
The Role of Proteomics in Biomarker Development for Improved Patient Diagnosis and Clinical Decision Making in Prostate Cancer

 , ,
, , 
Abstract
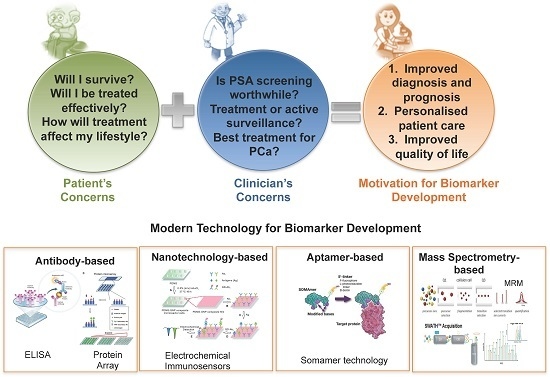
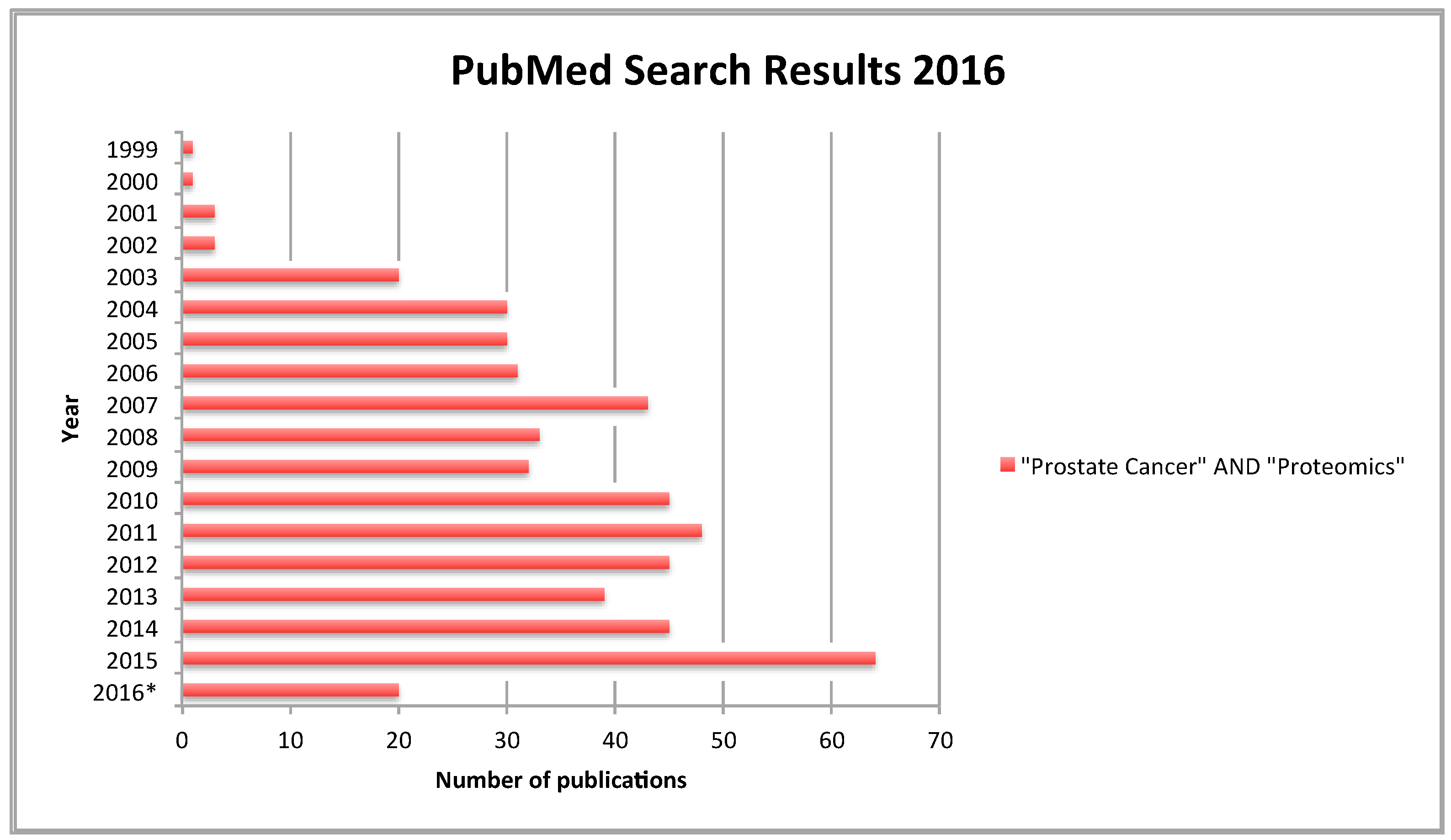
:
1. Introduction
2. Prostate Cancer
2.1. PSA Screening and Prostate Cancer Diagnosis
2.2. Disease Stratification and Curative Treatment Strategies
2.3. Impact of Curative Treatment on Patient Lifestyle
2.4. Impact of Curative Treatment on Patient Lifestyle
2.5. Clinical Need for Additional PCa Biomarkers
3. Newly Emerging Tests for Prostate Cancer
3.1. Tissue Based Prostate Cancer Biomarkers
3.2. Fluid Based Prostate Cancer Biomarkers
4. Proteomics to Answer Key Questions in Prostate Cancer
5. Biological Sources for Biomarker Discovery in Prostate Cancer
5.1. Tissue
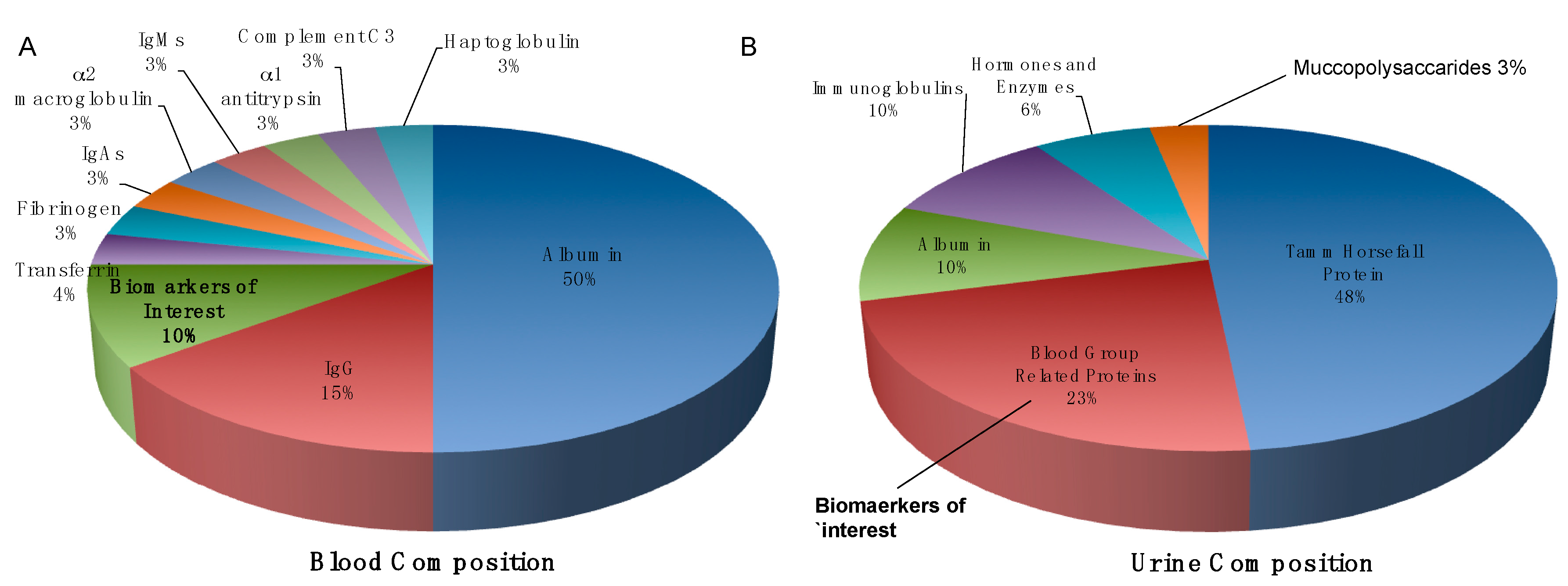
5.2. Blood
5.3. Urine
5.4. Semen
6. Proteomic Technology for Biomarker Discovery and Validation
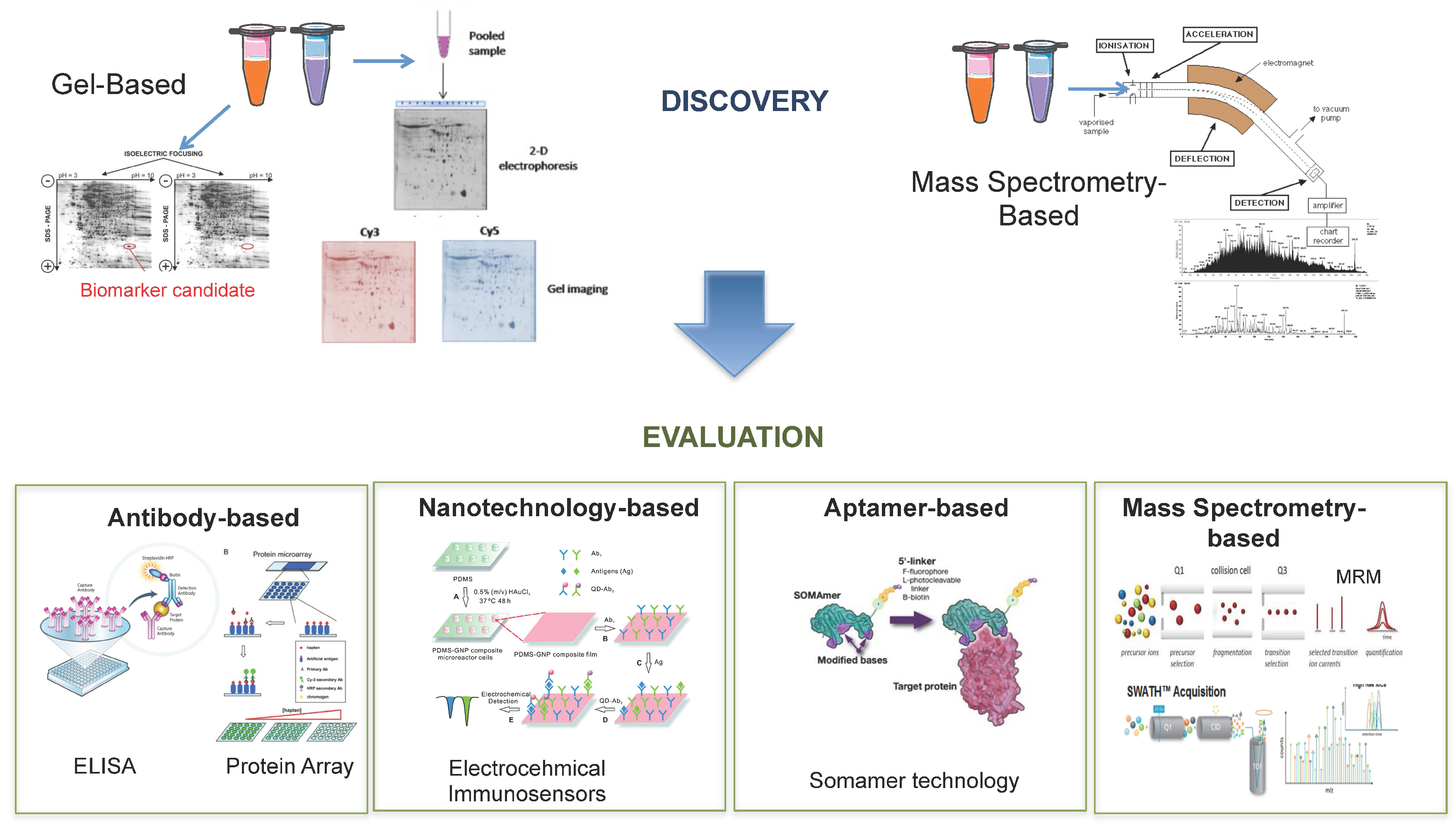
6.1. Biomarker Discovery
6.1.1. Gel-Based
6.1.2. Mass Spectrometry-Based
6.2. Biomarker Evaluation
6.2.1. Antibody-Based
6.2.2. Nanotechnology-Based
6.2.3. Aptamer-Based
6.2.4. Mass Spectrometry-Based
7. Clinical Evaluation of Prostate Cancer Biomarkers
7.1. Evaluation of Emerging Prostate Cancer Biomarkers
7.2. Overcoming Bottlenecks Associated with Clinical Evaluation of Prostate Cancer Biomarkers
7.3. Potential for Routine Use of MS Technologies for Clinical Diagnostics in PCa
8. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BPH | Benign Prostatic Hyperplasia |
| PCa | Prostate Cancer |
| AR | Androgen Receptor |
| PSA | Prostate Specific Antigen |
| US | United States |
| CE-IVD | European Conformity—In Vitro Diagnostics |
| CHRT | Combined hormone and radiation therapy |
| ADT | Androgen deprivation therapy |
| AS | Active surveillance |
| PRIAS | Prostate cancer research international active surveillance |
| SCAN | Scotland cancer research |
| CCO | Cancer care Ontario |
| GAP | Global action plan |
| fPSA | free PSA |
| hK2 | Hexokinase 2 |
| MSMB | microseminoprotein beta |
| MIC1 | Macrophage inhibitor cytokine 1 |
| DRE | Digital rectal exam |
| PSAV | PSA velocity |
| PSADT | PSA doubling time |
| p2PSA | [-2] proenzyme PSA |
| PHI | Prostate health index |
| PTEN | Phosphate and tensin homologue |
| FFPE | Fresh frozen paraffin embedded |
| RT-PCR | Real time polymerase chain reaction |
| CCP | Cell cycle progression |
| TURP | Transurethral resection of the prostate |
| PCA3 | Prostate cancer antigen 3 |
| CLIA | Clinical laboratory improvement amendments |
| LCM | Laser capture microdissection |
| RBC | Red blood cells |
| WBC | White blood cells |
| EPS | Expressed prostatic secretion |
| 2D-PAGE | Two dimensional poly-acrylamide gel electrophoresis |
| DIGE | Differential gel electrophoresis |
| SDS | Sodium dodecyl sulfate |
| MS | Mass spectrometry |
| LC-MS/MS | Liquid chromatography tandem mass spectrometry |
| SWATH | Sequential window acquisition of all theoretical ions |
| MRM | Multiple reaction monitoring |
| ELISA | Enzyme-linked |
| QD | Quantum dot |
| CNT | Carbon nanotube |
| SRM | Selected reaction monitoring |
| NCI EDRN | National Cancer Institute—The early detection research network |
| NCI CPTAC | National Cancer Institute—Cancer clinical proteomics research |
| ANOVA | Analysis of variance |
| FDR | False discovery rate |
| IMRT | Intensity modulated radiation therapy |
| IPSS | International prostate symptom score |
| SISCAPA | Stable isotope standard capture with anti-peptide antibodies |
| MALDI-MS | Matrix assisted laser desorption/ionization mass spectrometry |
References
- Vickers, A.J.; Edwards, K.; Cooperberg, M.R.; Mushlin, A.I. A simple schema for informed decision-making about prostate cancer screening. Ann. Intern. Med. 2015, 73, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.; Martin, R.M.; Evans, D.M.; Tilling, K.; Davey Smith, G.; Kemp, J.P.; Lane, J.A.; Hamdy, F.C.; Neal, D.E.; Donovan, J.L.; et al. Incorporating known genetic variants does not improve the accuracy of PSA testing to identify high risk prostate cancer on biopsy. PLoS ONE 2015, 10, e0136735. [Google Scholar] [CrossRef] [PubMed]
- Chamie, K.; Williams, S.B.; Hershman, D.L.; Wright, J.D.; Nguyen, P.L.; Hu, J.C. Population-based assessment of determining predictors for quality of prostate cancer surveillance. Cancer 2015, 121, 4150–4157. [Google Scholar] [CrossRef] [PubMed]
- Abate-shen, C.; Shen, M.M. Molecular genetics of prostate cancer. Genes Dev. 2000, 732, 2410–2434. [Google Scholar] [CrossRef]
- Morisot, A.; Bessaoud, F.; Landais, P.; Rébillard, X.; Trétarre, B.; Daurès, J.-P. Prostate cancer: Net survival and cause-specific survival rates after multiple imputation. BMC Med. Res. Methodol. 2015, 15, 54. [Google Scholar] [CrossRef] [PubMed]
- Dunn, M.W.; Kazer, M.W. Prostate cancer overview. Semin. Oncol. Nurs. 2011, 27, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Crook, J.; Ots, A.F. Prognostic factors for newly diagnosed prostate cancer and their role in treatment selection. Semin. Radiat. Oncol. 2013, 23, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Ehdaie, B.; Poon, B.Y.; Sjoberg, D.D.; Recabal, P.; Laudone, V.; Touijer, K.; Eastham, J.; Scardino, P.T. Variation of serum prostate-specific antigen in men with prostate cancer managed with active surveillance. BJU Int. 2015. [Google Scholar] [CrossRef] [PubMed]
- Etzioni, R.; Gulati, R.; Tsodikov, A.; Wever, E.M.; Penson, D.F.; Heijnsdijk, E.A.M.; Katcher, J.; Draisma, G.; Feuer, E.J.; de Koning, H.J.; et al. The prostate cancer conundrum revisited: Treatment changes and prostate cancer mortality declines. Cancer 2012, 118, 5955–6963. [Google Scholar] [CrossRef] [PubMed]
