Imaging Biomarkers of Tumour Proliferation and Invasion for Personalised Lung Cancer Therapy


Abstract
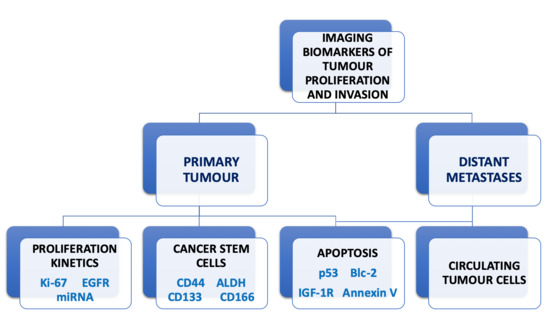
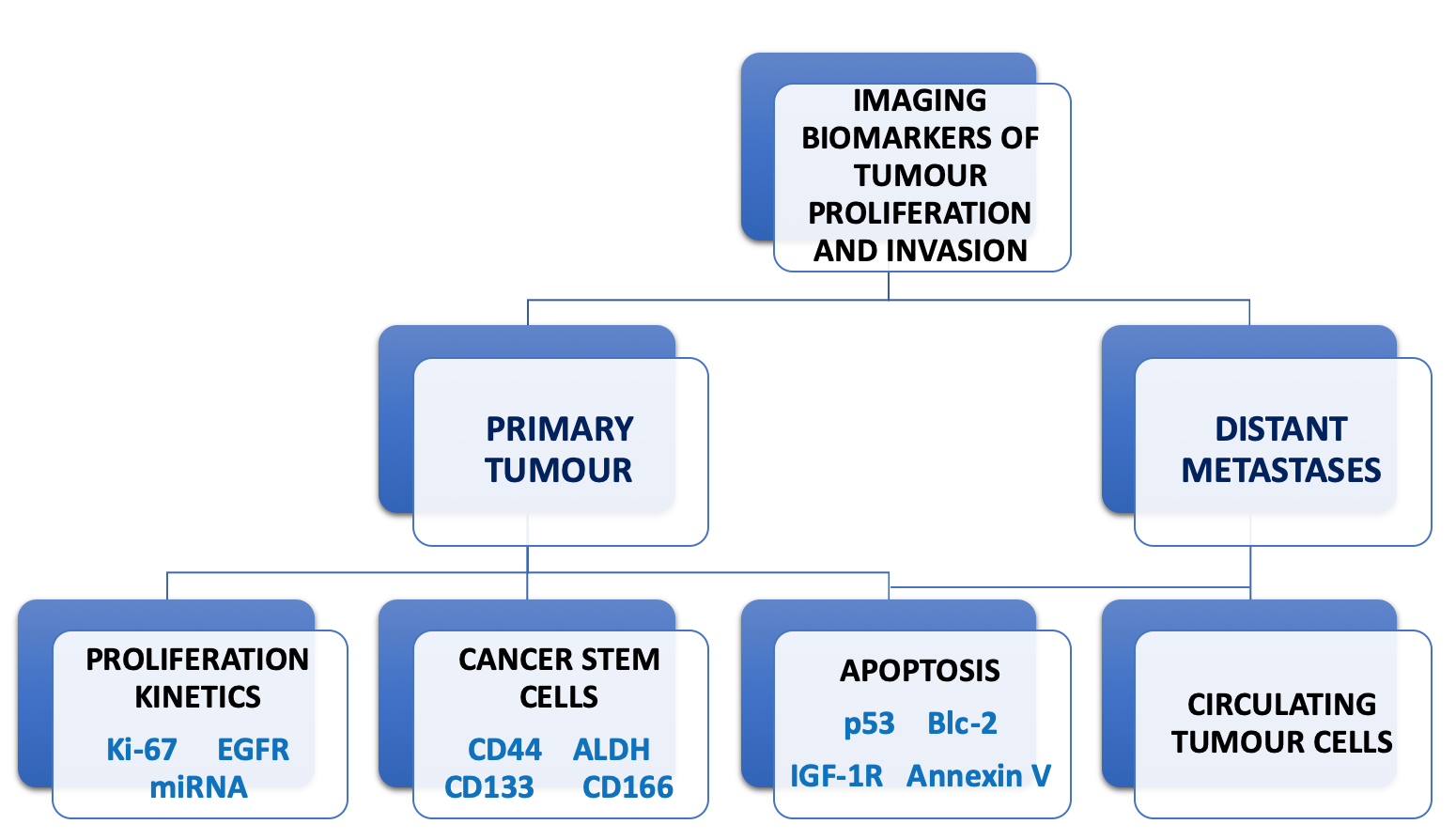
1. Introduction
2. Tumour Proliferation and Imaging Biomarkers
2.1. Tumour Proliferation
2.2. Imaging Biomarkers for Proliferation
2.2.1. Positron Emission Tomography (PET) Imaging Biomarkers
2.2.2. Single Photon Emission Computed Tomography (SPECT) Imaging Biomarkers
2.2.3. Magnetic Resonance Imaging (MRI) Biomarkers
2.3. Summary of Current Status for Proliferation Biomarkers
3. Cancer Stem Cells and Imaging Biomarkers
3.1. Cancer Stem Cells
3.2. Imaging Biomarkers for Cancer Stem Cells
3.3. Summary of Current Status for CSC Biomarkers
4. Circulating Tumour Cells and Imaging Biomarkers
4.1. Circulating Tumour Cells and Distant Metastasis
4.2. Circulating Tumour Cells as Biomarkers in NSCLC
4.3. Summary of Current Status for CTC Biomarkers
5. Imaging Biomarkers for Apoptosis
Summary of Current Status for Apoptosis Biomarkers
6. Conclusions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- European Society of Radiology (ESR). Medical imaging in personalised medicine: A white paper of the research committee of the European Society of Radiology (ESR). Insights Imag. 2015, 6, 141–155. [Google Scholar] [CrossRef]
- Gerdes, J.; Schwab, U.; Lemke, H.; Stein, H. Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation. Int. J. Cancer 1983, 31, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Warth, A.; Cortis, J.; Soltermann, A.; Meister, M.; Budczies, J.; Stenzinger, A.; Goeppert, B.; Thomas, M.; Herth, F.J.; Schirmacher, P.; et al. Tumour cell proliferation (Ki-67) in non-small cell lung cancer: A critical reappraisal of its prognostic role. Br. J. Cancer 2014, 111, 1222–1229. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Varella-Garcia, M.; Bunn, P.A., Jr.; Di Maria, M.V.; Veve, R.; Bremmes, R.M.; Barón, A.E.; Zeng, C.; Franklin, W.A. Epidermal growth factor receptor in non-small-cell lung carcinomas: Correlation between gene copy number and protein expression and impact on prognosis. J. Clin. Oncol. 2003, 21, 3798–3807. [Google Scholar] [CrossRef]
- Grandis, J.R.; Sok, J.C. Signaling through the epidermal growth factor receptor during the development of malignancy. Pharmacol. Ther. 2004, 102, 37–46. [Google Scholar] [CrossRef]
- Vokes, E.E.; Chu, E. Anti-EGFR therapies: Clinical experience in colorectal, lung, and head and neck cancers. Oncology 2006, 20 (Suppl. 2), 15–25. [Google Scholar]
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.F.; et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef]
- Hoang, T.; Myung, S.K.; Pham, T.T.; Kim, J.; Ju, W. Comparative Efficacy of Targeted Therapies in Patients with Non-Small Cell Lung Cancer: A Network Meta-Analysis of Clinical Trials. J. Clin. Med. 2020, 9, 1063. [Google Scholar] [CrossRef]
