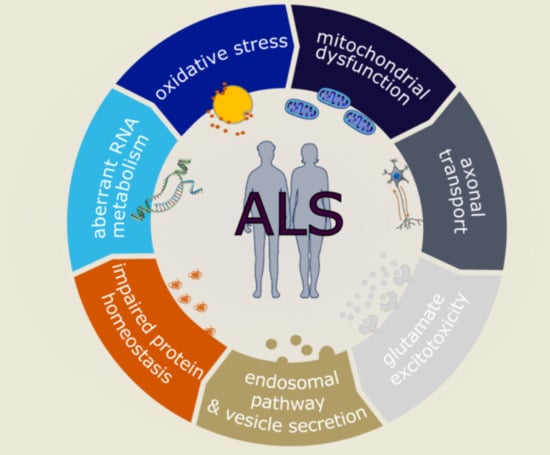
Molecular and Cellular Mechanisms Affected in ALS

 , ,
, ,  and
and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
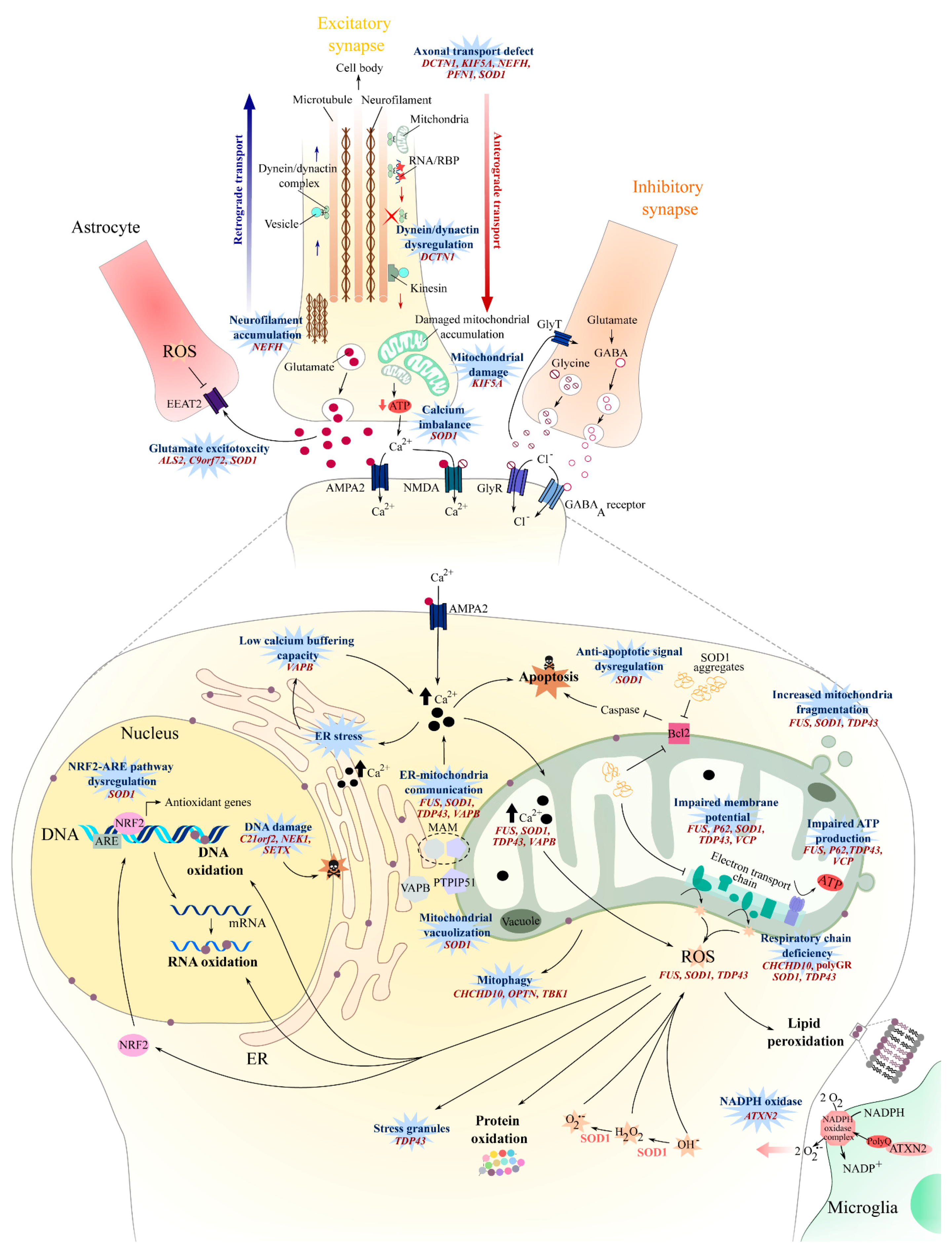
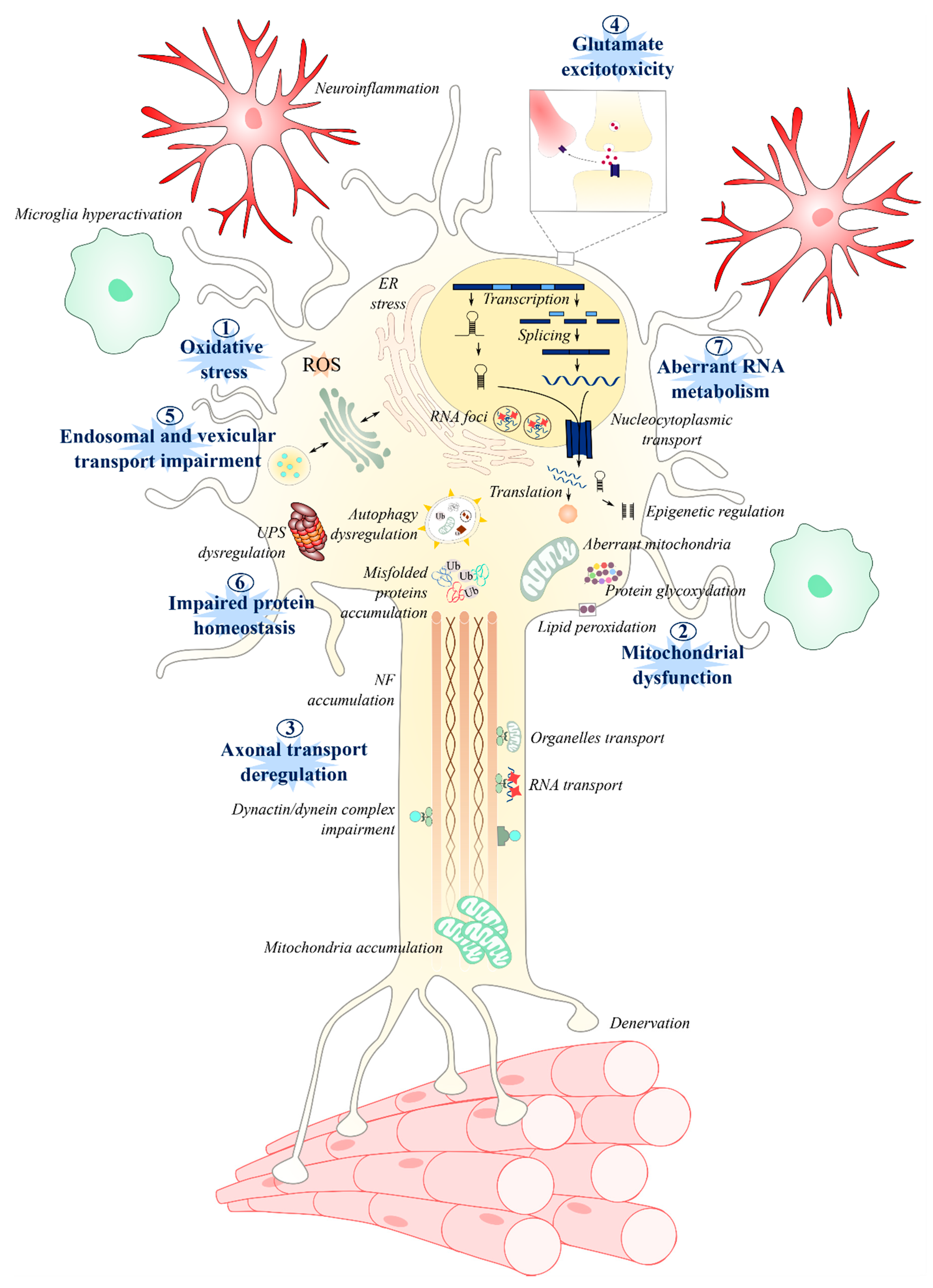
2. Oxidative Stress
3. Mitochondrial Dysfunction
4. Axonal Transport
5. Glutamate Excitotoxicity
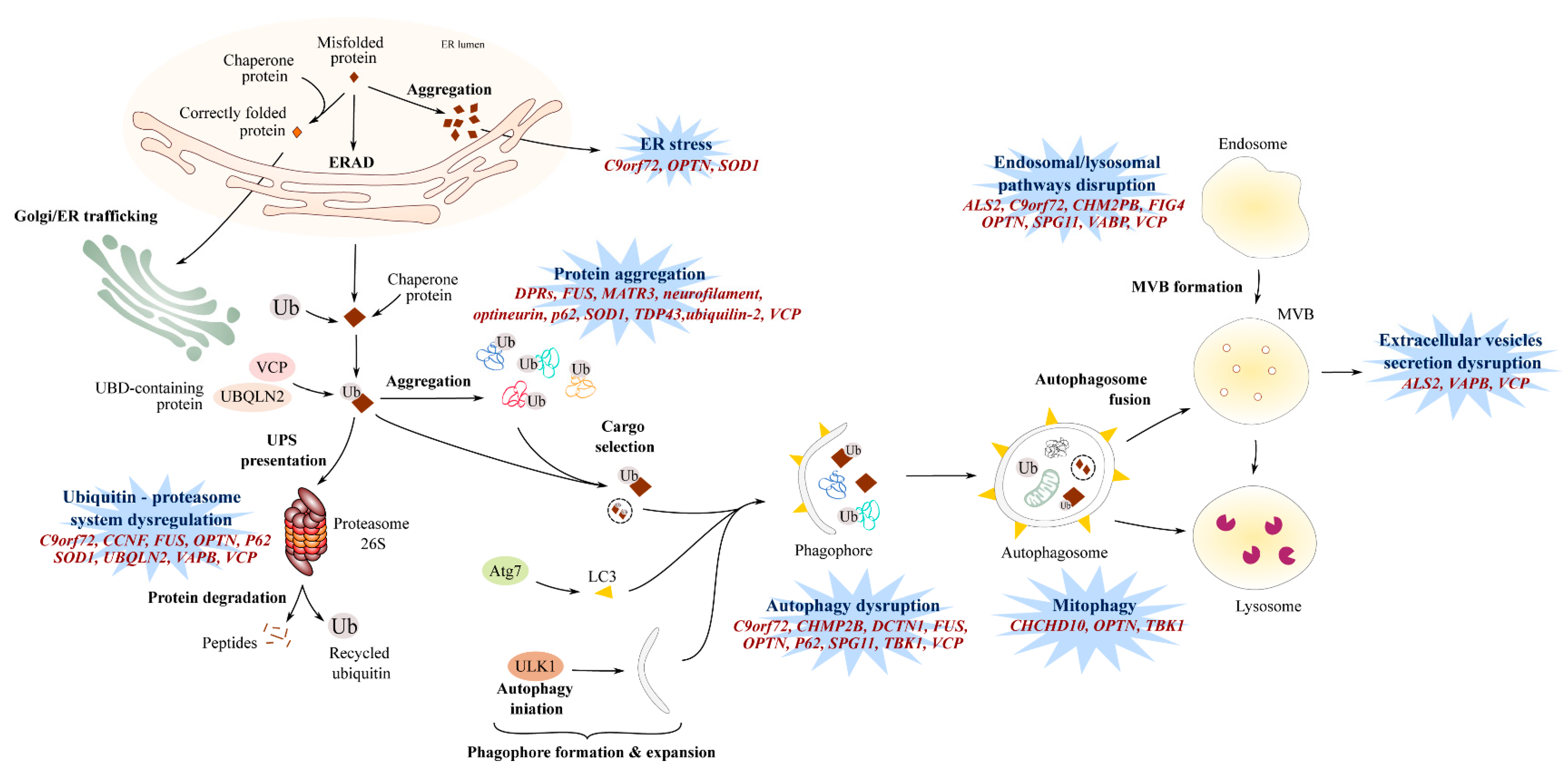
6. Endosomal Pathway and Vesicle Secretion
7. Impaired Protein Homeostasis
7.1. Proteasome and Autophagic Degradation Pathways
7.2. Endoplasmic Reticulum Stress
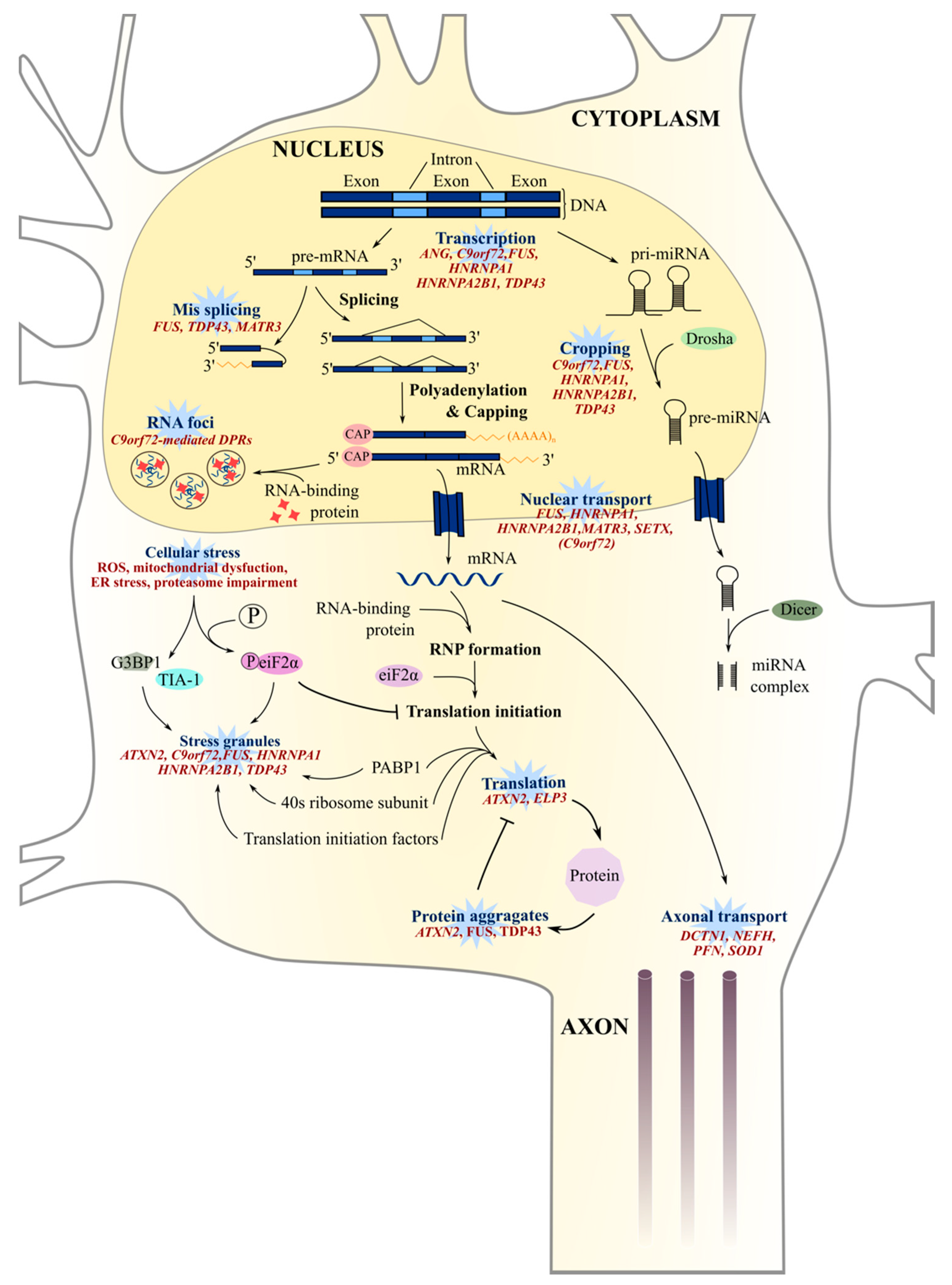
8. Aberrant RNA Metabolism
8.1. RNA Splicing and Translation
8.2. RNA Foci
8.3. Epigenetic Modulation
8.4. Stress Granules and Nucleocytoplasmic Transport
9. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef] [PubMed]
- Ragagnin, A.M.G.; Shadfar, S.; Vidal, M.; Jamali, M.S.; Atkin, J.D. Motor Neuron Susceptibility in ALS/FTD. Front. Neurosci. 2019, 13, 532. [Google Scholar] [CrossRef] [PubMed]
- Nicaise, C.; Mitrecic, D.; Pochet, R. Brain and spinal cord affected by amyotrophic lateral sclerosis induce differential growth factors expression in rat mesenchymal and neural stem cells. Neuropathol. Appl. Neurobiol. 2011, 37, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, C.; Pagnini, F.; Friede, T.; Young, C.A. Treatment of fatigue in amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst. Rev. 2018. [Google Scholar] [CrossRef]
- Turner, M.R.; Al-Chalabi, A.; Chio, A.; Hardiman, O.; Kiernan, M.C.; Rohrer, J.D.; Rowe, J.; Seeley, W.; Talbot, K. Genetic screening in sporadic ALS and FTD. J. Neurol. Neurosurg. Psychiatry 2017, 88, 1042–1044. [Google Scholar] [CrossRef]
- Vijayakumar, U.G.; Milla, V.; Cynthia Stafford, M.Y.; Bjourson, A.J.; Duddy, W.; Duguez, S.M.-R. A Systematic Review of Suggested Molecular Strata, Biomarkers and Their Tissue Sources in ALS. Front. Neurol. 2019, 10, 400. [Google Scholar] [CrossRef]
- Connolly, O.; Le Gall, L.; McCluskey, G.; Donaghy, C.G.; Duddy, W.J.; Duguez, S. A Systematic Review of Genotype–Phenotype Correlation across Cohorts Having Causal Mutations of Different Genes in ALS. J. Pers. Med. 2020, 10, 58. [Google Scholar] [CrossRef]
- Watson, M.R.; Lagow, R.D.; Xu, K.; Zhang, B.; Bonini, N.M. A drosophila model for amyotrophic lateral sclerosis reveals motor neuron damage by human SOD1. J. Biol. Chem. 2008, 283, 24972–24981. [Google Scholar] [CrossRef]
- Estes, P.S.; Boehringer, A.; Zwick, R.; Tang, J.E.; Grigsby, B.; Zarnescu, D.C. Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum. Mol. Genet. 2011, 20, 2308–2321. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, M.; Deng, J.; Chen, X.; Ye, Y.; Zhu, L.; Liu, J.; Ye, H.; Shen, Y.; Li, Y.; et al. Expression of human FUS protein in Drosophila leads to progressive neurodegeneration. Protein Cell 2011, 2, 477–486. [Google Scholar] [CrossRef]
- Xu, Z.; Poidevin, M.; Li, X.; Li, Y.; Shu, L.; Nelson, D.L.; Li, H.; Hales, C.M.; Gearing, M.; Wingo, T.S.; et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 7778–7783. [Google Scholar] [CrossRef] [PubMed]
- Therrien, M.; Parker, J.A. Worming forward: Amyotrophic lateral sclerosis toxicity mechanisms and genetic interactions in Caenorhabditis elegans. Front. Genet. 2014, 5, 85. [Google Scholar] [CrossRef] [PubMed]
- Sakowski, S.A.; Lunn, J.S.; Busta, A.S.; Oh, S.S.; Zamora-Berridi, G.; Palmer, M.; Rosenberg, A.A.; Philip, S.G.; Dowling, J.J.; Feldman, E.L. Neuromuscular effects of G93A-SOD1 expression in zebrafish. Mol. Neurodegener. 2012, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.A.B.; Drapeau, P. Loss and gain of FUS function impair neuromuscular synaptic transmission in a genetic model of ALS. Hum. Mol. Genet. 2013, 22, 4282–4292. [Google Scholar] [CrossRef] [PubMed]
- Ciura, S.; Lattante, S.; Le Ber, I.; Latouche, M.; Tostivint, H.; Brice, A.; Kabashi, E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Schmid, B.; Hruscha, A.; Hogl, S.; Banzhaf-Strathmann, J.; Strecker, K.; van der Zee, J.; Teucke, M.; Eimer, S.; Hegermann, J.; Kittelmann, M.; et al. Loss of ALS-associated TDP-43 in zebrafish causes muscle degeneration, vascular dysfunction, and reduced motor neuron axon outgrowth. Proc. Natl. Acad. Sci. USA 2013, 110, 4986–4991. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Pansarasa, O.; Bordoni, M.; Drufuca, L.; Diamanti, L.; Sproviero, D.; Trotti, R.; Bernuzzi, S.; La Salvia, S.; Gagliardi, S.; Ceroni, M.; et al. Lymphoblastoid cell lines as a model to understand amyotrophic lateral sclerosis disease mechanisms. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef]
- Boston-Howes, W.; Williams, E.O.; Bogush, A.; Scolere, M.; Pasinelli, P.; Trotti, D. Nordihydroguaiaretic acid increases glutamate uptake in vitro and in vivo: Therapeutic implications for amyotrophic lateral sclerosis. Exp. Neurol. 2008, 213, 229–237. [Google Scholar] [CrossRef]
- Moreno-Martet, M.; Mestre, L.; Loria, F.; Guaza, C.; Fernandez-Ruiz, J.; de Lago, E. Identification of receptors and enzymes for endocannabinoids in NSC-34 cells: Relevance for in vitro studies with cannabinoids in motor neuron diseases. Neurosci. Lett. 2012, 508, 67–72. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Van Damme, P.; Bogaert, E.; Dewil, M.; Hersmus, N.; Kiraly, D.; Scheveneels, W.; Bockx, I.; Braeken, D.; Verpoorten, N.; Verhoeven, K.; et al. Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity. Proc. Natl. Acad. Sci. USA 2007, 104, 14825–14830. [Google Scholar] [CrossRef] [PubMed]
- Volk, A.E.; Weishaupt, J.H.; Andersen, P.M.; Ludolph, A.C.; Kubisch, C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med. Genet. 2018, 30, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Boillée, S.; Vande Velde, C.; Cleveland, D.W. ALS: A Disease of Motor Neurons and Their Nonneuronal Neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef]
- Gupta, R.; Lan, M.; Mojsilovic-Petrovic, J.; Choi, W.H.; Safren, N.; Barmada, S.; Lee, M.J.; Kalb, R. The Proline/Arginine Dipeptide from Hexanucleotide Repeat Expanded C9ORF72 Inhibits the Proteasome. Eneuro 2017, 4. [Google Scholar] [CrossRef]
- Mitsumoto, H.; Santella, R.M.; Liu, X.; Bogdanov, M.; Zipprich, J.; Wu, H.-C.; Mahata, J.; Kilty, M.; Bednarz, K.; Bell, D.; et al. Oxidative Stress Biomarkers in Sporadic ALS; Taylor & Francis: Oxfordshire, UK, 2008; Volume 9, pp. 177–183. [Google Scholar]
- Chang, Y.; Kong, Q.; Shan, X.; Tian, G.; Ilieva, H.; Cleveland, D.W.; Rothstein, J.D.; Borchelt, D.R.; Wong, P.C.; Lin, C.G. Messenger RNA Oxidation Occurs Early in Disease Pathogenesis and Promotes Motor Neuron Degeneration in ALS. PLoS ONE 2008, 3, e2849. [Google Scholar] [CrossRef]
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.-F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; Mcloughlin, D.M.; et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content Europe PMC Funders Group. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar] [CrossRef]
- Kiaei, M.; Kipiani, K.; Calingasan, N.; Wille, E.; Chen, J.; Heissig, B.; Rafii, S.; Lorenzl, S.; Beal, M. Matrix metalloproteinase-9 regulates TNF-α and FasL expression in neuronal, glial cells and its absence extends life in a transgenic mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2007, 205, 74–81. [Google Scholar] [CrossRef]
- Laslo, P.; Lipski, J.; Nicholson, L.F.B.; Miles, G.B.; Funk, G.D. GluR2 AMPA Receptor Subunit Expression in Motoneurons at Low and High Risk for Degeneration in Amyotrophic Lateral Sclerosis. Exp. Neurol. 2001, 169, 461–471. [Google Scholar] [CrossRef]
- Foerster, B.R.; Pomper, M.G.; Callaghan, B.C.; Petrou, M.; Edden, R.A.E.; Mohamed, M.A.; Welsh, R.C.; Carlos, R.C.; Barker, P.B.; Feldman, E.L. An imbalance between excitatory and inhibitory neurotransmitters in amyotrophic lateral sclerosis revealed by use of 3-T proton magnetic resonance spectroscopy. JAMA Neurol. 2013, 70, 1009–1016. [Google Scholar] [CrossRef]
- Kumar, A.; Bala, L.; Kalita, J.; Misra, U.K.; Singh, R.L.; Khetrapal, C.L.; Babu, G.N. Metabolomic analysis of serum by (1) H NMR spectroscopy in amyotrophic lateral sclerosis. Clin. Chim. Acta 2010, 411, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Spreux-Varoquaux, O.; Bensimon, G.; Lacomblez, L.; Salachas, F.; Pradat, P.F.; Le Forestier, N.; Marouan, A.; Dib, M.; Meininger, V. Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: A reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. J. Neurol. Sci. 2002, 193, 73–78. [Google Scholar] [CrossRef]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.A.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Leigh, P.N.; Whitwell, H.; Garofalo, O.; Buller, J.; Swash, M.; Martin, J.E.; Gallo, J.M.; Weller, R.O.; Anderton, B.H. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain 1991, 114 Pt 2, 775–788. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Cykowski, M.D.; Dickson, D.W.; Powell, S.Z.; Arumanayagam, A.S.; Rivera, A.L.; Appel, S.H. Dipeptide repeat (DPR) pathology in the skeletal muscle of ALS patients with C9ORF72 repeat expansion. Acta Neuropathol. 2019, 138, 667–670. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.-C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef]