- Bangma, C.H.; Roemeling, S.; Schröder, F.H. Overdiagnosis and overtreatment of early detected prostate cancer. World J. Urol. 2007, 25, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Glaser, A.W.; Corner, J.L. Prostate cancer outcomes: The three questions. Eur. Urol. 2015, 67, 357–358. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.J.; Al-Ahmadie, H.a.; Gopalan, A.; Tickoo, S.K.; Scardino, P.T.; Reuter, V.E.; Samson, W.; Fine, M.D. Do prostatic transition zone tumors have a distinct morphology? Am. J. Surg. Pathol. 2008, 32, 1709–1714. [Google Scholar] [CrossRef] [PubMed]
- McNeal, J. Development of carcinoma in the prostate. Cancer 1969, 23, 24–34. [Google Scholar] [CrossRef]
- Selman, S.H. The McNeal prostate: A review. Urology 2011, 78, 1224–1228. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, C.E.; Bangma, C.H.; Bjartell, A.; Catto, J.W.F.; Culig, Z.; Grönberg, H.; Luo, J.; Visakorpi, T.; Rubin, M.A. The mutational landscape of prostate cancer. Eur. Urol. 2013, 64, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Rycaj, K.; Cho, E.J.; Liu, X.; Chao, H.-P.; Liu, B.; Li, Q.; Devkota, A.K.; Zhang, D.; Chen, X.; Moore, J.; et al. Longitudinal tracking of subpopulation dynamics and molecular changes during LNCaP cell castration and identification of inhibitors that could target the PSA-/lo castration-resistant cells. Oncotarget 2016, 7, 14220–14240. [Google Scholar] [PubMed]
- Shah, R.B.; Mehra, R.; Chinnaiyan, A.M.; Shen, R.; Ghosh, D.; Zhou, M.; Macvicar, G.R.; Varambally, S.; Harwood, J.; Bismar, T.A.; et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: Lessons from a rapid autopsy program. Cancer Res. 2004, 64, 9209–9216. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.S.; Eeles, R.; Wedge, D.C.; Van Loo, P.; Gundem, G.; Alexandrov, L.B.; Kremeyer, B.; Butler, A.; Lunch, A.G.; Camacho, N.; et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat. Genet. 2015, 47, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Koch, M.O.; Eble, J.N.; Ulbright, T.M.; Li, L.; Cheng, L. Heterogeneity of Gleason grade in multifocal adenocarcinoma of the prostate. Cancer 2004, 100, 2362–2366. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.; Neal, D.E. The genomic evolution of human prostate cancer. Br. J. Cancer 2015, 113, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.D.; Bennett, C.L.; Andriole, G.L.; Garnick, M.B.; Petrylak, D.P. The utility of prostate-specific antigen in the management of advanced prostate cancer. BJU Int. 2013, 112, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Vigna-Taglianti, R.; Russi, E.G.; Boriano, A.; Gianello, L.; Denaro, N.; Lucio, F.; Arena, G.; Buglione, M.; Pergolizzi, S.; Ricardi, U.; et al. Reliability of prostate-specific antigen-marker in determining biochemical failure during the first 2 years after external beam radiation therapy and hormone therapy in patients with non-operated prostate cancer. Urol. Oncol. 2014, 32, 30.e1–30.e7. [Google Scholar] [CrossRef] [PubMed]
- Cooperberg, M.R.; Lubeck, D.P.; Meng, M.V.; Mehta, S.S.; Carroll, P.R. The changing face of low-risk prostate cancer: Trends in clinical presentation and primary management. J. Clin. Oncol. 2004, 22, 2141–2149. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ramakrishnan, A.; Fletcher, S.; Prochownik, E.V.; Genetics, M. Predicting life expectancy in men diagnosed with prostate cancer. Eur. Urol. 2015, 2, 756–765. [Google Scholar]
- Vickers, A.J.; Cronin, A.M.; Roobol, M.J.; Hugosson, J.; Jones, J.S.; Kattan, M.W.; Klein, E.; Hamdy, F.; Neal, D.; Donovan, J.; et al. The relationship between prostate-specific antigen and prostate cancer risk: The Prostate Biopsy Collaborative Group. Clin. Cancer Res. 2010, 16, 4374–4381. [Google Scholar] [CrossRef] [PubMed]
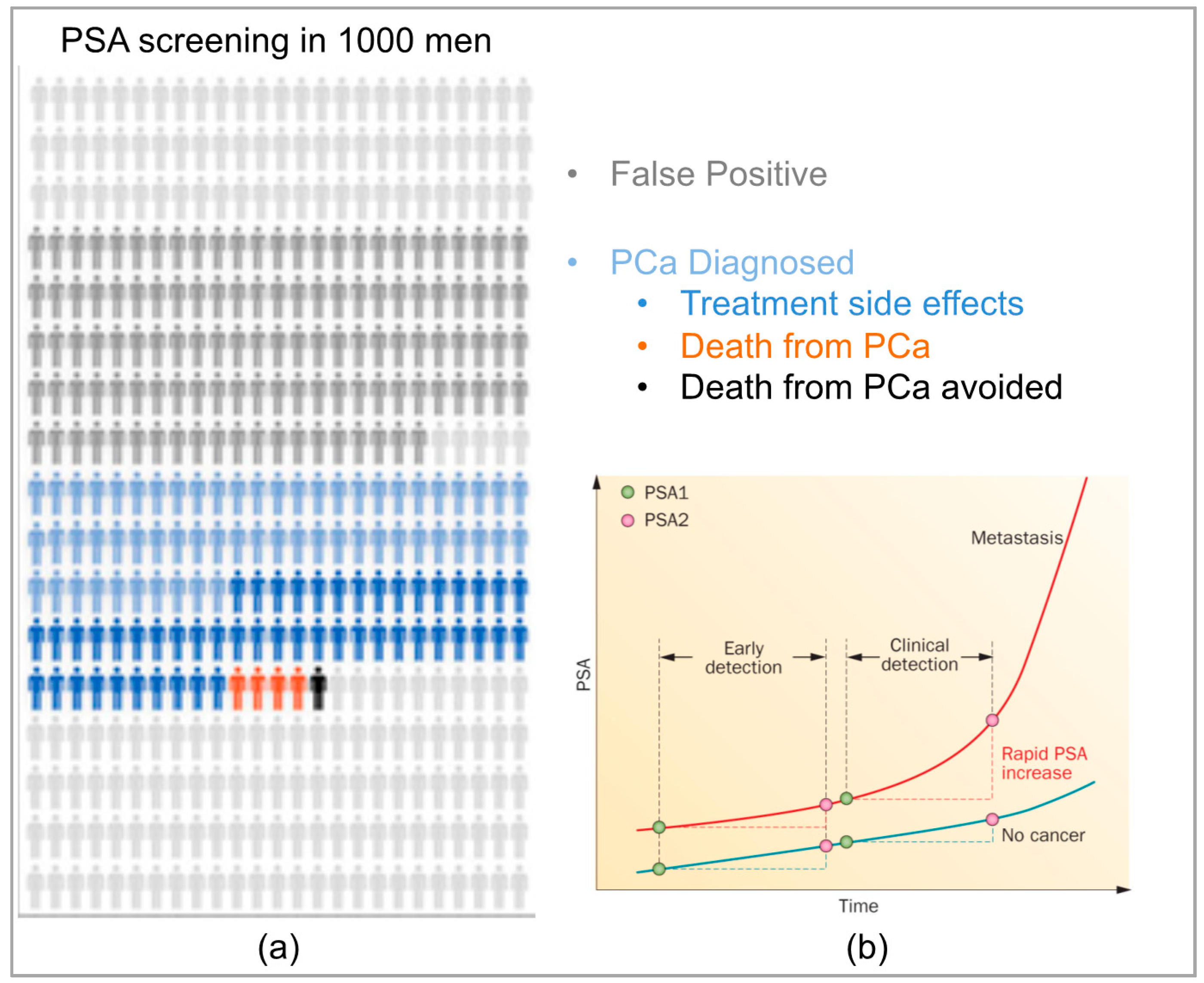
- Howrey, B.T.; Kuo, Y.-F.; Lin, Y.-L.; Goodwin, J.S. The impact of PSA screening on prostate cancer mortality and overdiagnosis of prostate cancer in the United States. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Auvinen, A.; Moss, S.; Tammela, T.L.J.; Taari, K.; Roobol, M.; Schroder, F.H.; Bangma, C.H.; Carlsson, S.; Aus, G.; Zappa, M. Absolute effect of prostate cancer screening: Balance of benefits and harms by center within the European Randomized Study of Prostate Cancer Screening. Clin. Cancer Res. 2016, 22, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Punnen, S.; Cooperberg, M.R.; D’Amico, A.V.; Karakiewicz, P.I.; Moul, J.W.; Scher, H.I.; Schlomm, T.; Freedlan, S.J. Management of biochemical recurrence after primary treatment of prostate cancer: A systematic review of the literature. Eur. Urol. 2013, 64, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Lipsitz, R.; Miller, T.; Janakiraman, S. Benefits and harms of prostate-specific antigen screening for prostate cancer: An evidence update for the, U.S. preventive services task force. Ann. Intern. Med. 2007, 149, 192–199. [Google Scholar] [CrossRef]
- Roobol, M.J.; Carlsson, S.V. Risk stratification in prostate cancer screening. Nat. Rev. Urol. 2013, 10, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, P.A. Gleason grading and prognostic factors in carcinoma of the prostate. Mod. Pathol. 2004, 17, 292–306. [Google Scholar] [CrossRef] [PubMed]
- Doshi, C.; Vacchio, M.; Attwood, K.; Murekeyisoni, C.; Mehedint, D.C.; Badkhshan, S.; Azabdaftari, G.; Sule, N.; Guro, K.A.; Mohler, J.L. Clinical significance of prospectively assigned Gleason tertiary pattern 4 in contemporary Gleason score 3 + 3 = 6 prostate cancer. Prostate 2016, 76, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I.; Zelefsky, M.J.; Sjoberg, D.D.; Nelson, J.B.; Egevad, L.; Magi-Galluzzi, C.; Vickers, A.J.; Parwani, A.V.; Reulter, V.E.; Fine, S.W. A contemporary prostate cancer grading system: A validated alternative to the gleason score. Eur. Urol. 2015, 69, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Egevad, L.; Mazzucchelli, R.; Montironi, R. Implications of the international society of urological pathology modified gleason grading system. Arch. Pathol. Lab. Med. 2012, 136, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I. A new contemporary prostate cancer grading system. Ann. Pathol. 2015, 35, 474–476. [Google Scholar] [CrossRef] [PubMed]