- Zugazagoitia, J.; Ramos, I.; Trigo, J.M.; Palka, M.; Gómez-Rueda, A.; Jantus-Lewintre, E.; Camps, C.; Isla, D.; Iranzo, P.; Ponce-Aix, S.; et al. Clinical utility of plasma-based digital next-generation sequencing in patients with advance-stage lung adenocarcinomas with insufficient tumor samples for tissue genotyping. Ann. Oncol. 2019, 30, 290–296. [Google Scholar] [CrossRef]
- Nana-Sinkam, S.P.; Geraci, M.W. MicroRNA in lung cancer. J. Thorac. Oncol. 2006, 1, 929–931. [Google Scholar] [CrossRef]
- Webster, R.J.; Giles, K.M.; Price, K.J.; Zhang, P.M.; Mattick, J.S.; Leedman, P.J. Regulation of epidermal growth factor receptor signaling in human cancer cells by microRNA-7. J. Biol. Chem. 2009, 284, 5731–5741. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.Y.; Yu, S.L.; Yang, P.C. MicroRNA in lung cancer. Br. J. Cancer 2010, 103, 1144–1148. [Google Scholar] [CrossRef]
- Szyszko, T.A.; Yip, C.; Szlosarek, P.; Goh, V.; Cook, G.J. The role of new PET tracers for lung cancer. Lung Cancer 2016, 94, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhou, Y.; Tang, X.; Yang, C.; Tian, Y.; Xie, R.; Chen, T.; Yang, J.; Jing, M.; Chen, F.; et al. EGFR mutation decreases FDG uptake in non‑small cell lung cancer via the NOX4/ROS/GLUT1 axis. Int. J. Oncol. 2019, 54, 370–380. [Google Scholar] [CrossRef]
- Caicedo, C.; Garcia-Velloso, M.J.; Lozano, M.D.; Labiano, T.; Vigil Diaz, C.; Lopez-Picazo, J.M.; Gurpide, A.; Zulueta, J.J.; Richter Echevarria, J.A.; Perez Gracia, J.L. Role of [¹⁸F]FDG PET in prediction of KRAS and EGFR mutation status in patients with advanced non-small-cell lung cancer. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 2058–2065. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Fan, J.; Xu, J.; Wu, F.; Huang, Q.; Guo, M.; Liao, T.; Liu, S.; Lan, X.; Liao, S.; et al. Value of 18F-FDG PET/CT for predicting EGFR mutations and positive ALK expression in patients with non-small cell lung cancer: A retrospective analysis of 849 Chinese patients. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 735–750. [Google Scholar] [CrossRef]
- Guan, J.; Xiao, N.J.; Chen, M.; Zhou, W.L.; Zhang, Y.W.; Wang, S.; Dai, Y.M.; Li, L.; Zhang, Y.; Li, Q.Y.; et al. 18F-FDG uptake for prediction EGFR mutation status in non-small cell lung cancer. Medicine 2016, 95, e4421. [Google Scholar] [CrossRef]
- Sun, X.; Xiao, Z.; Chen, G.; Han, Z.; Liu, Y.; Zhang, C.; Sun, Y.; Song, Y.; Wang, K.; Fang, F.; et al. A PET imaging approach for determining EGFR mutation status for improved lung cancer patient management. Sci. Transl. Med. 2018, 10, eaan8840. [Google Scholar] [CrossRef]
- Shields, A.F.; Grierson, J.R.; Dohmen, B.M.; Machulla, H.J.; Stayanoff, J.C.; Lawhorn-Crews, J.M.; Obradovich, J.E.; Muzik, O.; Mangner, T.J. Imaging proliferation in vivo with [F-18]FLT and positron emission tomography. Nat. Med. 1998, 4, 1334–1336. [Google Scholar] [CrossRef] [PubMed]
- Buck, A.K.; Halter, G.; Schirrmeister, H.; Kotzerke, J.; Wurziger, I.; Glatting, G.; Mattfeldt, T.; Neumaier, B.; Reske, S.N.; Hetzel, M. Imaging proliferation in lung tumours with PET: 18F-FLT versus 18F-FDG. J. Nucl. Med. 2003, 44, 1426–1431. [Google Scholar] [PubMed]