- Haeusler, A.R.; Donnelly, C.J.; Rothstein, J.D. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat. Rev. Neurosci. 2016, 17, 383–395. [Google Scholar] [CrossRef]
- Butti, Z.; Patten, S.A. RNA Dysregulation in Amyotrophic Lateral Sclerosis. Front. Genet. 2019, 9, 712. [Google Scholar] [CrossRef]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.-C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, F.; Riboldi, G.; Salani, S.; Nizzardo, M.; Simone, C.; Corti, S.; Hedlund, E. Cellular therapy to target neuroinflammation in amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 2014, 71, 999–1015. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Boncompagni, S.; Bonconpagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Betteridge, D.J. What is oxidative stress? Metabolism 2000, 49, 3–8. [Google Scholar] [CrossRef]
- Kandlur, A.; Satyamoorthy, K.; Gangadharan, G. Oxidative Stress in Cognitive and Epigenetic Aging: A Retrospective Glance. Front. Mol. Neurosci. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J.; Ince, P.G.; Falkous, G.; Mantle, D. Oxidative damage to protein in sporadic motor neuron disease spinal cord. Ann. Neurol. 1995, 38, 691–695. [Google Scholar] [CrossRef]
- Rosen, D.R. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.F.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A Comprehensive Review of Amyotrophic Lateral Sclerosis; Wolters Kluwer—Medknow Publications: Mumbai, India, 2015; Volume 6, p. 171. [Google Scholar]
- Shibata, N.; Nagai, R.; Uchida, K.; Horiuchi, S.; Yamada, S.; Hirano, A.; Kawaguchi, M.; Yamamoto, T.; Sasaki, S.; Kobayashi, M. Morphological evidence for lipid peroxidation and protein glycoxidation in spinal cords from sporadic amyotrophic lateral sclerosis patients. Brain Res. 2001, 917, 97–104. [Google Scholar] [CrossRef]
- Juarez, J.C.; Manuia, M.; Burnett, M.E.; Betancourt, O.; Boivin, B.; Shaw, D.E.; Tonks, N.K.; Mazar, A.P.; Donate, F. Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 7147–7152. [Google Scholar] [CrossRef]
- Crosby, K.; Crown, A.M.; Roberts, B.L.; Brown, H.; Ayers, J.I.; Borchelt, D.R. Loss of charge mutations in solvent exposed Lys residues of superoxide dismutase 1 do not induce inclusion formation in cultured cell models. PLoS ONE 2018, 13, e0206751. [Google Scholar] [CrossRef]
- Saccon, R.A.; Bunton-Stasyshyn, R.K.A.; Fisher, E.M.C.; Fratta, P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 2013, 136, 2342–2358. [Google Scholar] [CrossRef] [PubMed]
- Kirby, J.; Halligan, E.; Baptista, M.J.; Allen, S.; Heath, P.R.; Holden, H.; Barber, S.C.; Loynes, C.A.; Wood-Allum, C.A.; Lunec, J.; et al. Mutant SOD1 alters the motor neuronal transcriptome: Implications for familial ALS. Brain 2005, 128, 1686–1706. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.-C. The Nrf2-ARE Pathway. Ann. N. Y. Acad. Sci. 2008, 1147, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Hirano, A.; Kato, S.; Nagai, R.; Horiuchi, S.; Komori, T.; Umahara, T.; Asayama, K.; Kobayashi, M. Advanced glycation endproducts are deposited in neuronal hyaline inclusions: A study on familial amyotrophic lateral sclerosis with superoxide dismutase-1 mutation. Acta Neuropathol. 1999, 97, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Andrus, P.K.; Fleck, T.J.; Gurney, M.E.; Hall, E.D. Protein Oxidative Damage in a Transgenic Mouse Model of Familial Amyotrophic Lateral Sclerosis. J. Neurochem. 2002, 71, 2041–2048. [Google Scholar] [CrossRef]
- Hall, E.D.; Andrus, P.K.; Oostveen, J.A.; Fleck, T.J.; Gurney, M.E. Relationship of oxygen radical-induced lipid peroxidative damage to disease onset and progression in a transgenic model of familial ALS. J. Neurosci. Res. 1998, 53, 66–77. [Google Scholar] [CrossRef]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- van Blitterswijk, M.; Mullen, B.; Heckman, M.G.; Baker, M.C.; DeJesus-Hernandez, M.; Brown, P.H.; Murray, M.E.; Hsiung, G.Y.R.; Stewart, H.; Karydas, A.M.; et al. Ataxin-2 as potential disease modifier in C9ORF72 expansion carriers. Neurobiol. Aging 2014. [Google Scholar] [CrossRef]
- Chiò, A.; Calvo, A.; Moglia, C.; Canosa, A.; Brunetti, M.; Barberis, M.; Restagno, G.; Conte, A.; Bisogni, G.; Marangi, G.; et al. ATXN2 polyQ intermediate repeats are a modifier of ALS survival. Neurology 2015. [Google Scholar] [CrossRef]
- Bertoni, A.; Giuliano, P.; Galgani, M.; Rotoli, D.; Ulianich, L.; Adornetto, A.; Santillo, M.R.; Porcellini, A.; Avvedimento, V.E. Early and Late Events Induced by PolyQ-expanded Proteins. J. Biol. Chem. 2011, 286, 4727–4741. [Google Scholar] [CrossRef]
- Polci, R.; Peng, A.; Chen, P.-L.; Riley, D.J.; Chen, Y. NIMA-Related Protein Kinase 1 Is Involved Early in the Ionizing Radiation-Induced DNA Damage Response. Cancer Res. 2004, 64, 8800–8803. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Lin, H.; Wang, X.; Zuo, Q.; Qin, J.; Zhang, P. The NEK1 interactor, C21ORF2, is required for efficient DNA damage repair. Acta Biochim. Biophys. Sin. (Shanghai) 2015, 47, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Kannan, A.; Bhatia, K.; Branzei, D.; Gangwani, L. Combined deficiency of Senataxin and DNA-PKcs causes DNA damage accumulation and neurodegeneration in spinal muscular atrophy. Nucleic Acids Res. 2018, 46, 8326–8346. [Google Scholar] [CrossRef] [PubMed]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.-F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef] [PubMed]
- van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; Diekstra, F.P.; Pulit, S.L.; van der Spek, R.A.A.; Võsa, U.; de Jong, S.; Robinson, M.R.; et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1043–1048. [Google Scholar] [CrossRef]
- Chen, Y.-Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 1128–1135. [Google Scholar] [CrossRef]
- Kenna, K.P.; Van Doormaal, P.T.C.; Dekker, A.M.; Ticozzi, N.; Kenna, B.J.; Diekstra, F.P.; Van Rheenen, W.; Van Eijk, K.R.; Jones, A.R.; Keagle, P.; et al. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1037–1042. [Google Scholar] [CrossRef]
- Higelin, J.; Catanese, A.; Semelink-Sedlacek, L.L.; Oeztuerk, S.; Lutz, A.K.; Bausinger, J.; Barbi, G.; Speit, G.; Andersen, P.M.; Ludolph, A.C.; et al. NEK1 loss-of-function mutation induces DNA damage accumulation in ALS patient-derived motoneurons. Stem Cell Res. 2018, 30, 150–162. [Google Scholar] [CrossRef]
- Bennett, C.L.; Dastidar, S.G.; Ling, S.C.; Malik, B.; Ashe, T.; Wadhwa, M.; Miller, D.B.; Lee, C.; Mitchell, M.B.; van Es, M.A.; et al. Senataxin mutations elicit motor neuron degeneration phenotypes and yield TDP-43 mislocalization in ALS4 mice and human patients. Acta Neuropathol. 2018, 136, 425–443. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; Vercesi, A.E. Mitochondrial damage induced by conditions of oxidative stress. Free Radic. Biol. Med. 1999, 26, 463–471. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Cycle or a Double-Edged Sword? Antioxid. Redox Signal. 2007, 9, 2277–2294. [Google Scholar] [CrossRef] [PubMed]
- Reichmann, D.; Voth, W.; Jakob, U. Maintaining a Healthy Proteome during Oxidative Stress. Mol. Cell 2018, 69, 203–213. [Google Scholar] [CrossRef] [PubMed]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More Than Just a Powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Horie, Y.; Iwata, M. Mitochondrial alterations in dorsal root ganglion cells in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 2007, 114, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial Cristae Shape Determines Respiratory Chain Supercomplexes Assembly and Respiratory Efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Zerbes, R.M.; Bohnert, M.; Stroud, D.A.; von der Malsburg, K.; Kram, A.; Oeljeklaus, S.; Warscheid, B.; Becker, T.; Wiedemann, N.; Veenhuis, M.; et al. Role of MINOS in Mitochondrial Membrane Architecture: Cristae Morphology and Outer Membrane Interactions Differentially Depend on Mitofilin Domains. J. Mol. Biol. 2012, 422, 183–191. [Google Scholar] [CrossRef]
- Pánek, T.; Eliáš, M.; Vancová, M.; Lukeš, J.; Hashimi, H. Returning to the Fold for Lessons in Mitochondrial Crista Diversity and Evolution. Curr. Biol. 2020, 30, R575–R588. [Google Scholar] [CrossRef]
- Genin, E.C.; Plutino, M.; Bannwarth, S.; Villa, E.; Cisneros-Barroso, E.; Roy, M.; Ortega-Vila, B.; Fragaki, K.; Lespinasse, F.; Pinero-Martos, E.; et al. CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO Mol. Med. 2016, 8, 58–72. [Google Scholar] [CrossRef]