- Brimo, F.; Montironi, R.; Egevad, L.; Erbersdobler, A.; Lin, D.W.; Nelson, J.B.; Rubin, M.A.; van der Kwast, T.; Amin, M.; Epstein, J.I. Contemporary grading for prostate cancer: Implications for patient care. Eur. Urol. 2013, 63, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J.; Berney, D.M.; Fisher, G.; Mesher, D.; Møller, H.; Reid, J.E.; Perry, M.; Park, J.; Younus, A.; Gutlin, A. Prognostic value of a cell cycle progression signature for prostate cancer death in a conservatively managed needle biopsy cohort. Br. J. Cancer 2012, 106, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Jani, A.B.; Hellman, S. Early prostate cancer: Clinical decision-making. Lancet 2003, 361, 1045–1053. [Google Scholar] [CrossRef]
- Thompson, I.M.; Tangen, C.M.; Paradelo, J.; Lucia, M.S.; Miller, G.; Troyer, D.; Messing, E.; Forman, J.; Chin, J.; Swanson, G. Adjuvant radiotherapy for pathological T3N0M0 prostate cancer significantly reduces risk of metastases and improves survival: Long-term followup of a randomized clinical trial. J. Urol. 2009, 181, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Van der Kwast, T.H.; Bolla, M.; Van Poppel, H.; Van Cangh, P.; Vekemans, K.; Da Pozzo, L.; Bosset, J.F.; Kurth, K.H.; Schroder, F.H.; Collette, L. Identification of Patients With Prostate Cancer Who Benefit From Immediate Postoperative Radiotherapy: EORTC 22911. J. Clin. Oncol. 2007, 25, 4178–4186. [Google Scholar] [CrossRef] [PubMed]
- Blomberg, K.; Wengstrom, Y.; Sundberg, K.; Browall, M.; Isaksson, A.-K.; Nyman, M.H.; Langius-Eklof, A. Symptoms and self-care strategies during and six months after radiotherapy for prostate cancer—Scoping the perspectives of patients, professionals and literature. Eur. J. Oncol. Nurs. 2015, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chuu, C.-P.; Kokontis, J.M.; Hiipakka, R.A.; Fukuchi, J.; Lin, H.P.; Lin, C.Y.; Huo, C.; Su, L.C. Androgens as therapy for androgen receptor-positive castration-resistant prostate cancer. J. Biomed. Sci. 2011, 18, 63. [Google Scholar] [CrossRef] [PubMed]
- Snoeks, L.; Ogilvie, A. New treatment options for patients with metastatic prostate cancer. J. Med. 2013, 71, 290–294. [Google Scholar]
- Castagneto, B.; Stevani, I.; Ferraris, V.; Giorcelli, L.; Perachino, M. Anti-Androgen therapy suspension following prolonged clinical and biochemical response: Outcomes in a series of elderly patients with advanced prostate cancer. Eur. J. Cancer 2010, 47, 241–245. [Google Scholar]
- Flaig, T.W.; Potluri, R.C.; Ng, Y.; Todd, M.B.; Mehra, M. Treatment evolution for metastatic castration-resistant prostate cancer with recent introduction of novel agents: Retrospective analysis of real-world data. Cancer Med. 2015, 5, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Briganti, A.; Karnes, R.J.; Da Pozzo, L.F.; Cozzarini, C.; Capitanio, U.; Gallina, A.; Suardi, N.; Bianchi, M.; Tutolo, M.; Salonia, A. Combination of adjuvant hormonal and radiation therapy significantly prolongs survival of patients with pT2-4 pn+ prostate cancer: Results of a matched analysis. Int. Braz. J. Urol. 2011, 37, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, S.; Leapman, M.; Carroll, P.; Schröder, F.; Albertsen, P.C.; Ilic, D.; Barry, M.; Frosch, D.L.; Vickers, A. Who and when should we screen for prostate cancer? Interviews with key opinion leaders. BMC Med. 2015, 13, 288. [Google Scholar] [CrossRef] [PubMed]
- McCullough, A.R. Sexual dysfunction after radical prostatectomy. Rev. Urol. 2005, 7 (Suppl. S2), S3–S10. [Google Scholar] [PubMed]
- Canalichio, K.; Jaber, Y.; Wang, R. Surgery and hormonal treatment for prostate cancer and sexual function. Transl. Androl. Urol. 2015, 4, 103–109. [Google Scholar] [PubMed]
- Adejoro, O.; Gupta, P.; Ziegelmann, M.; Weight, C.; Konety, B. Effect of minimally invasive radical prostatectomy in older men. Urol. Oncol. 2016, 34, 234.e1–234.e11. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, A.N.; Cathcart, P.J.; Yap, T.; Hines, J.; Nathan, S.; Briggs, T.P.; Kelly, J.D.; Minhas, S. Recovery of baseline erectile function in men following radical prostatectomy for high-risk prostate cancer: A prospective analysis using validated measures. J. Sex. Med. 2016, 13, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Daly, P.E.; Dunne, M.T.; O’Shea, C.M.; Finn, M.A.F.; Armstrong, J.G. The effect of short term neo-adjuvant androgen deprivation on erectile function in patients treated with external beam radiotherapy for localised prostate cancer: An analysis of the 4- versus 8-month randomised trial. Radiother. Oncol. 2012, 104, 96–102. [Google Scholar] [CrossRef] [PubMed]
- DiBlasio, C.J.; Malcolm, J.B.; Derweesh, I.H.; Womack, J.H.; Kincade, M.C.; Mancini, J.G.; Ogles, M.L.; Lamar, K.D.; Patterson, A.L.; Wake, R.W. Patterns of sexual and erectile dysfunction and response to treatment in patients receiving androgen deprivation therapy for prostate cancer. BJU Int. 2008, 102, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Lund, L.; Svolgaard, N.; Poulsen, M.H. Prostate cancer: A review of active surveillance. Res. Rep. Urol. 2014, 6, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Bruinsma, S.M.; Bangma, C.H.; Carroll, P.R.; Leapman, M.S.; Rannikko, A.; Petrides, N.; Weerakoon, M.; Bokhorst, L.P.; Roobol, M.J. Active surveillance for prostate cancer: A narrative review of clinical guidelines. Nat. Rev. Urol. 2016, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bangma, C.H.; Bul, M.; van der Kwast, T.H.; Pickles, T.; Korfage, I.J.; Hoeks, C.M.; Steyerberg, E.W.; Jenster, G.; Kattan, M.W.; Bellardita, L. Active surveillance for low-risk prostate cancer. Crit. Rev. Oncol. Hematol. 2013, 85, 295–302. [Google Scholar] [CrossRef] [PubMed]
- McNeill, A.; McLaren, D. SCAN guideline for active surveillance (deferred radical treatment) of early, low-risk. Available online: http://www.scan.scot.nhs.uk/Documents/SCAN%20Protocol%20for%20Active%20Surveillance%20of%20Early%20Prostate%20Cancer%20-%2017072009.pdf (accessed online 14 July 2016).