- Shen, G.; Ma, H.; Pang, F.; Ren, P.; Kuang, A. Correlations of 18F-FDG and 18F-FLT uptake on PET with Ki-67 expression in patients with lung cancer: A meta-analysis. Acta Radiol. 2018, 59, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Kairemo, K.; Santos, E.B.; Macapinlac, H.A.; Subbiah, V. Early Response Assessment to Targeted Therapy Using 3′-deoxy-3′[(18)F]-Fluorothymidine (18F-FLT) PET/CT in Lung Cancer. Diagnostics 2020, 10, 26. [Google Scholar] [CrossRef]
- Zannetti, A.; Iommelli, F.; Speranza, A.; Salvatore, M.; Del Vecchio, S. 3′-deoxy-3′-18F-fluorothymidine PET/CT to guide therapy with epidermal growth factor receptor antagonists and Bcl-xL inhibitors in non-small cell lung cancer. J. Nucl. Med. 2012, 53, 443–450. [Google Scholar] [CrossRef]
- Iommelli, F.; De Rosa, V.; Gargiulo, S.; Panico, M.; Monti, M.; Greco, A.; Gramanzini, M.; Ortosecco, G.; Fonti, R.; Brunetti, A.; et al. Monitoring reversal of MET-mediated resistance to EGFR tyrosine kinase inhibitors in non-small cell lung cancer using 3′-deoxy-3′-[18F]-fluorothymidine positron emission tomography. Clin. Cancer Res. 2014, 20, 4806–4815. [Google Scholar] [CrossRef]
- Iommelli, F.; De Rosa, V.; Terlizzi, C.; Monti, M.; Panico, M.; Fonti, R.; Del Vecchio, S. Inositol Trisphosphate Receptor Type 3-mediated Enhancement of EGFR and MET Cotargeting Efficacy in Non-Small Cell Lung Cancer Detected by 18F-fluorothymidine. Clin. Cancer Res. 2018, 24, 3126–3136. [Google Scholar] [CrossRef]
- Oh, M.; Tanaka, T.; Kobayashi, M.; Furukawa, T.; Mori, T.; Kudo, T.; Fujieda, S.; Fujibayashi, Y. Radio-copper-labeled Cu-ATSM: An indicator of quiescent but clonogenic cells under mild hypoxia in a Lewis lung carcinoma model. Nucl. Med. Biol. 2009, 36, 419–426. [Google Scholar] [CrossRef]
- Xiao, Z.; Song, Y.; Kai, W.; Sun, X.; Shen, B. Evaluation of 99mTc-HYNIC-MPG as a novel SPECT radiotracer to detect EGFR-activating mutations in NSCLC. Oncotarget 2017, 8, 40732–40740. [Google Scholar] [CrossRef]
- Wang, Z.; Qiao, R.; Tang, N.; Lu, Z.; Wang, H.; Zhang, Z.; Xue, X.; Huang, Z.; Zhang, S.; Zhang, G.; et al. Active targeting theranostic iron oxide nanoparticles for MRI and magnetic resonance-guided focused ultrasound ablation of lung cancer. Biomaterials 2017, 127, 25–35. [Google Scholar] [CrossRef]
- Kelsey, C.R.; Marks, L.B.; Hollis, D.; Hubbs, J.L.; Ready, N.E.; D’Amico, T.A.; Boyd, J.A. Local recurrence after surgery for early stage lung cancer: An 11-year experience with 975 patients. Cancer 2009, 115, 5218–5227. [Google Scholar] [CrossRef] [PubMed]
- Carney, D.N.; Gazdar, A.F.; Bunn, P.A., Jr.; Guccion, J.G. Demonstration of the stem cell nature of clonogenic tumor cells from lung cancer patients. Stem Cells 1982, 1, 149–164. [Google Scholar] [PubMed]