- Genin, E.C.; Bannwarth, S.; Lespinasse, F.; Ortega-Vila, B.; Fragaki, K.; Itoh, K.; Villa, E.; Lacas-Gervais, S.; Jokela, M.; Auranen, M.; et al. Loss of MICOS complex integrity and mitochondrial damage, but not TDP-43 mitochondrial localisation, are likely associated with severity of CHCHD10-related diseases. Neurobiol. Dis. 2018, 119, 159–171. [Google Scholar] [CrossRef]
- Zhou, W.; Ma, D.; Sun, A.X.; Tran, H.-D.; Ma, D.; Singh, B.K.; Zhou, J.; Zhang, J.; Wang, D.; Zhao, Y.; et al. PD-linked CHCHD2 mutations impair CHCHD10 and MICOS complex leading to mitochondria dysfunction. Hum. Mol. Genet. 2019, 28, 1100–1116. [Google Scholar] [CrossRef]
- Purandare, N.; Somayajulu, M.; Hüttemann, M.; Grossman, L.I.; Aras, S. The cellular stress proteins CHCHD10 and MNRR1 (CHCHD2): Partners in mitochondrial and nuclear function and dysfunction. J. Biol. Chem. 2018, 293, 6517–6529. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Yang, M.; Chen, Y.; Chen, X.; Liu, J.; Sun, S.; Cheng, H.; Li, Y.; Bigio, E.H.; Mesulam, M.; et al. FUS Interacts with HSP60 to Promote Mitochondrial Damage. PLoS Genet. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wang, P.; Chen, X.; Cheng, H.; Liu, J.; Fushimi, K.; Zhu, L.; Wu, J.Y. FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc. Natl. Acad. Sci. USA 2018, 115, E9678–E9686. [Google Scholar] [CrossRef] [PubMed]
- Crozat, A.; Aman, P.; Mandahl, N.; Ron, D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 1993, 363, 640–644. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA Processing Protein, Cause Familial Amyotrophic Lateral Sclerosis Type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J.; Bosco, D.A.; LeClerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071. [Google Scholar] [CrossRef]
- Magrané, J.; Cortez, C.; Gan, W.-B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef]
- Bernardini, C.; Censi, F.; Lattanzi, W.; Barba, M.; Calcagnini, G.; Giuliani, A.; Tasca, G.; Sabatelli, M.; Ricci, E.; Michetti, F. Mitochondrial network genes in the skeletal muscle of amyotrophic lateral sclerosis patients. PLoS ONE 2013, 8, e57739. [Google Scholar] [CrossRef]
- Higgins, C.M.J.; Jung, C.; Xu, Z. ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci. 2003, 4, 16. [Google Scholar] [CrossRef]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H. Amyotrophic Lateral Sclerosis-Associated SOD1 Mutant Proteins Bind and Aggregate with Bcl-2 in Spinal Cord Mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.E.; Bowling, A.C.; Baik, M.J.; Gurney, M.; Brown, R.H.; Beal, M.F. Metabolic Dysfunction in Familial, but Not Sporadic, Amyotrophic Lateral Sclerosis. J. Neurochem. 2002, 71, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, F.R.; Manfredi, G.; Mawrin, C.; Beal, M.F.; Schon, E.A. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 2002, 80, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Dröse, S.; Brandt, U. Molecular Mechanisms of Superoxide Production by the Mitochondrial Respiratory Chain. In Mitochondrial Oxidative Phosphorylation; Springer: New York, NY, USA, 2012; pp. 145–169. [Google Scholar]
- Umoh, M.E.; Fournier, C.; Li, Y.; Polak, M.; Shaw, L.; Landers, J.E.; Hu, W.; Gearing, M.; Glass, J.D. Comparative analysis of C9orf72 and sporadic disease in an ALS clinic population. Neurology 2016, 87, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.R.; Boeve, B.F.F.; Boxer, A.L.L.; Baker, M.; Rutherford, N.J.J.; Nicholson, A.M.M.; Finch, N.A.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Choi, S.Y.; Lopez-Gonzalez, R.; Krishnan, G.; Phillips, H.L.; Li, A.N.; Seeley, W.W.; Yao, W.-D.; Almeida, S.; Gao, F.-B. C9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nat. Neurosci. 2019, 22, 851–862. [Google Scholar] [CrossRef]
- Onesto, E.; Colombrita, C.; Gumina, V.; Borghi, M.O.; Dusi, S.; Doretti, A.; Fagiolari, G.; Invernizzi, F.; Moggio, M.; Tiranti, V.; et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 2016, 4, 47. [Google Scholar] [CrossRef]
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 2019, 50, 274–289. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Holzbaur, E.L.F. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; Stevnsner, T.; Nilsen, H.; Bohr, V.A.; Fang, E.F. Mitophagy in neurodegeneration and aging. Neurochem. Int. 2017, 109, 202–209. [Google Scholar] [CrossRef]
- Damiano, M.; Starkov, A.A.; Petri, S.; Kipiani, K.; Kiaei, M.; Mattiazzi, M.; Flint Beal, M.; Manfredi, G. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J. Neurochem. 2006, 96, 1349–1361. [Google Scholar] [CrossRef]
- Sathasivam, S.; Grierson, A.J.; Shaw, P.J.; Shaw, P.J. Characterization of the caspase cascade in a cell culture model of SOD1-related familial amyotrophic lateral sclerosis: Expression, activation and therapeutic effects of inhibition. Neuropathol. Appl. Neurobiol. 2005, 31, 467–685. [Google Scholar] [CrossRef]
- Sutton, M.A.; Schuman, E.M. Dendritic Protein Synthesis, Synaptic Plasticity, and Memory. Cell 2006, 127, 49–58. [Google Scholar] [CrossRef]
- Yuan, A.; Rao, M.V.; Veeranna; Nixon, R.A. Neurofilaments and Neurofilament Proteins in Health and Disease. Cold Spring Harb. Perspect. Biol. 2017, 9, a018309. [Google Scholar] [CrossRef]
- Brady, S. Mice overexpressing the human neurofilament heavy gene as a model of ALS. Neurobiol. Aging 1995. [Google Scholar] [CrossRef]
- Prokop, A. Cytoskeletal organization of axons in vertebrates and invertebrates. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef] [PubMed]
- Figlewicz, D.A.; Krizus, A.; Martinoli, M.G.; Meininger, V.; Dib, M.; Rouleau, G.A.; Julien, J.P. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum. Mol. Genet. 1994, 3, 1757–1761. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.; Bhembre, N.; Bodas, S.; Veer, S.; Ghose, A.; Callan-Jones, A.; Pullarkat, P. The axonal actin-spectrin lattice acts as a tension buffering shock absorber. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Witke, W.; Podtelejnikov, A.V.; Di Nardo, A.; Sutherland, J.D.; Gurniak, C.B.; Dotti, C.; Mann, M. In mouse brain profilin I and profilin II associate with regulators of the endocytic pathway and actin assembly. EMBO J. 1998, 17, 967–976. [Google Scholar] [CrossRef]
- Wu, C.-H.; Fallini, C.; Ticozzi, N.; Keagle, P.J.; Sapp, P.C.; Piotrowska, K.; Lowe, P.; Koppers, M.; McKenna-Yasek, D.; Baron, D.M.; et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012, 488, 499–503. [Google Scholar] [CrossRef]
- Fil, D.; DeLoach, A.; Yadav, S.; Alkam, D.; MacNicol, M.; Singh, A.; Compadre, C.M.; Goellner, J.J.; O’Brien, C.A.; Fahmi, T.; et al. Mutant Profilin1 transgenic mice recapitulate cardinal features of motor neuron disease. Hum. Mol. Genet. 2017. [Google Scholar] [CrossRef]
- Tempes, A.; Weslawski, J.; Brzozowska, A.; Jaworski, J. Role of dynein-dynactin complex, kinesins, motor adaptors, and their phosphorylation in dendritogenesis. J. Neurochem. 2020. [Google Scholar] [CrossRef]
- Uchida, A.; Alami, N.H.; Brown, A. Tight Functional Coupling of Kinesin-1A and Dynein Motors in the Bidirectional Transport of Neurofilaments. Mol. Biol. Cell 2009, 20, 4997–5006. [Google Scholar] [CrossRef]
- Heisler, F.F.; Lee, H.K.; Gromova, K.V.; Pechmann, Y.; Schurek, B.; Ruschkies, L.; Schroeder, M.; Schweizer, M.; Kneussel, M. GRIP1 interlinks N-cadherin and AMPA receptors at vesicles to promote combined cargo transport into dendrites. Proc. Natl. Acad. Sci. USA 2014, 111, 5030–5035. [Google Scholar] [CrossRef]
- Nakajima, K.; Yin, X.; Takei, Y.; Seog, D.-H.; Homma, N.; Hirokawa, N. Molecular motor KIF5A is essential for GABA(A) receptor transport, and KIF5A deletion causes epilepsy. Neuron 2012, 76, 945–961. [Google Scholar] [CrossRef]
- Smith, B.N.; Ticozzi, N.; Fallini, C.; Gkazi, A.S.; Topp, S.; Kenna, K.P.; Scotter, E.L.; Kost, J.; Keagle, P.; Miller, J.W.; et al. Exome-wide Rare Variant Analysis Identifies TUBA4A Mutations Associated with Familial ALS. Neuron 2014, 84, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Puls, I.; Jonnakuty, C.; LaMonte, B.H.; Holzbaur, E.L.F.; Tokito, M.; Mann, E.; Floeter, M.K.; Bidus, K.; Drayna, D.; Oh, S.J.; et al. Mutant dynactin in motor neuron disease. Nat. Genet. 2003, 33, 455–456. [Google Scholar] [CrossRef] [PubMed]
- LaMonte, B.H.; Wallace, K.E.; Holloway, B.A.; Shelly, S.S.; Ascano, J.; Tokito, M.; Van Winkle, T.; Howland, D.S.; Holzbaur, E.L.F. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron 2002, 34, 715–727. [Google Scholar] [CrossRef]
- Miki, H.; Setou, M.; Kaneshiro, K.; Hirokawa, N. All kinesin superfamily protein, KIF, genes in mouse and human. Proc. Natl. Acad. Sci. USA 2001, 98, 7004–7011. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, A.; Kenna, K.P.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283.e6. [Google Scholar] [CrossRef] [PubMed]