- Morash, C.; Tey, R.; Agbassi, C.; Klotz, L. Active surveillance for the management of localized prostate cancer: Guideline recommendations. Can. Urol. Assoc. J. 2015, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Bruinsma, S.M.; Bangma, C.H.; Obbink, H.; Roobol, M.J. 1036 Active surveillance for low risk prostate cancer: The study protocol of the Movember Global Action Plan 3 (GAP3) project. Eur. Urol. Suppl. 2015, 14, e1036–e1036a. [Google Scholar] [CrossRef]
- Stephenson, A.J.; Bolla, M.; Briganti, A.; Cozzarini, C.; Moul, J.W.; Roach, M.; van Poppel, H.; Zietman, A. Postoperative radiation therapy for pathologically advanced prostate cancer after radical prostatectomy. Eur. Urol. 2012, 61, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.M.; Howard, L.E.; Sourbeer, K.N.; Amarasekara, H.S.; Chow, L.C.; Cockrell, D.C.; Hanyok, B.T.; Aronson, W.J.; Kane, C.J.; Terris, M.K. Predictors of time to metastasis in castration-resistant prostate cancer. Urology 2016. [Google Scholar] [CrossRef] [PubMed]
- Shariat, S.F.; Karam, J.A.; Walz, J.; Roehrborn, C.G.; Montorsi, F.; Margulis, V.; Saad, F.; Slawin, K.M.; Karakiewicz, P.I. Improved prediction of disease relapse after radical prostatectomy through a panel of preoperative blood-based biomarkers. Clin. Cancer Res. 2008, 14, 3785–3791. [Google Scholar] [CrossRef] [PubMed]
- Slawin, K.M. Radiation Therapy after radical prostatectomy: Why patience is a virtue! the case for salvage radiation therapy. Rev. Urol. 2002, 4, 90–94. [Google Scholar] [PubMed]
- Hall, J.S.; Iype, R.; Senra, J.; Taylor, J.; Armenoult, L.; Oguejiofor, K.; Yaoyong, L.; Stratford, I.; Stern, P.L.; O’Connor, M.J. Investigation of radiosensitivity gene signatures in cancer cell lines. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Peltola, M.T.; Niemelä, P.; Väisänen, V.; Viitanen, T.; Alanen, K.; Nurmi, M.; Pettersson, K. Intact and internally cleaved free prostate-specific antigen in patients with prostate cancer with different pathologic stages and grades. Urology 2011, 77, 1009.e1–1009.e8. [Google Scholar] [CrossRef] [PubMed]
- Artibani, W. Landmarks in prostate cancer diagnosis: The biomarkers. BJU Int. 2012, 110, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.H.; van Schaik, R.H.N.; Kurstjens, J.; Horninger, W.; Klocker, H.; Bektic, J.; Wildhagen, M.F.; Roobol, M.J.; Bangma, C.H.; Bartsch, G. Prostate-Specific Antigen (PSA) Isoform p2PSA in Combination with Total PSA and Free PSA Improves Diagnostic Accuracy in Prostate Cancer Detection. Eur. Urol. 2010, 57, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Boegemann, M.; Stephan, C.; Cammann, H.; Vincendeau, S.; Houlgatte, A.; Jung, K.; Blanchet, J.S.; Semjonow, A. The percentage of prostate-specific antigen (PSA) isoform [-2]proPSA and the Prostate Health Index improve the diagnostic accuracy for clinically relevant prostate cancer at initial and repeat biopsy compared with total PSA and percentage free PSA in men. BJU Int. 2015, 117, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Grönberg, H.; Adolfsson, J.; Aly, M.; Nordström, T.; Wiklund, P.; Brandberg, Y.; Thompson, J.; Wiklund, F.; Lindberg, J.; Clements, M. Prostate cancer screening in men aged 50–69 years (STHLM3): A prospective population-based diagnostic study. Lancet Oncol. 2015, 16, 1667–1676. [Google Scholar] [CrossRef]
- Taylor, K.L.; Hoffman, R.M.; Davis, K.M.; Luta, G.; Leimpeter, A.; Lobo, T.; Kelly, S.P.; Shan, J.; Aaronson, D.; Tomko, C.A. Treatment Preferences for Active Surveillance vs. Active Treatment Among Men with Low-Risk Prostate Cancer. Cancer Epidemiol. Biomark. Prev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Moschini, M.; Spahn, M.; Mattei, A.; Cheville, J.; Karnes, R.J. Incorporation of tissue-based genomic biomarkers into localized prostate cancer clinics. BMC Med. 2016, 14, 67. [Google Scholar] [CrossRef] [PubMed]
- Khor, L.Y.; Bae, K.; Paulus, R.; Al-Saleem, T.; Hammond, M.E.; Grignon, D.J.; Che, M.; Venkatesan, V.; Byhardt, R.W.; Rotman, M.; et al. MDM2 and Ki-67 predict for distant metastasis and mortality in men treated with radiotherapy and androgen deprivation for prostate cancer: RTOG 92-02. J. Clin. Oncol. 2009, 27, 3177–3184. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.; Yang, Z.H.; Kudahetti, S.; Møller, H.; Scardino, P.; Cuzick, J.; Berney, D.M. Prognostic value of Ki-67 for prostate cancer death in a conservatively managed cohort. Br. J. Cancer 2013, 108, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Pollack, A.; DeSilvio, M.; Khor, L.Y.; Li, R.; Al-Saleem, T.I.; Hammond, M.E.; Venkatesan, V.; Lawton, C.A.; Roach, M.; Shipley, W.U.; et al. Ki-67 staining is a strong predictor of distant metastasis and mortality for men with prostate cancer treated with radiotherapy plus androgen deprivation: Radiation therapy oncology group trial 92-02. J. Clin. Oncol. 2004, 22, 2133–2140. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J.; Yang, Z.H.; Fisher, G.; Tikishvili, E.; Stone, S.; Lanchbury, J.S.; Camacho, N.; Merson, S.; Brewer, D.; Cooper, C.S. Prognostic value of PTEN loss in men with conservatively managed localised prostate cancer. Br. J. Cancer 2013, 108, 2582–2589. [Google Scholar] [CrossRef] [PubMed]
- Lotan, T.L.; Carvalho, F.L.; Peskoe, S.B.; Hicks, J.L.; Good, J.; Fedor, H.; Humphreys, E.; Han, M.; Platz, E.A.; Squire, J.A. PTEN Loss is associated with upgrading of prostate cancer from biopsy to radical prostatectomy. Mod. Pathol. 2015, 2, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Badani, K.; Thompson, D.J.S.; Buerki, C.; Davicioni, E.; Garrison, J.; Ghadessi, M.; Mitra, A.P.; Wood, P.J.; Hornberger, J. Impact of a genomic classifier of metastatic risk on postoperative treatment recommendations for prostate cancer patients: A report from the DECIDE study group. Oncotarget 2013, 4, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Saini, S. PSA and beyond: Alternative prostate cancer biomarkers. Cell. Oncol. 2016, 39, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Erho, N.; Crisan, A.; Vergara, I.A.; Mitra, A.P.; Ghadessi, M.; Buerki, C.; Bergstralh, E.J.; Kollmeyer, T.; Fink, S.; Haddad, Z. Discovery and validation of a prostate cancer genomic classifier that predicts early metastasis following radical prostatectomy. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Knezevic, D.; Goddard, A.D.; Natraj, N.; Cherbavaz, D.B.; Clark-Langone, K.M.; Snable, J.; Watson, D.; Falzarano, S.M.; Magi-Galluzzi, C.; Klein, E.A.; et al. Analytical validation of the Oncotype DX prostate cancer assay—A clinical RT-PCR assay optimized for prostate needle biopsies. BMC Genom. 2013, 14, 690. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.A.; Cooperberg, M.R.; Magi-Galluzzi, C.; Simko, J.P.; Falzarano, S.M.; Maddala, T.; Chan, J.M.; Li, J.; Cowan, J.E.; Tsiatis, A.C. A 17-gene assay to predict prostate cancer aggressiveness in the context of gleason grade heterogeneity, tumor multifocality, and biopsy undersampling. Eur. Urol. 2014, 66, 550–660. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.D.; Scholz, M.C.; Kar, A.J.; Fegan, J.E.; Haregewoin, A.; Kaldate, R.R.; Brawer, M.K. Cell cycle progression score and treatment decisions in prostate cancer: Results from an ongoing registry. Curr. Med. Res. Opin. 2014, 30, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J.; Swanson, G.P.; Fisher, G.; Brothman, A.R.; Berney, D.M.; Reid, J.E.; Mesher, D.; Speights, V.O.; Stankiewicz, E.; Foster, C.S.; et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes for recurrence and death from prostate cancer: A retrospective study in two cohorts. Lancet Oncol. 2011, 141, 520–529. [Google Scholar]
- Cooperberg, M.R.; Simko, J.P.; Cowan, J.E.; Reid, J.E.; Djalilvand, A.; Bhatnagar, S.; Gutin, A.; Lanchbury, J.S.; Swanson, G.P.; Stone, S.; et al. Validation of a cell-cycle progression gene panel to improve risk stratification in a contemporary prostatectomy cohort. J. Clin. Oncol. 2013, 31, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Freedland, S.J.; Gerber, L.; Reid, J.; Welbourn, W.; Tikishvili, E.; Park, J.; Younus, A.; Gutlin, A.; Sangale, Z.; Lanchbury, J.S.; et al. Prognostic utility of cell cycle progression score in men with prostate cancer after primary external beam radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Bishoff, J.T.; Freedland, S.J.; Gerber, L.; Tennstedt, P.; Reid, J.; Welbourn, W.; Graefen, M.; Sangale, Z.; Tikishvili, E.; Park, J.; et al. Prognostic utility of the cell cycle progression score generated from biopsy in men treated with prostatectomy. J. Urol. 2014, 192, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J.; Stone, S.; Fisher, G.; Yang, Z.H.; North, B.V.; Berney, D.M.; Beltran, L.; Greenberg, D.; Moller, H.; Reid, J.E. Validation of an RNA cell cycle progression score for predicting death from prostate cancer in a conservatively managed needle biopsy cohort. Br. J. Cancer 2015, 113, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Bishoff, J.; Freedland, S.; Schlomm, T.; Reid, J.; Brawer, M.; Stone, S.; Cuzick, J. The CCP score provides significant prognostic information in gleason score <7 patients. In Proceedings of the AUA 2016—The American Urological Association Annual Meeting, San Diego, CA, USA, 6–10 May 2016.