- Moore, N.; Lyle, S. Quiescent, slow-cycling stem cell populations in cancer: A review of the evidence and discussion of significance. J. Oncol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Perrin, R.; Hill, R.P.; Dubrovska, A.; Kurth, I. Hypoxia as a biomarker for radioresistant cancer stem cells. Int. J. Radiat. Biol. 2014, 90, 636–652. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, M.C. Hollingsworth RE, Hurt EM. Cancer stem cell plasticity and tumor hierarchy. World J. Stem Cells 2015, 7, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Qiu, Q.; Khanna, A.; Todd, N.W.; Deepak, J.; Xing, L.; Wang, H.; Liu, Z.; Su, Y.; Stass, S.A.; et al. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol. Cancer Res. 2009, 7, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.L.; Fiscus, R.R.; Tung, J.W.; Tin, V.P.; Cheng, L.C.; Sihoe, A.D.; Fink, L.M.; Ma, Y.; Wong, M.P. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS ONE 2010, 5, e14062. [Google Scholar] [CrossRef]
- Zöller, M. CD44: Can a cancer-initiating cell profit from an abundantly expressed molecule? Nat. Rev. Cancer 2011, 11, 254–267. [Google Scholar] [CrossRef]
- Zakaria, N.; Yusoff, N.M.; Zakaria, Z.; Lim, M.N.; Baharuddin, P.J.; Fakiruddin, K.S.; Yahaya, B. Human non-small cell lung cancer expresses putative cancer stem cell markers and exhibits the transcriptomic profile of multipotent cells. BMC Cancer 2015, 15, 84. [Google Scholar] [CrossRef]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef]
- Qu, H.; Li, R.; Liu, Z.; Zhang, J.; Luo, R. Prognostic value of cancer stem cell marker CD133 expression in non-small cell lung cancer: A systematic review. Int. J. Clin. Exp. Pathol. 2013, 6, 2644–2650. [Google Scholar] [PubMed]
- Salnikov, A.V.; Gladkich, J.; Moldenhauer, G.; Volm, M.; Mattern, J.; Herr, I. CD133 is indicative for a resistance phenotype but does not represent a prognostic marker for survival of non-small cell lung cancer patients. Int. J. Cancer 2010, 126, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, Y.; Furukawa, T.; Kiyono, Y.; Watanabe, R.; Waki, A.; Mori, T.; Yoshii, H.; Oh, M.; Asai, T.; Okazawa, H.; et al. Copper-64-diacetyl-bis (N4-methylthiosemicarbazone) accumulates in rich regions of CD133+ highly tumorigenic cells in mouse colon carcinoma. Nucl. Med. Biol. 2010, 37, 395–404. [Google Scholar] [CrossRef]
- Yang, Y.; Hernandez, R.; Rao, J.; Yin, L.; Qu, Y.; Wu, J.; England, C.G.; Graves, S.A.; Lewis, C.M.; Wang, P.; et al. Targeting CD146 with a 64Cu-labeled antibody enables in vivo immunoPET imaging of high-grade gliomas. Proc. Natl. Acad. Sci. USA 2015, 112, E6525–E6534. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, L.; Wang, A.; Ma, Y.; Zhang, H.; Zhu, Y. Multifunctional fluorescent magnetic nanoparticles for lung cancer stem cells research. Colloids Surf. B Biointerfaces 2015, 134, 431–439. [Google Scholar] [CrossRef]
- Wang, A.; Chen, L.; Pu, K.; Zhu, Y. Identification of stem-like cells in non-small cell lung cancer cells with specific peptides. Cancer Lett. 2014, 351, 100–107. [Google Scholar] [CrossRef]
- Gomez-Casal, R.; Bhattacharya, C.; Ganesh, N.; Bailey, L.; Basse, P.; Gibson, M.; Epperly, M.; Levina, V. Non-small-cell lung cancer cells survived ionizing radiation treatment display cancer stem cell and epithelial–mesenchymal transition phenotypes. Mol. Cancer 2013, 12, 94. [Google Scholar] [CrossRef]