- Karle, K.N.; Möckel, D.; Reid, E.; Schöls, L. Axonal transport deficit in a KIF5A(-/-) mouse model. Neurogenetics 2012, 13, 169–179. [Google Scholar] [CrossRef]
- Warita, H.; Itoyama, Y.; Abe, K. Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene. Brain Res. 1999, 819, 120–131. [Google Scholar] [CrossRef]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef]
- Tallon, C.; Russell, K.A.; Sakhalkar, S.; Andrapallayal, N.; Farah, M.H. Length-dependent axo-terminal degeneration at the neuromuscular synapses of type II muscle in SOD1 mice. Neuroscience 2016, 312, 179–189. [Google Scholar] [CrossRef]
- Pantelidou, M.; Zographos, S.E.; Lederer, C.W.; Kyriakides, T.; Pfaffl, M.W.; Santama, N. Differential expression of molecular motors in the motor cortex of sporadic ALS. Neurobiol. Dis. 2007, 26, 577–589. [Google Scholar] [CrossRef]
- Landers, J.E.; Melki, J.; Meininger, V.; Glass, J.D.; van den Berg, L.H.; van Es, M.A.; Sapp, P.C.; van Vught, P.W.J.; McKenna-Yasek, D.M.; Blauw, H.M.; et al. Reduced expression of the Kinesin-Associated Protein 3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2009, 106, 9004–9009. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Pitt, D.; Werner, P.; Raine, C.S. Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med. 2000, 6, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Vaarmann, A.; Kovac, S.; Holmström, K.M.; Gandhi, S.; Abramov, A.Y. Dopamine protects neurons against glutamate-induced excitotoxicity. Cell Death Dis. 2013, 4, e455. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Tsai, G.; Kuncl, R.W.; Clawson, L.; Cornblath, D.R.; Drachman, D.B.; Pestronk, A.; Stauch, B.L.; Coyle, J.T. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann. Neurol. 1990, 28, 18–25. [Google Scholar] [CrossRef]
- Shaw, P.J.; Forrest, V.; Ince, P.G.; Richardson, J.P.; Wastell, H.J. CSF and Plasma Amino Acid Levels in Motor Neuron Disease: Elevation of CSF Glutamate in a Subset of Patients. Neurodegeneration 1995, 4, 209–216. [Google Scholar] [CrossRef]
- Wang, Y.; Qin, Z. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis 2010, 15, 1382–1402. [Google Scholar] [CrossRef]
- Netzahualcoyotzi, C.; Tapia, R. Degeneration of spinal motor neurons by chronic AMPA-induced excitotoxicity in vivo and protection by energy substrates. Acta Neuropathol. Commun. 2015, 3, 27. [Google Scholar] [CrossRef]
- Van Den Bosch, L.; Vandenberghe, W.; Klaassen, H.; Van Houtte, E.; Robberecht, W. Ca2+-permeable AMPA receptors and selective vulnerability of motor neurons. J. Neurol. Sci. 2000, 180, 29–34. [Google Scholar] [CrossRef]
- Gregory, J.M.; Livesey, M.R.; McDade, K.; Selvaraj, B.T.; Barton, S.K.; Chandran, S.; Smith, C. Dysregulation of AMPA receptor subunit expression in sporadic ALS post-mortem brain. J. Pathol. 2020, 250, 67–78. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed]
- Corona, J.C.; Tapia, R. AMPA receptor activation, but not the accumulation of endogenous extracellular glutamate, induces paralysis and motor neuron death in rat spinal cord in vivo. J. Neurochem. 2004, 89, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Konen, L.M.; Wright, A.L.; Royle, G.A.; Morris, G.P.; Lau, B.K.; Seow, P.W.; Zinn, R.; Milham, L.T.; Vaughan, C.W.; Vissel, B. A new mouse line with reduced GluA2 Q/R site RNA editing exhibits loss of dendritic spines, hippocampal CA1-neuron loss, learning and memory impairments and NMDA receptor-independent seizure vulnerability. Mol. Brain 2020, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Heath, P.R.; Tomkins, J.; Ince, P.G.; Shaw, P.J. Quantitative assessment of AMPA receptor mRNA in human spinal motor neurons isolated by laser capture microdissection. Neuroreport 2002, 13, 1753–1757. [Google Scholar] [CrossRef] [PubMed]
- Takuma, H.; Kwak, S.; Yoshizawa, T.; Kanazawa, I. Reduction of GluR2 RNA editing, a molecular change that increases calcium influx through AMPA receptors, selective in the spinal ventral gray of patients with amyotrophic lateral sclerosis. Ann. Neurol. 1999, 46, 806–815. [Google Scholar] [CrossRef]
- Ince, P.; Stout, N.; Shaw, P.; Slade, J.; Hunziker, W.; Heizmann, C.W.; Baimbridge, K.G. Parvalbumin and calbindin D-28k in the human motor system and in motor neuron disease. Neuropathol. Appl. Neurobiol. 1993, 19, 291–299. [Google Scholar] [CrossRef]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 2000, 406, 78–81. [Google Scholar] [CrossRef]
- Hideyama, T.; Kwak, S. When Does ALS Start? ADAR2?GluA2 Hypothesis for the Etiology of Sporadic ALS. Front. Mol. Neurosci. 2011, 4. [Google Scholar] [CrossRef]
- Aizawa, H.; Sawada, J.; Hideyama, T.; Yamashita, T.; Katayama, T.; Hasebe, N.; Kimura, T.; Yahara, O.; Kwak, S. TDP-43 pathology in sporadic ALS occurs in motor neurons lacking the RNA editing enzyme ADAR2. Acta Neuropathol. 2010, 120, 75–84. [Google Scholar] [CrossRef]
- Medina, L.; Figueredo-Cardenas, G.; Rothstein, J.D.; Reiner, A. Differential Abundance of Glutamate Transporter Subtypes in Amyotrophic Lateral Sclerosis (ALS)-Vulnerable versus ALS-Resistant Brain Stem Motor Cell Groups. Exp. Neurol. 1996, 142, 287–295. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef]
- Kong, Q.; Chang, L.-C.; Takahashi, K.; Liu, Q.; Schulte, D.A.; Lai, L.; Ibabao, B.; Lin, Y.; Stouffer, N.; Mukhopadhyay, C.D.; et al. Small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection. J. Clin. Investig. 2014, 124, 1255–1267. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Patel, S.; Regan, M.R.; Haenggeli, C.; Huang, Y.H.; Bergles, D.E.; Jin, L.; Dykes Hoberg, M.; Vidensky, S.; Chung, D.S.; et al. β-Lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 2005, 433, 73–77. [Google Scholar] [CrossRef]
- Bristol, L.A.; Rothstein, J.D. Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Ann. Neurol. 1996, 39, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Komori, T.; Iwata, M. Excitatory amino acid transporter 1 and 2 immunoreactivity in the spinal cord in amyotrophic lateral sclerosis. Acta Neuropathol. 2000, 100, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Milanese, M.; Zappettini, S.; Onofri, F.; Musazzi, L.; Tardito, D.; Bonifacino, T.; Messa, M.; Racagni, G.; Usai, C.; Benfenati, F.; et al. Abnormal exocytotic release of glutamate in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2011, 116, 1028–1042. [Google Scholar] [CrossRef] [PubMed]
- van Zundert, B.; Peuscher, M.H.; Hynynen, M.; Chen, A.; Neve, R.L.; Brown, R.H.; Constantine-Paton, M.; Bellingham, M.C. Neonatal Neuronal Circuitry Shows Hyperexcitable Disturbance in a Mouse Model of the Adult-Onset Neurodegenerative Disease Amyotrophic Lateral Sclerosis. J. Neurosci. 2008, 28, 10864–10874. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased Glutamate Transport by the Brain and Spinal Cord in Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar] [CrossRef]
- Vucic, S.; Nicholson, G.A.; Kiernan, M.C. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 2008, 131, 1540–1550. [Google Scholar] [CrossRef]
- Perry, T.L.; Krieger, C.; Hansen, S.; Eisen, A. Amyotrophic lateral sclerosis: Amino acid levels in plasma and cerebrospinal fluid. Ann. Neurol. 1990, 28, 12–17. [Google Scholar] [CrossRef]
- Perry, T.L.; Hansen, S.; Jones, K. Brain glutamate deficiency in amyotrophic lateral sclerosis. Neurology 1987, 37, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- Wuolikainen, A.; Moritz, T.; Marklund, S.L.; Antti, H.; Andersen, P.M. Disease-related changes in the cerebrospinal fluid metabolome in amyotrophic lateral sclerosis detected by GC/TOFMS. PLoS ONE 2011, 6, e17947. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Hung, S.-T.; Rocha, G.; Lin, S.; Linares, G.R.; Staats, K.A.; Seah, C.; Wang, Y.; Chickering, M.; Lai, J.; et al. Identification and therapeutic rescue of autophagosome and glutamate receptor defects in C9ORF72 and sporadic ALS neurons. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, B.T.; Livesey, M.R.; Zhao, C.; Gregory, J.M.; James, O.T.; Cleary, E.M.; Chouhan, A.K.; Gane, A.B.; Perkins, E.M.; Dando, O.; et al. C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun. 2018, 9, 347. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lin, S.; Staats, K.A.; Li, Y.; Chang, W.-H.; Hung, S.-T.; Hendricks, E.; Linares, G.R.; Wang, Y.; Son, E.Y.; et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 2018, 24, 313–325. [Google Scholar] [CrossRef]
- Xiao, S.; McKeever, P.M.; Lau, A.; Robertson, J. Synaptic localization of C9orf72 regulates post-synaptic glutamate receptor 1 levels. Acta Neuropathol. Commun. 2019, 7, 161. [Google Scholar] [CrossRef]