- Shipitsin, M.; Small, C.; Choudhury, S.; Giladi, E.; Friedlander, S.; Nardone, J.; Hussain, S.; Hurley, A.D.; Ernst, C.; Huang, Y.E. Identification of proteomic biomarkers predicting prostate cancer aggressiveness and lethality despite biopsy-sampling error. Br. J. Cancer 2014, 111, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shipitsin, M.; Small, C.; Giladi, E.; Siddiqui, S.; Choudhury, S.; Hussain, S.; Huang, Y.E.; Chang, H.; Rimm, D.L.; Berman, D.M.; et al. Automated quantitative multiplex immunofluorescence in situ imaging identifies phospho-S6 and phospho-PRAS40 as predictive protein biomarkers for prostate cancer lethality. Proteome Sci. 2014, 12, 40. [Google Scholar] [CrossRef] [PubMed]
- Blume-Jensen, P.; Berman, D.M.; Rimm, D.L.; Shipitsin, M.; Putzi, M.; Nifong, T.P.; Small, C.; Choudhury, S.; Capela, T.; Coupal, T.; et al. Development and clinical validation of an in situ biopsy-based multimarker assay for risk stratification in prostate cancer. Clin. Cancer Res. 2015, 21, 2591–2600. [Google Scholar] [CrossRef] [PubMed]
- Schalken, J.A.; Hessels, D.; Verhaegh, G. New targets for therapy in prostate cancer: Differential display code 3 (DD3PCA3), a highly prostate cancer-specific gene. Urology 2003, 62, 34–43. [Google Scholar] [CrossRef]
- Falzarano, S.M.; Ferro, M.; Bollito, E.; Klein, E.A.; Carrieri, G.; Magi-Galluzzi, C. Novel biomarkers and genomic tests in prostate cancer: A critical analysis. Minerva Urol. Nefrol. 2015, 67, 211–231. [Google Scholar] [PubMed]
- Marks, L.S.; Bostwick, D.G. Prostate cancer specificity of PCA3 gene testing: Examples from clinical practice. Revl. Urol. 2008, 10, 175–181. [Google Scholar]
- Pepe, P.; Aragona, F. PCA3 score vs PSA free/total accuracy in prostate cancer diagnosis at repeat saturation biopsy. Anticancer Res. 2011, 31, 4445–4449. [Google Scholar] [PubMed]
- Cornu, J.N.; Cancel-Tassin, G.; Egrot, C.; Gaffory, C.; Haab, F.; Cussenot, O. Urine TMPRSS2: ERG fusion transcript integrated with PCA3 score, genotyping, and Biological features are correlated to the Results of prostatic biopsies in men at risk of prostate cancer. Prostate 2013, 73, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Salami, S.A.; Schmidt, F.; Laxman, B.; Regan, M.; Rickman, D.S.; Scherr, D.; Bueti, G.; Siddiqui, J.; Tomlins, S.A.; We, J.T.; et al. Combining Urinary Detection of TMPRSS2:ERG and PCA3 with Serum PSA to Predict Diagnosis of Prostate Cancer. Urol. Oncol. 2010, 48, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Leyten, G.H.J.M.; Hessels, D.; Smit, F.P.; Jannink, S.A.; De Jong, H.; Melchers, W.J.G.; Cornel, E.B.; de Reijke, T.M.; Vergunst, H.; Kil, P.; et al. Identification of a candidate gene panel for the early diagnosis of prostate cancer. Clin. Cancer Res. 2015, 21, 3061–3070. [Google Scholar] [CrossRef] [PubMed]
- Van Neste, L.; Hendriks, R.J.; Dijkstra, S.; Trooskens, G.; Cornel, E.B.; Jannink, S.A.; de Jong, H.; Hessels, D.; Smit, F.P.; Melchers, W.J.; et al. Detection of high-grade prostate cancer using a urinary molecular biomarker-based risk score. Eur. Urol. 2016, 1–9. [Google Scholar] [CrossRef] [PubMed]
- McKiernan, J.; Donovan, M.J.; O’Neill, V.; Bentink, S.; Noerholm, M.; Belzer, S.; Skog, J.; Kattan, M.W.; Partin, A.; Andriole, G.; et al. A novel urine exosome gene expression assay to predict high-grade prostate cancer at initial biopsy. JAMA Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- McDunn, J. LC-MS/MS-based Metabolomic urinalysis for prostate cancer patient management. In Proceeding of the 5 th Annual Conference and Exhibition of Association for Mass Spectrometry: Applications to the Clinical Lab, San Diego, CA, USA, 9–13 February 2013.
- Bratt, O.; Lilja, H. Serum markers in prostate cancer detection. Curr. Opin. Urol. 2015, 25, 59–64. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.L.; Parsons, J.K. 4-Kallikrein test and kallikrein markers in prostate cancer screening. Urol. Clin. N. Am. 2016, 43, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Vedder, M.; de Bekker-Grob, E.; Lilja, H.G.; Vickers, A.J.; van Leenders, G.; Steyerberg, E.W.; Roobol, M.J. The added value of percentage of free to total prostate-specific antigen, PCA3, and a kallikrein panel to the ERSPC risk calculator for prostate cancer in prescreened men. Eur. Urol. 2015, 2, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Leapman, M.; Nguyen, H.G.; Cooperberg, M.R. Clinical utility of biomarkers in localized prostate cancer. Curr. Oncol. Rep. 2016, 18. [Google Scholar] [CrossRef] [PubMed]
- Kamat, A.M.; Vlahou, A.; Taylor, J.A.; Hudson, M.L.; Pesch, B.; Ingersoll, M.A.; Todenhofer, T.; van Rhijn, B.; Kassouf, W.; Grossman, H.B.; et al. Considerations on the use of urine markers in the management of patients with low-/intermediate-risk non-muscle invasive bladder cancer. Urol. Oncol. Semin. Orig. Investig. 2014, 32, 1061–1068. [Google Scholar]
- James, P. Protein identification in the post-genome era: The rapid rise of proteomics. Q. Rev. Biophys. 1997, 30, 279–331. [Google Scholar] [CrossRef] [PubMed]
- Langley, S.R.; Dwyer, J.; Drozdov, I.; Yin, X.; Mayr, M. Proteomics: From single molecules to biological pathways. Cardiovasc. Res. 2013, 97, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Schiess, R.; Wollscheid, B.; Aebersold, R. Targeted proteomic strategy for clinical biomarker discovery. Mol. Oncol. 2009, 3, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Chugh, S.; Sharma, P.; Kislinger, T.; Gramolini, A.O. Clinical proteomics: Getting to the heart of the matter. Circ. Cardiovasc. Genet. 2012, 5, 377. [Google Scholar] [CrossRef] [PubMed]
- Pin, E.; Fredolini, C.; Petricoin, E.F. The role of proteomics in prostate cancer research: Biomarker discovery and validation. Clin. Biochem. 2013, 46, 524–538. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Eisenberger, M.A.; Halabi, S.; Oudard, S.; Nanus, D.M.; Petrylak, D.P.; Sartor, A.O.; Acher, H.I. Biomarkers in the management and treatment of men with metastatic castration-resistant prostate cancer. Eur. Urol. 2012, 61, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.A.; De Lemos, J.A. Benchmarks for the assessment of novel cardiovascular biomarkers. Circulation 2007, 115, 949–952. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.K.; Vaughan, T.B.; Atkinson, T.; Zhu, H.; Kyprianou, N. Emerging biomarkers of prostate cancer (Review). Oncol. Rep. 2012, 28, 409–417. [Google Scholar] [PubMed]
- Rifai, N.; Gillette, M.A.; Carr, S.A. Protein biomarker discovery and validation: The long and uncertain path to clinical utility. Nat. Biotechnol. 2006, 24, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.P.; Spary, L.K.; Mason, M.D.; Tabi, Z.; Brewis, I.A.; Clayton, A. Prostate stromal cell proteomics analysis discriminates normal from tumour reactive stromal phenotypes. Oncotarget 2016, 7, 20124–20139. [Google Scholar] [CrossRef] [PubMed]
- Adeola, H.A.; Smith, M.; Kaestner, L.; Blackburn, J.M.; Zerbini, L.F. Novel potential serological prostate cancer biomarkers using CT100+ cancer antigen microarray platform in a multi-cultural south african cohort. Oncotarget 2016, 7, 13945–13964. [Google Scholar] [PubMed]
- Li, Q.; Li, Y.; Wang, Y.; Cui, Z.; Gong, L.; Qu, Z.; Zhong, Y.; Zhou, J.; Zhou, Y.; Gao, Y.; et al. Quantitative proteomic study of human prostate cancer cells with different metastatic potentials. Int. J. Oncol. 2016, 48, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Ino, Y.; Arakawa, N.; Ishiguro, H.; Uemura, H.; Kubota, Y.; Hirano, H.; Toda, T. Phosphoproteome analysis demonstrates the potential role of THRAP3 phosphorylation in androgen-independent prostate cancer cell growth. Proteomics 2016, 16, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Kazuno, S.; Furukawa, J.-I.; Shinohara, Y.; Murayama, K.; Fujime, M.; Ueno, T.; Fujimura, T. Glycosylation status of serum immunoglobulin G in patients with prostate diseases. Cancer Med. 2016, 5, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Stone, L. Prostate cancer: Proteomics provides a prognostic marker. Nat. Rev. Urol. 2016, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Davalieva, K.; Kostovska, I.M.; Kiprijanovska, S.; Markoska, K.; Kubelka-Sabit, K.; Filipovski, V.; Stavridis, S.; Stankov, O.; Komina, S.; Petrusevska, G.; et al. Proteomics analysis of malignant and benign prostate tissue by 2D DIGE/MS reveals new insights into proteins involved in prostate cancer. Prostate 2015, 75, 1586–1600. [Google Scholar] [CrossRef] [PubMed]
- Arner, P.; Henjes, F.; Schwenk, J.M.; Darmanis, S.; Dahlman, I.; Iresjo, B.M.; Naredi, P.; Agustsson, T.; Lundholm, K.; Nilsson, P. Circulating carnosine dipeptidase 1 associates with weight loss and poor prognosis in gastrointestinal cancer. PLoS ONE 2015, 10, e0123566. [Google Scholar] [CrossRef] [PubMed]
- Bergamini, S.; Bellei, E.; Bonetti, L.R.; Monari, E.; Cuoghi, A.; Borelli, F.; Sighinolfi, M.C.; Bianchi, G.; Ozben, T.; Tomasi, A. Inflammation: An important parameter in the search of prostate cancer biomarkers. Proteome Sci. 2014, 12, 32. [Google Scholar] [CrossRef] [PubMed]
- Pallua, J.D.; Schaefer, G.; Seifarth, C.; Becker, M.; Meding, S.; Rauser, S.; Walch, A.; Handler, M.; Netzer, M.; Popovscaia, M.; et al. MALDI-MS tissue imaging identification of biliverdin reductase B overexpression in prostate cancer. J. Proteom. 2013, 91, 500–514. [Google Scholar] [CrossRef] [PubMed]