- Hou, J.M.; Krebs, M.G.; Lancashire, L.; Sloane, R.; Backen, A.; Swain, R.K.; Priest, L.J.; Greystoke, A.; Zhou, C.; Morris, K.; et al. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J. Clin. Oncol. 2012, 30, 525–532. [Google Scholar] [CrossRef]
- Klein, C.A. Cancer. The metastasis cascade. Science. 2008, 321, 1785–1787. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Riethdorf, S.; Pantel, K. Circulating tumor cells and bone marrow micrometastasis. Clin. Cancer Res. 2008, 14, 5013–5021. [Google Scholar] [CrossRef]
- Krebs, M.G.; Hou, J.M.; Ward, T.H.; Blackhall, F.H.; Dive, C. Circulating tumour cells: Their utility in cancer management and predicting outcomes. Ther. Adv. Med. Oncol. 2010, 2, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Ostoros, G.; Cobo, M.; Ciuleanu, T.; Cole, R.; McWalter, G.; Walker, J.; Dearden, S.; Webster, A.; Milenkova, T.; et al. Gefitinib treatment in EGFR mutated caucasian NSCLC: Circulating-free tumor DNA as a surrogate for determination of EGFR status. J. Thorac. Oncol. 2014, 9, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Van de Stolpe, A.; Pantel, K.; Sleijfer, S.; Terstappen, L.W.; den Toonder, J.M. Circulating tumor cell isolation and diagnostics: Toward routine clinical use. Cancer Res. 2011, 71, 5955–5960. [Google Scholar] [CrossRef] [PubMed]
- Taenzer, A.; Alix-Panabières, C.; Wikman, H.; Pantel, K. Circulating tumor-derived biomarkers in lung cancer. J. Thorac. Dis. 2012, 4, 448–449. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.G.; Hou, J.M.; Sloane, R.; Lancashire, L.; Priest, L.; Nonaka, D.; Ward, T.H.; Backen, A.; Clack, G.; Hughes, A.; et al. Analysis of circulating tumor cells in patients with non-small cell lung cancer using epithelial marker-dependent and -independent approaches. J. Thorac. Oncol. 2012, 7, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Kulasinghe, A.; Kapeleris, J.; Kimberley, R.; Mattarollo, S.R.; Thompson, E.W.; Thiery, J.P.; Kenny, L.; O’Byrne, K.; Punyadeera, C. The prognostic significance of circulating tumor cells in head and neck and non-small-cell lung cancer. Cancer Med. 2018, 7, 5910–5919. [Google Scholar] [CrossRef]
- Wyckoff, J.B.; Jones, J.G.; Condeelis, J.S.; Segall, J.E. A critical step in metastasis: In vivo analysis of intravasation at the primary tumor. Cancer Res. 2000, 60, 2504–2511. [Google Scholar]
- He, W.; Wang, H.; Hartmann, L.C.; Cheng, J.X.; Low, P.S. In vivo quantitation of rare circulating tumor cells by multiphoton intravital flow cytometry. Proc. Natl. Acad. Sci. USA 2007, 104, 11760–11765. [Google Scholar] [CrossRef]
- Kuo, C.W.; Chueh, D.Y.; Chen, P. Real-time in vivo imaging of subpopulations of circulating tumor cells using antibody conjugated quantum dots. J. Nanobiotechnol. 2019, 17, 26. [Google Scholar] [CrossRef]
- Nieva, J.; Wendel, M.; Luttgen, M.S.; Marrinucci, D.; Bazhenova, L.; Kolatkar, A.; Santala, R.; Whittenberger, B.; Burke, J.; Torrey, M.; et al. High-definition imaging of circulating tumor cells and associated cellular events in non-small cell lung cancer patients: A longitudinal analysis. Phys. Biol. 2012, 9, 016004. [Google Scholar] [CrossRef][Green Version]