- Tang, D.; Sheng, J.; Xu, L.; Zhan, X.; Liu, J.; Jiang, H.; Shu, X.; Liu, X.; Zhang, T.; Jiang, L.; et al. Cryo-EM structure of C9ORF72–SMCR8–WDR41 reveals the role as a GAP for Rab8a and Rab11a. Proc. Natl. Acad. Sci. USA 2020, 117, 9876–9883. [Google Scholar] [CrossRef]
- Lai, C.; Xie, C.; Shim, H.; Chandran, J.; Howell, B.W.; Cai, H. Regulation of endosomal motility and degradation by amyotrophic lateral sclerosis 2/alsin. Mol. Brain 2009, 2, 23. [Google Scholar] [CrossRef]
- Blasco, H.; Mavel, S.; Corcia, P.; Gordon, P.H. The glutamate hypothesis in ALS: Pathophysiology and drug development. Curr. Med. Chem. 2014, 21, 3551–3575. [Google Scholar] [CrossRef]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar] [CrossRef]
- Thorley, M.; Malatras, A.; Duddy, W.J.; Le Gall, L.; Mouly, V.; Butler Browne, G.; Duguez, S.M.-R. Changes in communication between muscle stem cells and their environment with aging. J. Neuromuscul. Dis. 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bergamini, G.; Rajendran, L. Cell-to-cell Communication by Extracellular Vesicles: Focus on Microglia. Neuroscience 2019, 405, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Théry, C. Exosomes: Secreted vesicles and intercellular communications. F1000 Biol. Rep. 2011, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Basso, M.; Pozzi, S.; Tortarolo, M.; Fiordaliso, F.; Bisighini, C.; Pasetto, L.; Spaltro, G.; Lidonnici, D.; Gensano, F.; Battaglia, E.; et al. Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: Implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J. Biol. Chem. 2013, 288, 15699–15711. [Google Scholar] [CrossRef]
- Silverman, J.M.; Christy, D.; Shyu, C.C.; Moon, K.-M.; Fernando, S.; Gidden, Z.; Cowan, C.M.; Ban, Y.; Stacey, R.G.; Grad, L.I.; et al. CNS-derived extracellular vesicles from superoxide dismutase 1 (SOD1)G93A ALS mice originate from astrocytes and neurons and carry misfolded SOD1. J. Biol. Chem. 2019, 294, 3744–3759. [Google Scholar] [CrossRef]
- Sproviero, D.; La Salvia, S.; Colombo, F.; Zucca, S.; Pansarasa, O.; Diamanti, L.; Costa, A.; Lova, L.; Giannini, M.; Gagliardi, S.; et al. Leukocyte Derived Microvesicles as Disease Progression Biomarkers in Slow Progressing Amyotrophic Lateral Sclerosis Patients. Front. Neurosci. 2019, 13, 344. [Google Scholar] [CrossRef]
- Chen, Y.; Xia, K.; Chen, L.; Fan, D. Increased Interleukin-6 Levels in the Astrocyte-Derived Exosomes of Sporadic Amyotrophic Lateral Sclerosis Patients. Front. Neurosci. 2019, 13, 574. [Google Scholar] [CrossRef]
- Westergard, T.; Jensen, B.K.; Wen, X.; Cai, J.; Kropf, E.; Iacovitti, L.; Pasinelli, P.; Trotti, D. Cell-to-Cell Transmission of Dipeptide Repeat Proteins Linked to C9orf72 -ALS/FTD. Cell Rep. 2016, 17, 645–652. [Google Scholar] [CrossRef]
- Scaramozza, A.; Marchese, V.; Papa, V.; Salaroli, R.; Sorarù, G.; Angelini, C.; Cenacchi, G. Skeletal Muscle Satellite Cells in Amyotrophic Lateral Sclerosis. Ultrastruct. Pathol. 2014, 38, 1–8. [Google Scholar] [CrossRef]
- Parkinson, N.; Ince, P.G.; Smith, M.O.; Highley, R.; Skibinski, G.; Andersen, P.M.; Morrison, K.E.; Pall, H.S.; Hardiman, O.; Collinge, J.; et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 2006, 67, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, G.; Parkinson, N.J.; Brown, J.M.; Chakrabarti, L.; Lloyd, S.L.; Hummerich, H.; Nielsen, J.E.; Hodges, J.R.; Spillantini, M.G.; Thusgaard, T.; et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 2005, 37, 806–808. [Google Scholar] [CrossRef] [PubMed]
- Blanc, L.; Vidal, M. New insights into the function of Rab GTPases in the context of exosomal secretion. Small GTPases 2018, 9, 95–106. [Google Scholar] [CrossRef]
- Yang, Y.; Hentati, A.; Deng, H.X.; Dabbagh, O.; Sasaki, T.; Hirano, M.; Hung, W.Y.; Ouahchi, K.; Yan, J.; Azim, A.C.; et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 2001, 29, 160–165. [Google Scholar] [CrossRef]
- Sellier, C.; Campanari, M.-L.; Julie Corbier, C.; Gaucherot, A.; Kolb-Cheynel, I.; Oulad-Abdelghani, M.; Ruffenach, F.; Page, A.; Ciura, S.; Kabashi, E.; et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016, 35, 1276–1297. [Google Scholar] [CrossRef] [PubMed]
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.A.K.; Levina, V.; Halloran, M.A.; Gleeson, P.A.; Blair, I.P.; Soo, K.Y.; et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Iwano, T.; Kunii, M.; Matsuda, S.; Mizuguchi, R.; Jung, Y.; Hagiwara, H.; Yoshihara, Y.; Yuzaki, M.; Harada, R.; et al. Rab8a and Rab8b are essential for several apical transport pathways but insufficient for ciliogenesis. J. Cell Sci. 2014, 127, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Seto, S.; Sugaya, K.; Tsujimura, K.; Nagata, T.; Horii, T.; Koide, Y. Rab39a interacts with phosphatidylinositol 3-kinase and negatively regulates autophagy induced by lipopolysaccharide stimulation in macrophages. PLoS ONE 2013, 8, e83324. [Google Scholar] [CrossRef] [PubMed]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef]
- O’Rourke, J.G.; Bogdanik, L.; Yáñez, A.; Lall, D.; Wolf, A.J.; Muhammad, A.K.M.G.; Ho, R.; Carmona, S.; Vit, J.P.; Zarrow, J.; et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 2016, 351, 1324–1329. [Google Scholar] [CrossRef]
- Aoki, Y.; Manzano, R.; Lee, Y.; Dafinca, R.; Aoki, M.; Douglas, A.G.L.; Varela, M.A.; Sathyaprakash, C.; Scaber, J.; Barbagallo, P.; et al. C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain 2017, 140, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Hantan, D.; Yamamoto, Y.; Sakisaka, T. VAP-B binds to Rab3GAP1 at the ER: Its implication in nuclear envelope formation through the ER-Golgi intermediate compartment. Kobe J. Med. Sci. 2014, 60, E48–E56. [Google Scholar] [PubMed]
- Lev, S.; Ben Halevy, D.; Peretti, D.; Dahan, N. The VAP protein family: From cellular functions to motor neuron disease. Trends Cell Biol. 2008, 18, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Rocha, N.; Kuijl, C.; van der Kant, R.; Janssen, L.; Houben, D.; Janssen, H.; Zwart, W.; Neefjes, J. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7–RILP–p150Glued and late endosome positioning. J. Cell Biol. 2009, 185, 1209–1225. [Google Scholar] [CrossRef] [PubMed]
- Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. Int. J. Mol. Sci. 2020, 21, 2576. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef]
- Bug, M.; Meyer, H. Expanding into new markets--VCP/p97 in endocytosis and autophagy. J. Struct. Biol. 2012, 179, 78–82. [Google Scholar] [CrossRef]
- Chow, C.Y.; Landers, J.E.; Bergren, S.K.; Sapp, P.C.; Grant, A.E.; Jones, J.M.; Everett, L.; Lenk, G.M.; McKenna-Yasek, D.M.; Weisman, L.S.; et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 2009, 84, 85–88. [Google Scholar] [CrossRef]
- Martyn, C.; Li, J. Fig4 deficiency: A newly emerged lysosomal storage disorder? Prog. Neurobiol. 2013, 101–102, 35–45. [Google Scholar] [CrossRef]
- Branchu, J.; Boutry, M.; Sourd, L.; Depp, M.; Leone, C.; Corriger, A.; Vallucci, M.; Esteves, T.; Matusiak, R.; Dumont, M.; et al. Loss of spatacsin function alters lysosomal lipid clearance leading to upper and lower motor neuron degeneration. Neurobiol. Dis. 2017, 102, 21–37. [Google Scholar] [CrossRef]
- Pérez-Brangulí, F.; Mishra, H.K.; Prots, I.; Havlicek, S.; Kohl, Z.; Saul, D.; Rummel, C.; Dorca-Arevalo, J.; Regensburger, M.; Graef, D.; et al. Dysfunction of spatacsin leads to axonal pathology in SPG11-linked hereditary spastic paraplegia. Hum. Mol. Genet. 2014, 23, 4859–4874. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Gill, C.; Phelan, J.P.; Hatzipetros, T.; Kidd, J.D.; Tassinari, V.R.; Levine, B.; Wang, M.Z.; Moreno, A.; Thompson, K.; Maier, M.; et al. SOD1-positive aggregate accumulation in the CNS predicts slower disease progression and increased longevity in a mutant SOD1 mouse model of ALS. Sci. Rep. 2019. [Google Scholar] [CrossRef] [PubMed]
- Feneberg, E.; Gray, E.; Ansorge, O.; Talbot, K.; Turner, M.R. Towards a TDP-43-Based Biomarker for ALS and FTLD. Mol. Neurobiol. 2018, 55, 7789–7801. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Jansen-West, K.; Xu, Y.-F.; Gendron, T.F.; Bieniek, K.F.; Lin, W.-L.; Sasaguri, H.; Caulfield, T.; Hubbard, J.; Daughrity, L.; et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014, 128, 505–524. [Google Scholar] [CrossRef]
- Picchiarelli, G.; Dupuis, L. Role of RNA Binding Proteins with prion-like domains in muscle and neuromuscular diseases. Cell Stress 2020, 4, 76–91. [Google Scholar] [CrossRef]
- Hirano, A. Familial Amyotrophic Lateral Sclerosis. Arch. Neurol. 1967, 16, 232. [Google Scholar] [CrossRef]
- Sun, C.N.; Araoz, C.; Lucas, G.; Morgan, P.N.; White, H.J. Amyotrophic lateral sclerosis. Inclusion bodies in a case of the classic sporadic form. Ann. Clin. Lab. Sci. 1975, 5, 38–44. [Google Scholar]
- Takahashi, K. Hereditary Amyotrophic Lateral Sclerosis. Arch. Neurol. 1972, 27, 292. [Google Scholar] [CrossRef]