- Leymarie, N.; Griffin, P.J.; Jonscher, K.; Kolarich, D.; Orlando, R.; McComb, M.; Zaia, J.; Aguilan, J.; Alley, W.R.; Altmann, F.; et al. Interlaboratory study on differential analysis of protein glycosylation by mass spectrometry: The ABRF glycoprotein research multi-institutional study 2012. Mol. Cell. Proteom. 2013, 12, 2935–2951. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; He, H.; Zhang, Y.; Yang, D.-L.; Huang, J.-H.; Zhu, Y.; Mo, R.; Chen, G.; Yang, S.; Chen, Y.; et al. An integrative proteomics and interaction network-based classifier for prostate cancer diagnosis. PLoS ONE 2013, 8, e63941. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.D.; Zhang, Y.Q.; He, H.C.; Dai, Q.S.; Qin, G.Q.; Chen, J.H.; Cai, C.; Fu, X.; Bi, X.C.; Zhu, J.G.; et al. Identification of novel serological tumor markers for human prostate cancer using integrative transcriptome and proteome analysis. Med. Oncol. 2012, 29, 2877–2888. [Google Scholar] [CrossRef] [PubMed]
- Endoh, K.; Nishi, M.; Ishiguro, H.; Uemura, H.; Miyagi, Y.; Aoki, I.; Hirano, H.; Kubota, Y.; Ryo, A. Identification of phosphorylated proteins involved in the oncogenesis of prostate cancer via Pin1-proteomic analysis. Prostate 2012, 72, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-L.; Huang, H.-J.; Ou, B.-Y.; Chow, N.-H.; Chen, Y.-W.; Tzai, T.-S.; Wu, C.J.; Chen, S.H. Urinary CD14 as a potential biomarker for benign prostatic hyperplasia—Discovery by combining MALDI-TOF-based biostatistics and ESI-MS/MS-based stable-isotope labeling. Proteom. Clin. Appl. 2011, 5, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Alaiya, A.A.; Al-Mohanna, M.; Aslam, M.; Shinwari, Z.; Al-Mansouri, L.; Al-Rodayan, M.; Al-Eid, M.; Ahmad, I.; Hanash, K.; Tulbah, A. Proteomics-based signature for human benign prostate hyperplasia and prostate adenocarcinoma. Int. J. Oncol. 2011, 38, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- True, L.D.; Zhang, H.; Ye, M.; Huang, C.-Y.; Nelson, P.S.; von Haller, P.D.; Tjoelker, L.W.; Kim, J.S.; Qian, W.J.; Smith, P.D.; et al. CD90/THY1 is overexpressed in prostate cancer-associated fibroblasts and could serve as a cancer biomarker. Mod. Pathol. 2010, 23, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Valmu, L.; Ravela, S.; Stenman, U.H. Proteomic analysis of pancreatic secretory trypsin inhibitor/tumor-associated trypsin inhibitor from urine of patients with pancreatitis or prostate cancer. Methods Mol. Biol. 2010, 641, 347–357. [Google Scholar] [PubMed]
- Thoenes, L.; Hoehn, M.; Kashirin, R.; Ogris, M.; Arnold, G.J.; Wagner, E.; Guenther, M. In vivo chemoresistance of prostate cancer in metronomic cyclophosphamide therapy. J. Proteom. 2010, 73, 1342–1354. [Google Scholar] [CrossRef] [PubMed]
- Van der Deen, M.; Akech, J.; Wang, T.; FitzGerald, T.J.; Altieri, D.C.; Languino, L.R.; Lian, J.B.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S. The cancer-related Runx2 protein enhances cell growth and responses to androgen and TGFbeta in prostate cancer cells. J. Cell. Biochem. 2010, 109, 828–837. [Google Scholar] [PubMed]
- Sardana, G.; Jung, K.; Stephan, C.; Diamandis, E.P. Proteomic analysis of conditioned media from the PC3, LNCaP, and 22Rv1 prostate cancer cell lines: Discovery and validation of candidate prostate cancer biomarkers. J. Proteome Res. 2008, 7, 3329–3338. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Murayama, Y.; Pan, Y.; Taima, T.; Fujimura, T.; Murayama, K.; Sadilek, M.; Egawa, S.; Ueno, S.; Ito, A.; et al. Haptoglobin-beta chain defined by monoclonal antibody RM2 as a novel serum marker for prostate cancer. Int. J. Cancer 2008, 123, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Ummanni, R.; Junker, H.; Zimmermann, U.; Venz, S.; Teller, S.; Giebel, J.; Scharf, C.; Woenckhaus, C.; Dombrowski, F.; Walther, R. Prohibitin identified by proteomic analysis of prostate biopsies distinguishes hyperplasia and cancer. Cancer Lett. 2008, 266, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Casale, G.P.; Tian, J.; Wehbi, N.K.; Abrahams, N.A.; Kaleem, Z.; Smith, L.M.; Johansson, S.L.; Elkahwaji, J.E.; Hemstreet, G.P. Quantitative fluorescence imaging analysis for cancer biomarker discovery: Application to beta-catenin in archived prostate specimens. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Ruan, W.; Sassoon, A.; An, F.; Simko, J.P.; Liu, B. Identification of clinically significant tumor antigens by selecting phage antibody library on tumor cells in situ using laser capture microdissection. Mol. Cell. Proteom. 2006, 5, 2364–2373. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B.; Pourian, M.R.; Chuan, Y.-C.; Byman, I.; Bergh, A.; Pang, S.T.; Norstedt, G.; Bergman, T.; Pousette, A. Proteomic comparison of prostate cancer cell lines LNCaP-FGC and LNCaP-r reveals heatshock protein 60 as a marker for prostate malignancy. Prostate 2006, 66, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.W.; Mobley, J.A.; Evans, J.E.; Carmody, J.F.; Ho, S.M. Mass profiling-directed isolation and identification of a stage-specific serologic protein biomarker of advanced prostate cancer. Proteomics 2005, 5, 2927–2238. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Y.; Lamond, A.I. A perspective on proteomics in cell biology. Trends Cell Biol. 2014, 24, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Seluanov, A.; Vaidya, A.; Gorbunova, V. Establishing primary adult fibroblast cultures from rodents. J. Vis. Exp. 2010, 5, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Lexander, H.; Hellman, U.; Palmberg, C.; Auer, G.; Hellström, M.; Franzén, B.; Jornvall, H.; Egevad, L. Evaluation of two sample preparation methods for prostate proteome analysis. Proteomics 2006, 6, 3918–3925. [Google Scholar] [CrossRef] [PubMed]
- Micke, P.; Ohshima, M.; Tahmasebpoor, S.; Ren, Z.-P.; Ostman, A.; Pontén, F.; Botling, J. Biobanking of fresh frozen tissue: RNA is stable in nonfixed surgical specimens. Lab. Investig. 2006, 86, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, W.; Ju, Z.; Tamboli, P.; Jonasch, E.; Mills, G.B.; Lu, Y.; Hennessy, B.T.; Tsavachiisou, D. An efficient procedure for protein extraction from formalin-fixed, paraffin-embedded tissues for reverse phase protein arrays. Proteome Sci. 2012, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, M.S.; Dalmas, D.A.; Boyce, R.W.; Thomas, H.C.; Frazier, K.S. Protein extraction of formalin-fixed, paraffin-embedded tissue enables robust proteomic profiles by mass spectrometry. J. Histochem. Cytochem. 2009, 57, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Malhotra, L.; Dickerson, R.; Chaffee, S.; Sen, C.K.; Roy, S. Laser capture microdissection: Big data from small samples. Gynecol. Oncol. 2015, 136, 554–561. [Google Scholar]
- Miller, R.A.; Winrow, C.J.; Spellman, D.S.; Song, Q.; Reiss, D.R.; Conway, J.P.; Taylor, R.R.; Coleman, P.J.; Hendrickson, R.C.; Renger, J.J. Quantitative proteomics in laser capture microdissected sleep nuclei from rat brain. J. Neurogenet. 2014, 28, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Staunton, L.; Tonry, C.; Lis, R.; Finn, S.; O’Leary, J.; Loda, M.; Bowden, M.; Pennington, S.R. Profiling the tumor microenvironment proteome in prostate cancer using laser capture microdissection coupled to LC-MS—A technical report. EuPA Open Proteom 2015, 10, 19–23. [Google Scholar] [CrossRef]
- Sluss, P.M.; Lewandrowski, K.B. Laboratory reference values. N. Engl. J. Med. 2004, 351, 2461. [Google Scholar]
- Anderson, N.L. The clinical plasma proteome: A survey of clinical assays for proteins in plasma and serum. Clin. Chem. 2010, 56, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Zhonghao, Y.; Kastenmüller, G.; He, Y.; Belcredi, P.; Möller, G.; Prehn, C.; Mendes, J.; Wahl, S.; Roemisch-Margl, W.; Ceglarek, U.; et al. Differences between human plasma and serum metabolite profiles. PLoS ONE 2011, 6, 1–6. [Google Scholar]
- Ray, S.; Reddy, P.J.; Jain, R.; Gollapalli, K.; Moiyadi, A.; Srivastava, S. Proteomic technologies for the identification of disease biomarkers in serum: Advances and challenges ahead. Proteomics 2011, 11, 2139–2161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Sun, H.; Yan, G.; Han, Y.; Wang, X. Serum proteomics in biomedical research : A systematic review. Appl. Biochem. Biotechnol. 2013, 170, 774–786. [Google Scholar] [CrossRef] [PubMed]
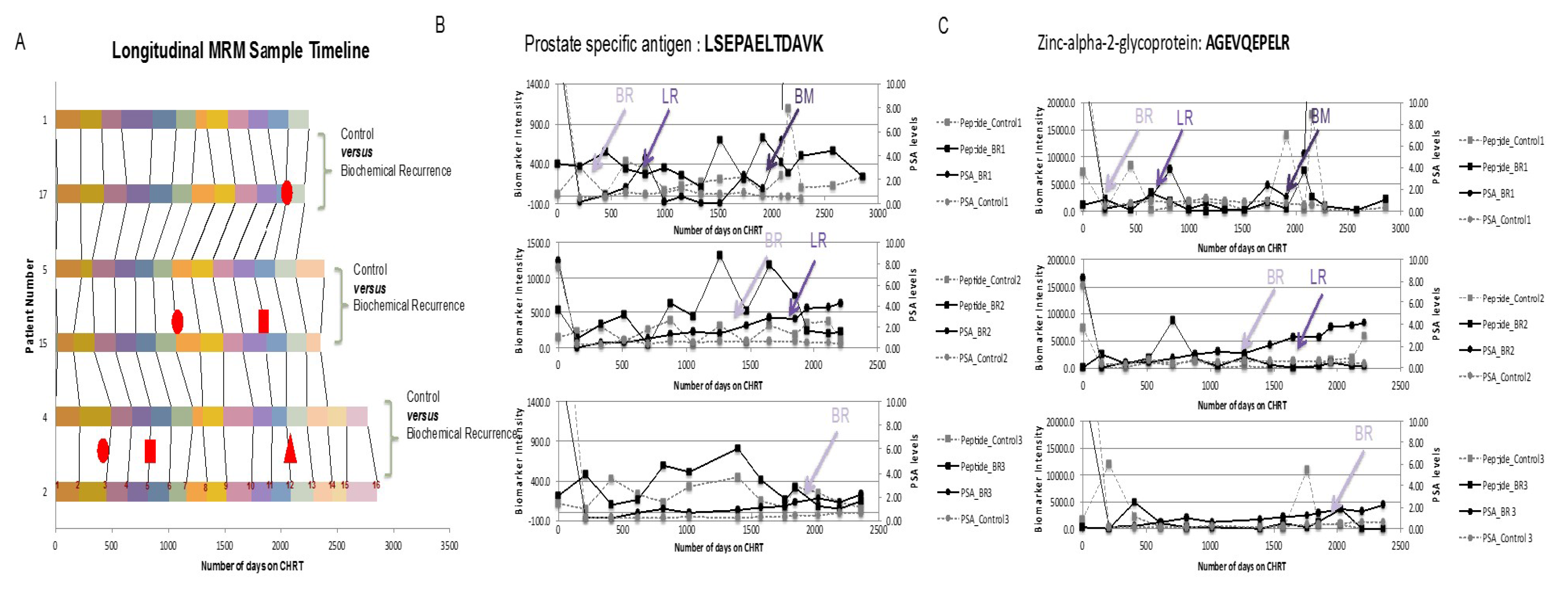
- Morrissey, B.; O’ Shea, C.; Armstrong, J.; Rooney, C.; Staunton, L.; Sheehan, M.; Shannon, A.M.; Pennington, S.R. Development of a label-free LC-MS/MS strategy to approach the identification of candidate protein biomarkers of disease recurrence in prostate cancer patients in a clinical trial of combined hormone and radiation therapy. Proteom. Clin. Appl. 2013, 7, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Keshishian, H.; Addona, T.; Burgess, M.; Kuhn, E.; Carr, S.A. Quantitative, Multiplexed Assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteom. 2008, 6, 2212–2229. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, R.L. Considerations for the use of blood plasma and serum for proteomic analysis. Internet J. Genom. Proteom. 2005, 1, 1–5. [Google Scholar]
- Millioni, R.; Tolin, S.; Puricelli, L.; Sbrignadello, S.; Fadini, G.P.; Tessari, P.; Arrigoni, G. High abundance proteins depletion vs low abundance proteins enrichment: Comparison of methods to reduce the plasma proteome complexity. PLoS ONE 2011, 6, e19603. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, S.; Jafari, A.; Moradpoor, R.; Ghasemi, E.; Khalkhal, E. Human urine proteomics : Analytical techniques and clinical applications in renal diseases. Int. J. Proteom. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Percy, A.J.; Yang, J.; Hardie, D.B.; Chambers, A.G.; Tamura-Wells, J.; Borchers, C.H. Precise quantitation of 136 urinary proteins by LC/MRM-MS using stable isotope labeled peptides as internal standards for biomarker discovery and/or verification studies. Methods 2015, 81, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Suárez, E.; Siwy, J.; Zürbig, P.; Mischak, H. Urine as a source for clinical proteome analysis: From discovery to clinical application. Biochim. Biophys. Acta 2013, 1844, 884–898. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Ignatchenko, V.; Yao, C.Q.; Kalatskaya, I.; Nyalwidhe, J.O.; Lance, R.S.; Gramolini, A.O.; Troyer, D.A.; Stein, L.D.; Boutros, P.C.; et al. Identification of differentially expressed proteins in direct expressed prostatic secretions of men with organ-confined versus extracapsular prostate cancer. Mol. Cell. Proteom. 2012, 11, 1870–1884. [Google Scholar] [CrossRef] [PubMed]