- Nair, V.S.; Keu, K.V.; Luttgen, M.S.; Kolatkar, A.; Vasanawala, M.; Kuschner, W.; Bethel, K.; Iagaru, A.H.; Hoh, C.; Shrager, J.B.; et al. An observational study of circulating tumor cells and (18)F-FDG PET uptake in patients with treatment-naive non-small cell lung cancer. PLoS ONE 2013, 8, e67733. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, A.D.; Holdgaard, P.C.; Spindler, K.L.; Pallisgaard, N.; Jakobsen, A. The correlation between cell-free DNA and tumour burden was estimated by PET/CT in patients with advanced NSCLC. Br. J. Cancer 2014, 110, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Morbelli, S.; Alama, A.; Ferrarazzo, G.; Coco, S.; Genova, C.; Rijavec, E.; Bongioanni, F.; Biello, F.; Dal Bello, M.G.; Barletta, G.; et al. Circulating Tumor DNA Reflects Tumor Metabolism Rather Than Tumor Burden in Chemotherapy-Naive Patients with Advanced Non-Small Cell Lung Cancer: 18F-FDG PET/CT Study. J. Nucl. Med. 2017, 58, 1764–1769. [Google Scholar] [CrossRef] [PubMed]
- Bayarri-Lara, C.I.; de Miguel Pérez, D.; Cueto Ladrón de Guevara, A.; Rodriguez Fernández, A.; Puche, J.L.; Sánchez-Palencia Ramos, A.; Ruiz Zafra, J.; Giraldo Ospina, C.F.; Delgado-Rodríguez, M.; Expósito Ruiz, M.; et al. Association of circulating tumour cells with early relapse and 18F-fluorodeoxyglucose positron emission tomography uptake in resected non-small-cell lung cancers. Eur. J. Cardio-Thorac. Surg. 2017, 52, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Monterisi, S.; Castello, A.; Toschi, L.; Federico, D.; Rossi, S.; Veronesi, G.; Lopci, E. Preliminary data on circulating tumor cells in metastatic NSCLC patients candidate to immunotherapy. Am. J. Nucl. Med. Mol. Imaging 2019, 9, 282–295. [Google Scholar]
- Mason, J.; Blyth, B.; MacManus, M.P.; Martin, O.A. Treatment for non-small-cell lung cancer and circulating tumor cells. Lung Cancer Manag. 2017, 6, 129–139. [Google Scholar] [CrossRef]
- Martin, O.A.; Anderson, R.L.; Russell, P.A.; Cox, R.A.; Ivashkevich, A.; Swierczak, A.; Doherty, J.P.; Jacobs, D.H.; Smith, J.; Siva, S.; et al. Mobilization of viable tumor cells into the circulation during radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 395–403. [Google Scholar] [CrossRef]
- Brown, J.M.; Attardi, L.D. The role of apoptosis in cancer development and treatment response. Nat. Rev. Cancer 2005, 5, 231–237. [Google Scholar] [CrossRef]
- Borner, M.M.; Brousset, P.; Pfanner-Meyer, B.; Bacchi, M.; Vonlanthen, S.; Hotz, M.A.; Altermatt, H.J.; Schlaifer, D.; Reed, J.C.; Betticher, D.C. Expression of apoptosis regulatory proteins of the Bcl-2 family and p53 in primary resected non-small-cell lung cancer. Br. J. Cancer 1999, 79, 952–958. [Google Scholar] [CrossRef]
- Maki, R.G. Small is beautiful: Insulin-like growth factors and their role in growth, development, and cancer. J. Clin. Oncol. 2010, 28, 4985–4995. [Google Scholar] [CrossRef]