- Shibata, N.; Hirano, A.; Kobayashi, M.; Siddique, T.; Deng, H.-X.; Hung, W.-Y.; Kato, T.; Asayama, K. Intense Superoxide Dismutase-1 Immunoreactivity in Intracytoplasmic Hyaline Inclusions of Familial Amyotrophic Lateral Sclerosis with Posterior Column Involvement. J. Neuropathol. Exp. Neurol. 1996, 55, 481–490. [Google Scholar] [CrossRef]
- Matsumoto, G.; Wada, K.; Okuno, M.; Kurosawa, M.; Nukina, N. Serine 403 Phosphorylation of p62/SQSTM1 Regulates Selective Autophagic Clearance of Ubiquitinated Proteins. Mol. Cell 2011, 44, 279–289. [Google Scholar] [CrossRef]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H.; et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 2007, 61, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.-X.; Chen, W.; Hong, S.-T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.R.; Sumner, C.J.; Caviston, J.P.; Tokito, M.K.; Ranganathan, S.; Ligon, L.A.; Wallace, K.E.; LaMonte, B.H.; Harmison, G.G.; Puls, I.; et al. A motor neuron disease–associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J. Cell Biol. 2006, 172, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Ayaki, T.; Ito, H.; Fukushima, H.; Inoue, T.; Kondo, T.; Ikemoto, A.; Asano, T.; Shodai, A.; Fujita, T.; Fukui, S.; et al. Immunoreactivity of valosin-containing protein in sporadic amyotrophic lateral sclerosis and in a case of its novel mutant. Acta Neuropathol. Commun. 2014, 2, 172. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Pioro, E.P.; Boehringer, A.; Chia, R.; Feit, H.; Renton, A.E.; Pliner, H.A.; Abramzon, Y.; Marangi, G.; Winborn, B.J.; et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 2014, 17, 664–666. [Google Scholar] [CrossRef]
- Chattopadhyay, M.; Nwadibia, E.; Strong, C.D.; Gralla, E.B.; Valentine, J.S.; Whitelegge, J.P. The Disulfide Bond, but Not Zinc or Dimerization, Controls Initiation and Seeded Growth in Amyotrophic Lateral Sclerosis-linked Cu,Zn Superoxide Dismutase (SOD1) Fibrillation. J. Biol. Chem. 2015, 290, 30624–30636. [Google Scholar] [CrossRef]
- Lang, L.; Kurnik, M.; Danielsson, J.; Oliveberg, M. Fibrillation precursor of superoxide dismutase 1 revealed by gradual tuning of the protein-folding equilibrium. Proc. Natl. Acad. Sci. USA 2012, 109, 17868–17873. [Google Scholar] [CrossRef]
- Chan, P.K.; Chattopadhyay, M.; Sharma, S.; Souda, P.; Gralla, E.B.; Borchelt, D.R.; Whitelegge, J.P.; Valentine, J.S. Structural similarity of wild-type and ALS-mutant superoxide dismutase-1 fibrils using limited proteolysis and atomic force microscopy. Proc. Natl. Acad. Sci. USA 2013, 110, 10934–10939. [Google Scholar] [CrossRef]
- Ivanova, M.I.; Sievers, S.A.; Guenther, E.L.; Johnson, L.M.; Winkler, D.D.; Galaleldeen, A.; Sawaya, M.R.; Hart, P.J.; Eisenberg, D.S. Aggregation-triggering segments of SOD1 fibril formation support a common pathway for familial and sporadic ALS. Proc. Natl. Acad. Sci. USA 2014, 111, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Abdolvahabi, A.; Shi, Y.; Chuprin, A.; Rasouli, S.; Shaw, B.F. Stochastic Formation of Fibrillar and Amorphous Superoxide Dismutase Oligomers Linked to Amyotrophic Lateral Sclerosis. ACS Chem. Neurosci. 2016, 7, 799–810. [Google Scholar] [CrossRef] [PubMed]
- McAlary, L.; Aquilina, J.A.; Yerbury, J.J. Susceptibility of Mutant SOD1 to Form a Destabilized Monomer Predicts Cellular Aggregation and Toxicity but Not In vitro Aggregation Propensity. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.S.A.; Antonyuk, S.V.; Hasnain, S.S. A faulty interaction between SOD1 and hCCS in neurodegenerative disease. Sci. Rep. 2016, 6, 27691. [Google Scholar] [CrossRef] [PubMed]
- Elam, J.S.; Taylor, A.B.; Strange, R.; Antonyuk, S.; Doucette, P.A.; Rodriguez, J.A.; Hasnain, S.S.; Hayward, L.J.; Valentine, J.S.; Yeates, T.O.; et al. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat. Struct. Biol. 2003, 10, 461–467. [Google Scholar] [CrossRef]
- Ayers, J.I.; Cashman, N.R. Prion-like mechanisms in amyotrophic lateral sclerosis. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 337–354. [Google Scholar]
- Baumer, K.M.; Koone, J.C.; Shaw, B.F. Kinetic Variability in Seeded Formation of ALS-Linked SOD1 Fibrils Across Multiple Generations. ACS Chem. Neurosci. 2020, 11, 304–313. [Google Scholar] [CrossRef]
- Healy, E.F.; Roth-Rodriguez, A.; Toledo, S. A model for gain of function in superoxide dismutase. Biochem. Biophys. Rep. 2020, 21, 100728. [Google Scholar] [CrossRef]
- Vogler, T.O.; Wheeler, J.R.; Nguyen, E.D.; Hughes, M.P.; Britson, K.A.; Lester, E.; Rao, B.; Betta, N.D.; Whitney, O.N.; Ewachiw, T.E.; et al. TDP-43 and RNA form amyloid-like myo-granules in regenerating muscle. Nature 2018, 563, 508–513. [Google Scholar] [CrossRef]
- Rhoads, S.N.; Monahan, Z.T.; Yee, D.S.; Shewmaker, F.P. The Role of Post-Translational Modifications on Prion-Like Aggregation and Liquid-Phase Separation of FUS. Int. J. Mol. Sci. 2018, 19, 886. [Google Scholar] [CrossRef]
- Pokrishevsky, E.; Grad, L.I.; Cashman, N.R. TDP-43 or FUS-induced misfolded human wild-type SOD1 can propagate intercellularly in a prion-like fashion. Sci. Rep. 2016, 6, 22155. [Google Scholar] [CrossRef]
- Sproviero, D.; La Salvia, S.; Giannini, M.; Crippa, V.; Gagliardi, S.; Bernuzzi, S.; Diamanti, L.; Ceroni, M.; Pansarasa, O.; Poletti, A.; et al. Pathological Proteins Are Transported by Extracellular Vesicles of Sporadic Amyotrophic Lateral Sclerosis Patients. Front. Neurosci. 2018, 12, 487. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H.; Kadowaki, H.; Nagai, A.; Maruyama, T.; Yokota, T.; Fukutomi, H.; Noguchi, T.; Matsuzawa, A.; Takeda, K.; Ichijo, H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008, 22, 1451–1464. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Scotter, E.L.; Nishimura, A.L.; Troakes, C.; Mitchell, J.C.; Kathe, C.; Urwin, H.; Manser, C.; Miller, C.C.; Hortobágyi, T.; et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum. Mol. Genet. 2013, 22, 2676–2688. [Google Scholar] [CrossRef] [PubMed]
- Teyssou, E.; Chartier, L.; Amador, M.-D.-M.; Lam, R.; Lautrette, G.; Nicol, M.; Machat, S.; Da Barroca, S.; Moigneu, C.; Mairey, M.; et al. Novel UBQLN2 mutations linked to amyotrophic lateral sclerosis and atypical hereditary spastic paraplegia phenotype through defective HSP70-mediated proteolysis. Neurobiol. Aging 2017, 58, 239.e11–239.e20. [Google Scholar] [CrossRef] [PubMed]
- Saeki, Y. Ubiquitin recognition by the proteasome. J. Biochem. 2017, 161, 113–124. [Google Scholar] [CrossRef]
- Ryan, T.A.; Tumbarello, D.A. Optineurin: A Coordinator of Membrane-Associated Cargo Trafficking and Autophagy. Front. Immunol. 2018, 9, 1024. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef]
- Chen, H.-J.; Anagnostou, G.; Chai, A.; Withers, J.; Morris, A.; Adhikaree, J.; Pennetta, G.; de Belleroche, J.S. Characterization of the properties of a novel mutation in VAPB in familial amyotrophic lateral sclerosis. J. Biol. Chem. 2010, 285, 40266–40281. [Google Scholar] [CrossRef]
- Tsai, P.C.; Liao, Y.C.; Chen, P.L.; Guo, Y.C.; Chen, Y.H.; Jih, K.Y.; Lin, K.P.; Soong, B.W.; Tsai, C.P.; Lee, Y.C. Investigating CCNF mutations in a Taiwanese cohort with amyotrophic lateral sclerosis. Neurobiol. Aging 2018, 62, 243–e1. [Google Scholar] [CrossRef]
- Türk, M.; Haaker, G.; Winter, L.; Just, W.; Nickel, F.T.; Linker, R.A.; Chevessier, F.; Schröder, R. C9ORF72-ALS: P62- and ubiquitin-aggregation pathology in skeletal muscle. Muscle Nerve 2014, 50, 454–455. [Google Scholar] [CrossRef]
- Shibata, N.; Hirano, A.; Kobayashi, M.; Sasaki, S.; Kato, T.; Matsumoto, S.; Shiozawa, Z.; Komori, T.; Ikemoto, A.; Umahara, T. Cu/Zn superoxide dismutase-like immunoreactivity in Lewy body-like inclusions of sporadic amyotrophic lateral sclerosis. Neurosci. Lett. 1994, 179, 149–152. [Google Scholar] [CrossRef]
- Hjerpe, R.; Bett, J.S.; Keuss, M.J.; Solovyova, A.; McWilliams, T.G.; Johnson, C.; Sahu, I.; Varghese, J.; Wood, N.; Wightman, M.; et al. UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell 2016, 166, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, J.; Pandey, U.B.; et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Kabashi, E.; Agar, J.N.; Taylor, D.M.; Minotti, S.; Durham, H.D. Focal dysfunction of the proteasome: A pathogenic factor in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2004, 89, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.L.; Warraich, S.T.; Yang, S.; Solski, J.A.; Fernando, R.; Rouleau, G.A.; Nicholson, G.A.; Blair, I.P. UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 2527.e3–2527.e10. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef]
- de Araujo, M.E.G.; Liebscher, G.; Hess, M.W.; Huber, L.A. Lysosomal size matters. Traffic 2020, 21, 60–75. [Google Scholar] [CrossRef]