- Davalieva, K.; Kiprijanovska, S.; Komina, S.; Petrusevska, G.; Zografska, N.C.; Polenakovic, M. Proteomics analysis of urine reveals acute phase response proteins as candidate diagnostic biomarkers for prostate cancer. Proteome Sci. 2015, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Truong, M.; Yang, B.; Jarrard, D.F. Toward the detection of prostate cancer in urine: A critical analysis. J. Urol. 2013, 189, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Jedinak, A.; Curatolo, A.; Zurakowski, D.; Dillon, S.; Bhasin, M.K.; Libermann, T.A.; Roy, R.; Sachdev, M.; Loughlin, K.R.; Moses, M.A. Novel non-invasive biomarkers that distinguish between benign prostate hyperplasia and prostate cancer. BMC Cancer 2015, 15, 259. [Google Scholar] [CrossRef] [PubMed]
- Katafigiotis, I.; Tyritzis, S.I.; Stravodimos, K.G.; Alamanis, C.; Pavlakis, K.; Vlahou, A.; Makridakis, M.; Katafigioti, A.; Garbis, D.S.; Constantinides, C.A. Zinc alpha 2-glycoprotein as a potential novel urine biomarker for the early diagnosis of prostate cancer. BJU Int. 2012, 110, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Mrozinski, P.; Zolotarjova, N.; Chen, H. Application note: Human serum and plasma protein depletion–novel high-capacity affinity column for the removal of the “Top 14” abundant proteins. Available online: https://www.agilent.com/cs/library/applications/5989-7839EN.pdf (accessed online 14 July 2016).
- Owen, D.H.; Katz, D.F. A review of the physical and chemical properties of human semen and the formulation of a semen simulant. J. Androl. 2005, 26, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Amaral, A.; Castillo, J.; Ramalho-Santos, J.; Oliva, R. The combined human sperm proteome: Cellular pathways and implications for basic and clinical science. Hum. Reprod. Update 2014, 20, 40–62. [Google Scholar] [CrossRef] [PubMed]
- Bartoov, B.; Eltes, F.; Reichart, M.; Langzam, J.; Lederman, H.; Zabludovsky, N. Quantitative ultramorphological analysis of human sperm: Fifteen years of experience in the diagnosis and management of male factor infertility. Arch. Androl. 1999, 43, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Pizzol, D.; Ferlin, A.; Garolla, A.; Lenzi, A.; Bertoldo, A.; Foresta, C. Genetic and molecular diagnostics of male infertility in the clinical practice. Front. Biosci. 2014, 19, 291–303. [Google Scholar] [CrossRef]
- Liu, D.Y.; Baker, H.W. Evaluation and assessment of semen for IVF/ICSI. Asian J. Androl. 2002, 4, 281–285. [Google Scholar] [PubMed]
- Neuhaus, J.; Schiffer, E.; von Wilcke, P.; Bauer, H.W.; Leung, H.; Siwy, J.; Ulrici, W.; Paasch, U.; Horn, L.C.; Stolzenburg, J.U. Seminal plasma as a source of prostate cancer peptide biomarker candidates for detection of indolent and advanced disease. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, J.; Schiffer, E.; von Wilcke, P.; Bauer, H.W.; Leung, H.; Siwy, J.; Ulrici, W.; Paasch, U.; Horn, L.C.; Stolzenburg, J.U. Seminal plasma as a source of prostate cancer peptide biomarker candidates for detection of indolent and advanced disease. PLoS ONE 2013, 8, e67514. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.W.; Thompson, H.S. Proteomics of semen and its constituents. Proteom. Clin. Appl. 2007, 1, 861–875. [Google Scholar] [CrossRef] [PubMed]
- Drake, R.R.; Elschenbroich, S.; Lopez-perez, O.; Kim, Y.; Ignatchenko, V.; Ignatchenko, A.; Nyalwidhe, J.O.; Basu, G.; Wilkins, C.E.; Gjurich, B.; et al. In-depth proteomic analyses of direct expressed prostatic secretions research articles. J. Proteome Res. 2010, 9, 2109–2116. [Google Scholar] [CrossRef] [PubMed]
- Junker, H.; Venz, S.; Zimmermann, U.; Thiele, A.; Scharf, C.; Walther, R. Stage-related alterations in renal cell carcinoma—Comprehensive quantitative analysis by 2D-DIGE and protein network analysis. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, P.H. High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [PubMed]
- Timms, J.F.; Cramer, R. Difference gel electrophoresis. Proteomics 2008, 8, 4886–4897. [Google Scholar] [CrossRef] [PubMed]
- Karp, N.A.; Feret, R.; Rubtsov, D.V.; Lilley, K.S. Comparison of DIGE and post-stained gel electrophoresis with both traditional and SameSpots analysis for quantitative. Proteomics 2008, 8, 948–960. [Google Scholar] [CrossRef] [PubMed]
- Minden, J.S.; Dowd, S.R.; Meyer, H.E.; Stuhler, K. Difference gel electrophoresis. Electrophoresis 2009, 30, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Geisler, C.; Gaisa, N.T.; Pfister, D.; Fuessel, S.; Kristiansen, G.; Braunschweig, T.; Gostek, S.; Beine, B.; Diehl, H.C.; Jackson, A.M.; et al. Identification and validation of potential new biomarkers for prostate cancer diagnosis and prognosis using 2D-DIGE and MS. Biomed. Res. Int. 2015, 2015, 454256. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.C.; Downes, M.R.; O’Donoghue, N.; O’Keane, C.; O’Neill, A.; Fan, Y.; Fitzpatrick, J.M.; Watson, R.W. 2D-DIGE as a strategy to identify serum markers for the progression of prostate cancer. J. Proteome Res. 2009, 8, 942–957. [Google Scholar] [CrossRef] [PubMed]
- Ummanni, R.; Duscharla, D.; Barett, C.; Venz, S.; Schlomm, T.; Heinzer, H.; Walther, R.; Bokemeyer, C.; Brummendorf, T.H.; Murthy, P.V. Prostate cancer-associated autoantibodies in serum against tumor-associated antigens as potential new biomarkers. J. Proteom. 2015, 119, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Fredolini, C.; Liotta, L.A.; Petricoin, E.F. Application of proteomic technologies for prostate cancer detection, prognosis, and tailored therapy. Crit. Rev. Clin. Lab. Sci. 2010, 47, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Forner, F.; Foster, L.J.; Toppo, S. Mass spectrometry data analysis in the proteomics era. Curr. Bioinform. 2007, 2, 63–93. [Google Scholar] [CrossRef]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Meissner, F.; Mann, M. Quantitative shotgun proteomics: Considerations for a high-quality workflow in immunology. Nat. Immunol. 2014, 15, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Kalli, A.; Smith, G.T.; Sweredoski, M.J.; Hess, S. Evaluation and optimization of mass spectrometric settings during data-dependent acquisition mode: Focus on LTQ-Orbitrap mass analyzers. J. Proteome Res. 2013, 12, 3071–3086. [Google Scholar] [CrossRef] [PubMed]
- Kelstrup, C.D.; Young, C.; Lavallee, R.; Nielsen, M.L.; Olsen, J.V. Optimized fast and sensitive acquisition methods for shotgun proteomics on a quadrupole orbitrap mass spectrometer. J. Proteome Res. 2012, 11, 3487–3497. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Liotta, L.A.; Petricoin, E.F. Cancer metabolism and mass spectrometry-based proteomics. Cancer Lett. 2013, 356, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Crutchfield, C.A.; Thomas, S.N.; Sokoll, L.J.; Chan, D.W. Advances in mass spectrometry—Based clinical biomarker discovery. Clin. Proteom. 2016, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hou, G.; Lou, X.; Sun, Y.; Xu, S.; Zi, J.; Wang, Q.; Zhour, B.; Han, B.; Wu, L.; Zhao, X.; et al. Biomarker Discovery and Verification of Esophageal Squamous Cell Carcinoma Using Integration of SWATH/MRM. J. Proteome Res. 2015, 14, 3793–3803. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.Y.K.; Etheridge, N.; Nouwens, A.S.; Dodd, P.R. SWATH analysis of the synaptic proteome in Alzheimer’s disease. Neurochem. Int. 2015, 87, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Yang, L.; Luo, J.; Guo, L.; Wang, Z.; Yang, X.; Jin, W.; Fang, Y.; Ye, J.; Shan, B.; et al. SWATH enables precise label-free quantification on proteome scale. Proteomics 2015, 15, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Guo, T.; Koh, C.C.; Gillessen, S.; Joerger, M.; Jochum, W.; Aebersold, R. Minimal sample requirement for highly multiplexed protein quantification in cell lines and tissues by PCT-SWATH mass spectrometry. Proteomics 2015, 15, 3711–3721. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, J.; Sethi, A.; Li, Q.K.; Chen, L.; Collins, B.; Gillet, L.C.; Wollscheid, B.; Zhang, H.; Aebersold, R. Glycoproteomic analysis of prostate cancer tissues by SWATH mass spectrometry discovers N-acylethanolamine acid amidase and protein tyrosine kinase 7 as signatures for tumor aggressiveness. Mol. Cell. Proteom. 2014, 13, 1753–1768. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Buil, A.; Collins, B.C.; Gillet, L.C.J.; Blum, L.C.; Cheng, L.Y.; Vitek, O.; Mouritsen, J.; Lachance, G.; Spector, T.D.; et al. Quantitative variability of 342 plasma proteins in a human twin population. Mol. Syst. Biol. 2015, 11, 786. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hüttenhain, R.; Surinova, S.; Gillet, L.C.J.; Mouritsen, J.; Brunner, R.; Navarro, P.; Aebersold, R. Quantitative measurements of N-linked glycoproteins in human plasma by SWATH-MS. Proteomics 2013, 13, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Leng, S.X.; McElhaney, J.E.; Walston, J.D.; Xie, D.; Fedarko, N.S.; Kuchel, G.A. Elisa and multiplex technologies for cytokine measurement in inflammation and aging research. J. Gerontol. 2013, 63, 879–884. [Google Scholar] [CrossRef]