- Yeo, C.D.; Park, K.H.; Park, C.K.; Lee, S.H.; Kim, S.J.; Yoon, H.K.; Lee, Y.S.; Lee, E.J.; Lee, K.Y.; Kim, T.J. Expression of insulin-like growth factor 1 receptor (IGF-1R) predicts poor responses to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in non-small cell lung cancer patients harboring activating EGFR mutations. Lung Cancer 2015, 87, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Bie, F.; Wang, Y.; Chen, X.; Yan, T.; Du, J. Prognostic value of IGF-1R in lung cancer: A PRISMA-compliant meta-analysis. Medicine 2019, 98, e15467. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, B.; McLarty, K.; Kersemans, V.; Reilly, R.M. The level of insulin growth factor-1 receptor expression is directly correlated with the tumor uptake of (111)In-IGF-1(E3 R) in vivo and the clonogenic survival of breast cancer cells exposed in vitro to trastuzumab (Herceptin). Nucl. Med. Biol. 2008, 35, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Link, J.M.; Stekhova, S.; Yagle, K.J.; Smith, C.; Krohn, K.A.; Tait, J.F. Site-specific labeling of annexin V with F-18 for apoptosis imaging. Bioconjug. Chem. 2008, 19, 1684–1688. [Google Scholar] [CrossRef] [PubMed]
- Lahorte, C.M.; Vanderheyden, J.L.; Steinmetz, N.; Van de Wiele, C.; Dierckx, R.A.; Slegers, G. Apoptosis-detecting radioligands: Current state of the art and future perspectives. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 887–919. [Google Scholar] [CrossRef] [PubMed]
- Höglund, J.; Shirvan, A.; Antoni, G.; Gustavsson, S.Å.; Långström, B.; Ringheim, A.; Sörensen, J.; Ben-Ami, M.; Ziv, I. 18F-ML-10, a PET tracer for apoptosis: First human study. J. Nucl. Med. 2011, 52, 720–725. [Google Scholar] [CrossRef]
- Marcu, L.G.; Moghaddasi, L.; Bezak, E. Imaging of Tumor Characteristics and Molecular Pathways with PET: Developments Over the Last Decade Toward Personalized Cancer Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2018, 102, 1165–1182. [Google Scholar] [CrossRef]
- Marcu, L.G.; Reid, P.; Bezak, E. The Promise of Novel Biomarkers for Head and Neck Cancer from an Imaging Perspective. Int. J. Mol. Sci. 2018, 19, 2511. [Google Scholar] [CrossRef]

{kind=link}
| Study Aim [Ref] | Study Type | Proliferation Marker/ Targeting Agent | Comments |
|---|---|---|---|
| Positron Emission Tomography | |||
| Proliferation imaging with 18F-FLT vs. 18F-FDG [Buck et al. (2003)] [22] | Prospective study (26 patients with pulmonary nodules) | Proliferation marker: Ki-67 Targeting agent: 18F-FLT 18F-FDG | A highly significant correlation (p < 0.0001) and a high correlation coefficient (r = 0.92) was observed between 18F-FLT uptake and Ki-67 index, while the correlation coefficient between Ki-67 and 18F-FDG was weak (r = 0.59). No FLT uptake was detected in non-proliferating tumours. |