- Wallroth, A.; Haucke, V. Phosphoinositide conversion in endocytosis and the endolysosomal system. J. Biol. Chem. 2018, 293, 1526–1535. [Google Scholar] [CrossRef]
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 5. [Google Scholar] [CrossRef]
- Goode, A.; Butler, K.; Long, J.; Cavey, J.; Scott, D.; Shaw, B.; Sollenberger, J.; Gell, C.; Johansen, T.; Oldham, N.J.; et al. Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy 2016, 12, 1094–1104. [Google Scholar] [CrossRef]
- Soo, K.Y.; Sultana, J.; King, A.; Atkinson, R.; Warraich, S.T.; Sundaramoorthy, V.; Blair, I.; Farg, M.A.; Atkin, J.D. ALS-associated mutant FUS inhibits macroautophagy which is restored by overexpression of Rab1. Cell Death Discov. 2015, 1, 15030. [Google Scholar] [CrossRef] [PubMed]
- Sundaramoorthy, V.; Walker, A.K.; Tan, V.; Fifita, J.A.; McCann, E.P.; Williams, K.L.; Blair, I.P.; Guillemin, G.J.; Farg, M.A.; Atkin, J.D. Defects in optineurin- and myosin VI-mediated cellular trafficking in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2015. [Google Scholar] [CrossRef] [PubMed]
- Barmada, S.J.; Serio, A.; Arjun, A.; Bilican, B.; Daub, A.; Ando, D.M.; Tsvetkov, A.; Pleiss, M.; Li, X.; Peisach, D.; et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 2014, 10, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Waite, A.J.; Bäumer, D.; East, S.; Neal, J.; Morris, H.R.; Ansorge, O.; Blake, D.J. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol. Aging 2014, 35, 1779.e5–1779.e13. [Google Scholar] [CrossRef] [PubMed]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.-M.; Le Masson, G. Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef]
- Allen, S.P.; Hall, B.; Castelli, L.M.; Francis, L.; Woof, R.; Siskos, A.P.; Kouloura, E.; Gray, E.; Thompson, A.G.; Talbot, K.; et al. Astrocyte adenosine deaminase loss increases motor neuron toxicity in amyotrophic lateral sclerosis. Brain 2019, 142, 586–605. [Google Scholar] [CrossRef]
- Boivin, M.; Pfister, V.; Gaucherot, A.; Ruffenach, F.; Negroni, L.; Sellier, C.; Charlet-Berguerand, N. Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. EMBO J. 2020, 39, e100574. [Google Scholar] [CrossRef]
- Stolz, A.; Wolf, D.H. Endoplasmic reticulum associated protein degradation: A chaperone assisted journey to hell. Biochim. Biophys. Acta Mol. Cell Res. 2010, 1803, 694–705. [Google Scholar] [CrossRef]
- Atkin, J.D.; Farg, M.A.; Walker, A.K.; McLean, C.; Tomas, D.; Horne, M.K. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 30, 400–407. [Google Scholar] [CrossRef]
- Vijayalakshmi, K.; Alladi, P.A.; Ghosh, S.; Prasanna, V.K.; Sagar, B.C.; Nalini, A.; Sathyaprabha, T.N.; Raju, T.R. Evidence of endoplasmic reticular stress in the spinal motor neurons exposed to CSF from sporadic amyotrophic lateral sclerosis patients. Neurobiol. Dis. 2011, 41, 695–705. [Google Scholar] [CrossRef]
- Yamanaka, T.; Nishiyama, R.; Shimogori, T.; Nukina, N. Proteomics-Based Approach Identifies Altered ER Domain Properties by ALS-Linked VAPB Mutation. Sci. Rep. 2020, 10, 7610. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.; Chalhoub, A.; Schooley, A.; Zhang, W.; Ngsee, J.K. A mutation in VAPB that causes amyotrophic lateral sclerosis also causes a nuclear envelope defect. J. Cell Sci. 2012, 125, 2831–2836. [Google Scholar] [CrossRef] [PubMed]
- Cadoni, M.P.L.; Biggio, M.L.; Arru, G.; Secchi, G.; Orrù, N.; Clemente, M.G.; Sechi, G.; Yamoah, A.; Tripathi, P.; Orrù, S.; et al. VAPB ER-Aggregates, A Possible New Biomarker in ALS Pathology. Cells 2020, 9, 164. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.C.; Ebmeier, C.C.; Podell, E.R.; Heimiller, J.; Taatjes, D.J.; Cech, T.R. FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes Dev. 2012, 26, 2690–2695. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J. Biol. Chem. 2001, 276, 36337–36343. [Google Scholar] [CrossRef] [PubMed]
- Colombrita, C.; Onesto, E.; Megiorni, F.; Pizzuti, A.; Baralle, F.E.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J. Biol. Chem. 2012. [Google Scholar] [CrossRef]
- Godena, V.K.; Romano, G.; Romano, M.; Appocher, C.; Klima, R.; Buratti, E.; Baralle, F.E.; Feiguin, F. TDP-43 Regulates Drosophila Neuromuscular Junctions Growth by Modulating Futsch/MAP1B Levels and Synaptic Microtubules Organization. PLoS ONE 2011, 6, e17808. [Google Scholar] [CrossRef]
- Herzog, J.J.; Deshpande, M.; Shapiro, L.; Rodal, A.A.; Paradis, S. TDP-43 misexpression causes defects in dendritic growth. Sci. Rep. 2017, 7, 15656. [Google Scholar] [CrossRef]
- Feiguin, F.; Godena, V.K.; Romano, G.; D’Ambrogio, A.; Klima, R.; Baralle, F.E. Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 2009, 583, 1586–1592. [Google Scholar] [CrossRef]
- Reber, S.; Stettler, J.; Filosa, G.; Colombo, M.; Jutzi, D.; Lenzken, S.C.; Schweingruber, C.; Bruggmann, R.; Bachi, A.; Barabino, S.M.; et al. Minor intron splicing is regulated by FUS and affected by ALS-associated FUS mutants. EMBO J. 2016, 35, 1504–1521. [Google Scholar] [CrossRef]
- Mori, K.; Lammich, S.; Mackenzie, I.R.A.; Forné, I.; Zilow, S.; Kretzschmar, H.; Edbauer, D.; Janssens, J.; Kleinberger, G.; Cruts, M.; et al. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 2013, 125, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, B.; Hartmann, H.; May, S.; Mö hl, C.; Ederle, H.; Michaelsen, M.; Schludi, M.H.; Dormann, D.; Edbauer, D. Cytoplasmic poly-GA aggregates impair nuclear import of TDP-43 in C9orf72 ALS/FTLD. Hum. Mol. Genet. 2017, 26, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Huang, R.; Yang, Y.; Chen, K.; Song, W.; Pan, P.L.; Li, J.P.; Shang, H.F. Ataxin-2 intermediate-length polyglutamine: A possible risk factor for Chinese patients with amyotrophic lateral sclerosis. Neurobiol. Aging 2011. [Google Scholar] [CrossRef]
- Jutzi, D.; Akinyi, M.V.; Mechtersheimer, J.; Frilander, M.J.; Ruepp, M.-D. The emerging role of minor intron splicing in neurological disorders. Cell Stress 2018, 2, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef]
- Leblond, C.S.; Gan-Or, Z.; Spiegelman, D.; Laurent, S.B.; Szuto, A.; Hodgkinson, A.; Dionne-Laporte, A.; Provencher, P.; de Carvalho, M.; Orrù, S.; et al. Replication study of MATR3 in familial and sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2016, 37, 209.e17–209.e21. [Google Scholar] [CrossRef]
- Salton, M.; Elkon, R.; Borodina, T.; Davydov, A.; Yaspo, M.-L.; Halperin, E.; Shiloh, Y. Matrin 3 binds and stabilizes mRNA. PLoS ONE 2011, 6, e23882. [Google Scholar] [CrossRef]
- Yang, C.; Wang, H.; Qiao, T.; Yang, B.; Aliaga, L.; Qiu, L.; Tan, W.; Salameh, J.; McKenna-Yasek, D.M.; Smith, T.; et al. Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2014, 111, E1121–E1129. [Google Scholar] [CrossRef]
- Kamelgarn, M.; Chen, J.; Kuang, L.; Arenas, A.; Zhai, J.; Zhu, H.; Gal, J. Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 2004–2014. [Google Scholar] [CrossRef]
- Yu, Y.; Chi, B.; Xia, W.; Gangopadhyay, J.; Yamazaki, T.; Winkelbauer-Hurt, M.E.; Yin, S.; Eliasse, Y.; Adams, E.; Shaw, C.E.; et al. U1 snRNP is mislocalized in ALS patient fibroblasts bearing NLS mutations in FUS and is required for motor neuron outgrowth in zebrafish. Nucleic Acids Res. 2015, 43, 3208–3218. [Google Scholar] [CrossRef]
- Rogelj, B.; Easton, L.E.; Bogu, G.K.; Stanton, L.W.; Rot, G.; Curk, T.; Zupan, B.; Sugimoto, Y.; Modic, M.; Haberman, N.; et al. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci. Rep. 2012, 2, 603. [Google Scholar] [CrossRef] [PubMed]
- Orozco, D.; Tahirovic, S.; Rentzsch, K.; Schwenk, B.M.; Haass, C.; Edbauer, D. Loss of fused in sarcoma (FUS) promotes pathological Tau splicing. EMBO Rep. 2012, 13, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Moloney, E.B.; de Winter, F.; Verhaagen, J. ALS as a distal axonopathy: Molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front. Neurosci. 2014, 8, 252. [Google Scholar] [CrossRef] [PubMed]
- Boehringer, A.; Garcia-Mansfield, K.; Singh, G.; Bakkar, N.; Pirrotte, P.; Bowser, R. ALS Associated Mutations in Matrin 3 Alter Protein-Protein Interactions and Impede mRNA Nuclear Export. Sci. Rep. 2017, 7, 14529. [Google Scholar] [CrossRef]
- Coelho, M.B.; Attig, J.; Bellora, N.; König, J.; Hallegger, M.; Kayikci, M.; Eyras, E.; Ule, J.; Smith, C.W.J. Nuclear matrix protein Matrin3 regulates alternative splicing and forms overlapping regulatory networks with PTB. EMBO J. 2015, 34, 653–668. [Google Scholar] [CrossRef]
- Zhang, X.; Yamashita, S.; Hara, K.; Doki, T.; Tawara, N.; Ikeda, T.; Misumi, Y.; Zhang, Z.; Matsuo, Y.; Nagai, M.; et al. A mutant MATR3 mouse model to explain multisystem proteinopathy. J. Pathol. 2019, 249, 182–192. [Google Scholar] [CrossRef]
