| Age at CSF analysis (±SD) | 68.02 (±8.04) | 67.86 (±7.50) | 0.906 |
| Age at amyloid-PET (±SD) | 66.76 (±7.34) | 67.36 (±8.73) | 0.587 |
| Time from CSF to amyloid-PET (±SD) * | 0.562 (±0.78) | −0.18 (±0.88) | 0.034 |
| Disease duration (±SD) | 3.03 (±3.31) | 2.61 (±1.84) | 0.951 |
| Sex (women/men) | 54/41 | 28/25 | 0.638 |
| Familiality (%) | 41.40 | 50.00 | 0.902 |
| Education (±SD) | 10.56 (±5.10) | 9.37 (±4.20) | 0.205 |
| TIB (±SD) | 108.70 (±8.02) | 104 (±4.03) | 0.292 |
| MMSE (±SD) | 19.55 (±5.44) | 20.32 (±5.68) | 0.400 |
| HDRS (±SD) | 28.45 (±5.61) | 30.75 (±6.18) | 0.525 |
| ApoE ε4+ (%) | 24.48 | 3.69 | 0.008 |
| Amyloid-PET + | Concordance [95% C.I.] % | ||
|---|---|---|---|
| CSF Aβ+ | AD | 9/23 (39.13%) | 39.13% [19.94–63.40] |
| FTD | 1/12 (8.33%) | 76.92% [54.02–99.83] | |
| Total | 10/36 (27.28%) | 52.78% [36.47–69.09] | |
| Aβ42/40 + | Concordance [95% C.I.] % | ||
| Aβ1–42 + | AD | 28/40 (70.00%) | 92.50% [84.34–100] |
| FTD | 4/27 (7.41%) | 85.19% [71.79–98.59] | |
| Total | 32/67 (47.76%) | 89.55% [82.23–96.88] |
| Sensitivity [95% C.I.] | Specificity [95% C.I.] | Accuracy [95% C.I.] | |
|---|---|---|---|
| (%) | (%) | (%) | |
| Aβ positivity (n = 148) | 81.05 [74.74–87.37] | 77.36 [70.62–84.10] | 79.73 [73.25–86.21] |
| CSF Aβ (n = 126) | 64.56 [56.20–72.91] | 80.85 [73.98–87.72] | 70.63 [62.68–78.59] |
| Aβ1–42 (n = 126) | 60.76 [52.23–69.29] | 82.98 [76.42–89.54] | 69.05 [60.98–77.12] |
| Aβ42/40 (n = 67) | 77.50 [67.50–87.50] | 81.48 [72.18–90.78] | 79.10 [69.37–88.84] |
| Amyloid-PET (n = 58) | 89.74 [81.94–97.55] | 78.95 [68.46–89.44] | 86.21 [77.33–95.08] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giacomucci, G.; Mazzeo, S.; Bagnoli, S.; Casini, M.; Padiglioni, S.; Polito, C.; Berti, V.; Balestrini, J.; Ferrari, C.; Lombardi, G.; et al. Matching Clinical Diagnosis and Amyloid Biomarkers in Alzheimer’s Disease and Frontotemporal Dementia. J. Pers. Med. 2021, 11, 47. https://doi.org/10.3390/jpm11010047
Giacomucci G, Mazzeo S, Bagnoli S, Casini M, Padiglioni S, Polito C, Berti V, Balestrini J, Ferrari C, Lombardi G, et al. Matching Clinical Diagnosis and Amyloid Biomarkers in Alzheimer’s Disease and Frontotemporal Dementia. Journal of Personalized Medicine. 2021; 11(1):47. https://doi.org/10.3390/jpm11010047
Chicago/Turabian StyleGiacomucci, Giulia, Salvatore Mazzeo, Silvia Bagnoli, Matteo Casini, Sonia Padiglioni, Cristina Polito, Valentina Berti, Juri Balestrini, Camilla Ferrari, Gemma Lombardi, and et al. 2021. "Matching Clinical Diagnosis and Amyloid Biomarkers in Alzheimer’s Disease and Frontotemporal Dementia" Journal of Personalized Medicine 11, no. 1: 47. https://doi.org/10.3390/jpm11010047
APA StyleGiacomucci, G., Mazzeo, S., Bagnoli, S., Casini, M., Padiglioni, S., Polito, C., Berti, V., Balestrini, J., Ferrari, C., Lombardi, G., Ingannato, A., Sorbi, S., Nacmias, B., & Bessi, V. (2021). Matching Clinical Diagnosis and Amyloid Biomarkers in Alzheimer’s Disease and Frontotemporal Dementia. Journal of Personalized Medicine, 11(1), 47. https://doi.org/10.3390/jpm11010047