| PET imaging for EGFR mutation evaluation and response to treatment [Sun et al. (2018)] [20] | Preclinical rodent model; Clinical NSCLC study | Proliferation marker: EGFR Targeting agent: 18F-MPG | A greater response to EGFR-TKI was found in patients with SUVmax ≥ 2.23 (81.58% vs. 6.06%). Median progression-free survival was also longer (348 days) in the cohort with SUVmax ≥ 2.23 than in SUVmax < 2.23 (183 days). 18F-MPG PET for quantification of EGFR-activating mutation status could identify patients sensitive to EGFR-TKIs. |
| Evaluation of the role of 64Cu-ATSM in PET imaging [Oh et al. (2009)] [28] | In vivo mice study (Lewis lung carcinoma tumour cells implanted in mice) | Proliferation markers: Ki-67 BrdU Targeting agent: 64Cu-ATSM 18F-FDG | Tumour regions with high 18F-FDG but low 64Cu-ATSM uptake correlated with increase in Ki-67. On the other hand, the number of BrdU-positive cells were positively correlated with 64Cu-ATSM uptake and negatively related to 18F-FDG accumulation. This suggests that cells in regions with high 64Cu-ATSM uptake were quiescent, yet were sensitive to progression factors, like quiescent CSCs. |
| Single Photon Emission Computed Tomography | |||
| Evaluation of 99mTc-HYNIC-MPG for detection of EGFR-activating mutations [Xiao et al. (2017)] [29] | In vitro cell line study (human NSCLC cell lines EGFR+/- and wild-type); In vivo animal xenograft model | Proliferation marker: EGFR Targeting agent: 99mTc-HYNIC-MPG | 99mTc-HYNIC-MPG uptake was the highest in the cell line with exon 19 deletion (PC9), probably due to the activating mutations in EGFR tyrosine kinase domain. SPECT imaging with 99mTc-HYNIC-MPG could potentially identify NSCLC patients that would benefit the most from targeted therapies with EGFR-TKIs. |
| Magnetic Resonance Imaging | |||
| EGFR targeting with active iron oxide NP for MRI [Wang et al. (2017)] [30] | H460 lung cancer cells (in vitro) and tumour-bearing rats (H460 lung xenografts) in vivo. | Proliferation marker: EGFR Targeting agent: Anti-EGFR-polyethylene glycol-superparamagnetic iron oxide (anti-EGFR-PEG-SPIO) | Both in vitro and in vivo MRI studies showed the potential of anti-EGFR-labeled iron oxide nanoparticles to identify and target lung cells that overexpress EGFR. The study had both imaging and therapeutic (theranostic) goals achieved with anti-EGFR targeting based on magnetic nanoparticles using MRI and focused ultrasound ablation. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcu, L.G. Imaging Biomarkers of Tumour Proliferation and Invasion for Personalised Lung Cancer Therapy. J. Pers. Med. 2020, 10, 222. https://doi.org/10.3390/jpm10040222
Marcu LG. Imaging Biomarkers of Tumour Proliferation and Invasion for Personalised Lung Cancer Therapy. Journal of Personalized Medicine. 2020; 10(4):222. https://doi.org/10.3390/jpm10040222
Chicago/Turabian StyleMarcu, Loredana G. 2020. "Imaging Biomarkers of Tumour Proliferation and Invasion for Personalised Lung Cancer Therapy" Journal of Personalized Medicine 10, no. 4: 222. https://doi.org/10.3390/jpm10040222
APA StyleMarcu, L. G. (2020). Imaging Biomarkers of Tumour Proliferation and Invasion for Personalised Lung Cancer Therapy. Journal of Personalized Medicine, 10(4), 222. https://doi.org/10.3390/jpm10040222