- Simpson, C.L.; Lemmens, R.; Miskiewicz, K.; Broom, W.J.; Hansen, V.K.; van Vught, P.W.J.; Landers, J.E.; Sapp, P.; Van Den Bosch, L.; Knight, J.; et al. Variants of the elongator protein 3 ( ELP3 ) gene are associated with motor neuron degeneration. Hum. Mol. Genet. 2009, 18, 472–481. [Google Scholar] [CrossRef]
- Bento-Abreu, A.; Jager, G.; Swinnen, B.; Rué, L.; Hendrickx, S.; Jones, A.; Staats, K.A.; Taes, I.; Eykens, C.; Nonneman, A.; et al. Elongator subunit 3 (ELP3) modifies ALS through tRNA modification. Hum. Mol. Genet. 2018, 27, 1276–1289. [Google Scholar] [CrossRef]
- Greenway, M.J.; Andersen, P.M.; Russ, C.; Ennis, S.; Cashman, S.; Donaghy, C.; Patterson, V.; Swingler, R.; Kieran, D.; Prehn, J.; et al. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat. Genet. 2006, 38, 411–413. [Google Scholar] [CrossRef]
- Wu, D.; Yu, W.; Kishikawa, H.; Folkerth, R.D.; Iafrate, A.J.; Shen, Y.; Xin, W.; Sims, K.; Hu, G. Angiogenin loss-of-function mutations in amyotrophic lateral sclerosis. Ann. Neurol. 2007, 62, 609–617. [Google Scholar] [CrossRef]
- Kirby, J.; Highley, J.R.; Cox, L.; Goodall, E.F.; Hewitt, C.; Hartley, J.A.; Hollinger, H.C.; Fox, M.; Ince, P.G.; Mcdermott, C.J.; et al. Lack of unique neuropathology in amyotrophic lateral sclerosis associated with p.K54E angiogenin (ANG) mutation. Neuropathol. Appl. Neurobiol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Aladesuyi Arogundade, O.; Stauffer, J.E.; Saberi, S.; Diaz-Garcia, S.; Malik, S.; Basilim, H.; Rodriguez, M.J.; Ohkubo, T.; Ravits, J. Antisense RNA foci are associated with nucleoli and TDP-43 mislocalization in C9orf72-ALS/FTD: A quantitative study. Acta Neuropathol. 2019, 137, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.R.; Selvaraj, B.T.; Barton, S.K.; McDade, K.; Abrahams, S.; Chandran, S.; Smith, C.; Gregory, J.M. Improved detection of RNA foci in C9orf72 amyotrophic lateral sclerosis post-mortem tissue using BaseScopeTM shows a lack of association with cognitive dysfunction. Brain Commun. 2020, 2. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Hur, J.; Lunn, J.S.; Paez-Colasante, X.; Bender, D.E.; Yung, R.; Sakowski, S.A.; Feldman, E.L. Expression of microRNAs in human post-mortem amyotrophic lateral sclerosis spinal cords provides insight into disease mechanisms. Mol. Cell. Neurosci. 2016, 71, 34–45. [Google Scholar] [CrossRef]
- Porta, S.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M.-Y. Drosha Inclusions Are New Components of Dipeptide-Repeat Protein Aggregates in FTLD-TDP and ALS C9orf72 Expansion Cases. J. Neuropathol. Exp. Neurol. 2015, 74, 380–387. [Google Scholar] [CrossRef]
- Sobue, G.; Mcalary, L.; Taylor, J.P.; Purice, M.D. Linking hnRNP Function to ALS and FTD Pathology. Front. Neurosci. 2018, 1, 326. [Google Scholar] [CrossRef]
- Kawahara, Y.; Mieda-Sato, A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 3347–3352. [Google Scholar] [CrossRef]
- Morlando, M.; Dini Modigliani, S.; Torrelli, G.; Rosa, A.; Di Carlo, V.; Caffarelli, E.; Bozzoni, I. FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. EMBO J. 2012, 31, 4502–4510. [Google Scholar] [CrossRef]
- Waller, R.; Wyles, M.; Heath, P.R.; Kazoka, M.; Wollff, H.; Shaw, P.J.; Kirby, J. Small RNA Sequencing of Sporadic Amyotrophic Lateral Sclerosis Cerebrospinal Fluid Reveals Differentially Expressed miRNAs Related to Neural and Glial Activity. Front. Neurosci. 2017, 11, 731. [Google Scholar] [CrossRef]
- Campos-Melo, D.; Droppelmann, C.A.; He, Z.; Volkening, K.; Strong, M.J. Altered microRNA expression profile in amyotrophic lateral sclerosis: A role in the regulation of NFL mRNA levels. Mol. Brain 2013, 6, 26. [Google Scholar] [CrossRef]
- Salinas, J.; Lin, H.; Aparico, H.J.; Huan, T.; Liu, C.; Rong, J.; Beiser, A.; Himali, J.J.; Freedman, J.E.; Larson, M.G.; et al. Whole blood microRNA expression associated with stroke: Results from the Framingham Heart Study. PLoS ONE 2019, 14, e0219261. [Google Scholar] [CrossRef] [PubMed]
- Boissart, C.; Nissan, X.; Giraud-Triboult, K.; Peschanski, M.; Benchoua, A. miR-125 potentiates early neural specification of human embryonic stem cells. Development 2012, 139, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Jauhari, A.; Singh, T.; Pandey, A.; Singh, P.; Singh, N.; Srivastava, A.K.; Pant, A.B.; Parmar, D.; Yadav, S. Differentiation Induces Dramatic Changes in miRNA Profile, Where Loss of Dicer Diverts Differentiating SH-SY5Y Cells Toward Senescence. Mol. Neurobiol. 2017, 54, 4986–4995. [Google Scholar] [CrossRef] [PubMed]
- Ranjith Sama, R.K.; Ward, C.L.; Kaushansky, L.J.; Lemay, N.; Ishigaki, S.; Urano, F.; Bosco, D.A. FUS/TLS Assembles Into Stress Granules and Is a Prosurvival Factor During Hyperosmolar Stress. J. Cell. Physiol. 2013, 228, 2222–2231. [Google Scholar] [CrossRef]
- Guil, S.; Cáceres, J.F. The multifunctional RNA-binding protein hnRNP A1 is required for processing of miR-18a. Nat. Struct. Mol. Biol. 2007, 14, 591–596. [Google Scholar] [CrossRef]
- Zhang, K.; Daigle, J.G.; Cunningham, K.M.; Coyne, A.N.; Ruan, K.; Grima, J.C.; Bowen, K.E.; Wadhwa, H.; Yang, P.; Rigo, F.; et al. Stress Granule Assembly Disrupts Nucleocytoplasmic Transport. Cell 2018, 173, 958–971.e17. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, N.C.; Wang, Y.-D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef]
- Buchan, J.R.; Kolaitis, R.-M.; Taylor, J.P.; Parker, R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef]
- Uversky, V.N. The roles of intrinsic disorder-based liquid-liquid phase transitions in the “Dr. Jekyll–Mr. Hyde” behavior of proteins involved in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Autophagy 2017, 13, 2115–2162. [Google Scholar] [CrossRef]
- Vanneste, J.; Vercruysse, T.; Boeynaems, S.; Sicart, A.; Van Damme, P.; Daelemans, D.; Van Den Bosch, L. C9orf72-generated poly-GR and poly-PR do not directly interfere with nucleocytoplasmic transport. Sci. Rep. 2019, 9, 15728. [Google Scholar] [CrossRef]
- Brenner, D.; Weishaupt, J.H. Update on amyotrophic lateral sclerosis genetics. Curr. Opin. Neurol. 2019, 32, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, M.C.N.; Muotri, A.R.; Mu, Y.; Smith, A.M.; Cezar, G.G.; Gage, F.H. Non-Cell-Autonomous Effect of Human SOD1G37R Astrocytes on Motor Neurons Derived from Human Embryonic Stem Cells. Cell Stem Cell 2008, 3, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Hooten, K.G.; Beers, D.R.; Zhao, W.; Appel, S.H. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 364–375. [Google Scholar] [CrossRef]
- Brites, D.; Vaz, A.R. Microglia centered pathogenesis in ALS: Insights in cell interconnectivity. Front. Cell. Neurosci. 2014, 8, 117. [Google Scholar] [CrossRef]
- Mougeot, J.-L.C.; Li, Z.; Price, A.E.; Wright, F.A.; Brooks, B.R. Microarray analysis of peripheral blood lymphocytes from ALS patients and the SAFE detection of the KEGG ALS pathway. BMC Med. Genom. 2011, 4, 74. [Google Scholar] [CrossRef]
- Pradat, P.-F.; Barani, A.; Wanschitz, J.; Dubourg, O.; Lombès, A.; Bigot, A.; Mouly, V.; Bruneteau, G.; Salachas, F.; Lenglet, T.; et al. Abnormalities of satellite cells function in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2011, 12, 264–271. [Google Scholar] [CrossRef]
- Scaricamazza, S.; Salvatori, I.; Giacovazzo, G.; Loeffler, J.P.; Renè, F.; Rosina, M.; Quessada, C.; Proietti, D.; Heil, C.; Rossi, S.; et al. Skeletal-Muscle Metabolic Reprogramming in ALS-SOD1G93A Mice Predates Disease Onset and Is A Promising Therapeutic Target. iScience 2020, 23, 101087. [Google Scholar] [CrossRef]
- Loeffler, J.P.; Picchiarelli, G.; Dupuis, L.; Gonzalez De Aguilar, J.L. The role of skeletal muscle in amyotrophic lateral sclerosis. Brain Pathol. 2016, 26, 227–236. [Google Scholar] [CrossRef]
- Morgan, S.; Duguez, S.; Duddy, W. Personalized Medicine and Molecular Interaction Networks in Amyotrophic Lateral Sclerosis (ALS): Current Knowledge. J. Pers. Med. 2018, 8, 44. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Gall, L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duddy, W.J.; Duguez, S. Molecular and Cellular Mechanisms Affected in ALS. J. Pers. Med. 2020, 10, 101. https://doi.org/10.3390/jpm10030101
Le Gall L, Anakor E, Connolly O, Vijayakumar UG, Duddy WJ, Duguez S. Molecular and Cellular Mechanisms Affected in ALS. Journal of Personalized Medicine. 2020; 10(3):101. https://doi.org/10.3390/jpm10030101
Chicago/Turabian StyleLe Gall, Laura, Ekene Anakor, Owen Connolly, Udaya Geetha Vijayakumar, William J. Duddy, and Stephanie Duguez. 2020. "Molecular and Cellular Mechanisms Affected in ALS" Journal of Personalized Medicine 10, no. 3: 101. https://doi.org/10.3390/jpm10030101
APA StyleLe Gall, L., Anakor, E., Connolly, O., Vijayakumar, U. G., Duddy, W. J., & Duguez, S. (2020). Molecular and Cellular Mechanisms Affected in ALS. Journal of Personalized Medicine, 10(3), 101. https://doi.org/10.3390/jpm10030101