- Kragstrup, T.W.; Vorup-Jensen, T.; Deleuran, B.; Hvid, M. A simple set of validation steps identifies and removes false results in a sandwich enzyme-linked immunosorbent assay caused by anti-animal IgG antibodies in plasma from arthritis patients. Springerplus 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Addona, T.A.; Shi, X.; Keshishian, H.; Mani, D.R.; Burgess, M.; Gillette, M.A.; Clauser, K.R.; Shen, D.; Lewis, G.D.; Farrell, L.A.; et al. A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nat. Biotechnol. 2012, 29, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gonzalez, M.; Jara-Acevedo, R.; Matarraz, S.; Jara-Acevedo, M.; Paradinas, S.; Sayagües, J.M.; Orfao, A.; Fuentes, M. Nanotechniques in proteomics: Protein microarrays and novel detection platforms. Eur. J. Pharm. Sci. 2012, 45, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Reis, B.S.; Jungbluth, A.A.; Frosina, D.; Holz, M.; Ritter, E.; Nakayama, E.; Ishida, T.; Obata, Y.; Carver, B.; Scher, H.; et al. Prostate cancer progression correlates with increased humoral immune response to a human endogenous retrovirus GAG protein. Clin. Cancer Res. 2013, 19, 6112–6125. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-M.; Noh, H.B.; Park, D.S.; Ryu, S.H.; Koo, J.S.; Shim, Y.B. Immunosensors for detection of Annexin II and MUC5AC for early diagnosis of lung cancer. Biosens. Bioelectron. 2009, 25, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Brazhnik, K.; Sokolova, Z.; Baryshnikova, M.; Bilan, R.; Efimov, A.; Nabiev, I.; Sukhanova, A. Quantum dot-based lab-on-a-bead system for multiplexed detection of free and total prostate-specific antigens in clinical human serum samples. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Chandra, H.; Srivastava, S. Nanotechniques in proteomics: Current status, promises and challenges. Biosens. Bioelectron. 2010, 25, 2389–4201. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.P.; Lee, B.Y.; Lee, J.; Hong, S.; Sim, S.J. Enhancement of sensitivity and specificity by surface modification of carbon nanotubes in diagnosis of prostate cancer based on carbon nanotube field effect transistors. Biosens. Bioelectron. 2009, 24, 3372–3378. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Hong, S.; Singh, R.; Jang, J. Single-walled carbon nanotube based transparent immunosensor for detection of a prostate cancer biomarker osteopontin. Anal. Chim. Acta 2015, 869, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Kim, H.; Kim, H.S.; Um, S.H.; Choi, J.W.; Oh, B.K. MMP-2 detective silicon nanowire biosensor using enzymatic cleavage reaction. J. Biomed. Nanotechnol. 2013, 9, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Sosic, A.; Meneghello, A.; Antognoli, A.; Cretaio, E.; Gatto, B. Development of a Multiplex Sandwich Aptamer Microarray for the Detection of VEGF165 and Thrombin. Sensors 2013, 13, 13425–13428. [Google Scholar] [CrossRef] [PubMed]
- Khezrian, S.; Salimi, A.; Teymourian, H.; Hallaj, R. Biosensors and Bioelectronics Label-free electrochemical IgE aptasensor based on covalent attachment of aptamer onto multiwalled carbon nanotubes/ionic liquid/chitosan nanocomposite modified electrode. Biosens. Bioelectron. 2013, 43, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, S.; Vaught, J.D.; Bock, C.; Gold, L.; Katilius, E.; Keeney, T.R.; Kim, N.; Saccomano, N.A.; Wilcox, S.K.; Zichi, D.; et al. From SOMAmer-based biomarker discovery to diagnostic and clinical applications: A SOMAmer-based, streamlined multiplex proteomic assay. PLoS ONE 2011, 6, e26332. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Ahn, Y.H.; Ji, E.S.; Lee, J.Y.; Kim, J.Y.; An, H.J.; Yoo, J.S. Quantitative analysis of low-abundance serological proteins with peptide affinity-based enrichment and pseudo-multiple reaction monitoring by hybrid quadrupole time-of-flight mass spectrometry. Anal. Chim. Acta 2015, 882, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Kavosi, B.; Salimi, A.; Hallaj, R.; Moradi, F. Ultrasensitive electrochemical immunosensor for PSA biomarker detection in prostate cancer cells using gold nanoparticles/PAMAM dendrimer loaded with enzyme linked aptamer as integrated triple signal amplification strategy. Biosens. Bioelectron. 2015, 74, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Nahid, P.; Bliven-sizemore, E.; Jarlsberg, L.G.; Mary, A.; Groote, D.; Johnson, J.L.; Muzanyi, G.; Engle, M.; Weiner, M.; Janjic, N.; et al. Treatment response in pulmonary tuberculosis. Tuberculosis 2015, 94, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Ostroff, R.M.; Bigbee, W.L.; Franklin, W.; Gold, L.; Mehan, M.; Miller, Y.E.; Pass, H.I.; Rom, W.N.; Siegfried, J.M.; Stewart, A.; et al. Unlocking biomarker discovery: Large scale application of aptamer proteomic technology for early detection of lung cancer. PLoS ONE 2010, 5, e15003. [Google Scholar] [CrossRef] [PubMed]
- Hathout, Y.; Brody, E.; Clemens, P.R.; Cripe, L.; DeLisle, R.K.; Furlong, P.; Gordish-Dressman, H.; Hache, L.; Henricson, E.; Hoffman, E.P.; et al. Large-scale serum protein biomarker discovery in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2015, 112, 7153–7158. [Google Scholar] [CrossRef] [PubMed]
- Mehan, M.R.; Williams, S.A.; Siegfried, J.M.; Bigbee, W.L.; Weissfeld, J.L.; Wilson, D.O.; Pass, H.I.; Rom, W.N.; Mulet, T.; Meister, M.; et al. Validation of a blood protein signature for non-small cell lung cancer. Clin. Proteom. 2014, 11. [Google Scholar] [CrossRef] [PubMed]
- Domanski, D.; Percy, A.J.; Yang, J.; Chambers, A.G.; Hill, J.S.; Freue, G.V.C.; Borchers, C.H. MRM-based multiplexed quantitation of 67 putative cardiovascular disease biomarkers in human plasma. Proteomics 2012, 12, 1222–1243. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Tomazela, D.M.; Frewen, B.; Maclean, B.; Merrihew, G.; Peterman, S.; Maccoss, M.J. Expediting the development of targeted SRM assays: Using data from shotgun proteomics to automate method development. J. Proteome Res. 2009, 8, 2733–2739. [Google Scholar] [CrossRef] [PubMed]
- Ebhardt, H.A.; Sabidó, E.; Hüttenhain, R.; Collins, B.; Aebersold, R. Range of protein detection by selected/multiple reaction monitoring mass spectrometry in an unfractionated human cell culture lysate. Proteomics 2012, 12, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Wasinger, V.C.; Zeng, M.; Yau, Y. Current status and advances in quantitative proteomic mass spectrometry. Int. J. Proteom. 2013, 2013, 180605. [Google Scholar] [CrossRef] [PubMed]
- Hüttenhain, R.; Soste, M.; Selevsek, N.; Röst, H.; Sethi, A.; Carapito, C.; Farrah, T.; Deutsch, E.W.; Kusebauch, U.; Moritz, R.L.; et al. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Yocum, A.K.; Khan, A.P.; Zhao, R.; Chinnaiyan, A.M. Development of selected reaction monitoring-MS methodology to measure peptide biomarkers in prostate cancer. Proteomics 2010, 10, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Lee, Y.; Lee, M.Y.; Shin, J.; Han, J.M.; Yang, E.G.; Yu, M.H.; Kim, S.; Hwang, D.; Lee, C. Multiple reaction monitoring of multiple low-abundance transcription factors in whole lung cancer cell lysates. J. Proteome Res. 2013, 12, 2582–2596. [Google Scholar] [CrossRef] [PubMed]
- Percy, A.J.; Chambers, A.G.; Yang, J.; Borchers, C.H. Multiplexed MRM-based quantitation of candidate cancer biomarker proteins in undepleted and non-enriched human plasma. Proteomics 2013, 13, 2202–2215. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Wang, X.; Yang, W.; Eshghi, S.T.; Sun, S.; Hoti, N.; Chen, L.; Yang, S.; Passay, J.; Rubin, A.; et al. Integrated proteomic and glycoproteomic analyses of prostate cancer cells reveal glycoprotein alteration in protein abundance and glycosylation. Mol. Cell. Proteom. 2015, 14, 2753–2763. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, A.; Poisson, L.M.; Rajendiran, T.M.; Amjad, P.; Li, Y.; Nyati, M.K.; Ahsan, A.; Kalyana-Sundaram, S.; Han, B.; Cao, X. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009, 457, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Florentinus, A.K.; Bowden, P.; Sardana, G.; Diamandis, E.P.; Marshall, J.G. Identification and quantification of peptides and proteins secreted from prostate epithelial cells by unbiased liquid chromatography tandem mass spectrometry using goodness of fit and analysis of variance. J. Proteom. 2012, 75, 1303–1317. [Google Scholar] [CrossRef] [PubMed]
- Sardana, G.; Marshall, J.; Diamandis, E.P. Discovery of candidate tumor markers for prostate cancer via proteomic analysis of cell culture-conditioned medium. Clin. Chem. 2007, 53, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Saraon, P.; Cretu, D.; Musrap, N.; Karagiannis, G.S.; Batruch, I.; Drabovich, A.P.; van der Kwast, T.; Mizokami, A.; Morrissey, C.; Jarvi, K.; et al. Quantitative proteomics reveals that enzymes of the ketogenic pathway are associated with prostate cancer progression. Mol. Cell. Proteom. 2013, 12, 1589–1601. [Google Scholar] [CrossRef] [PubMed]
- Miyake, H.; Pollak, M.; Gleave, M.E. Castration-induced up-regulation of insulin-like growth factor binding protein-5 potentiates insulin-like growth factor-I activity and accelerates progression to androgen independence in prostate cancer models. Cancer Res. 2000, 60, 3058–3064. [Google Scholar] [PubMed]
- Brooks, J.D.; Wei, W.; Hawley, S.; Auman, H.; Newcomb, L.; Boyer, H.; Fazli, L.; Simko, J.; Hurtado-Coll, A.; Troyer, D.A.; et al. Evaluation of ERG and SPINK1 by immunohistochemical staining and clinicopathological outcomes in a multi-institutional radical prostatectomy cohort of 1067 patients. PLoS ONE 2015, 10, e0132343. [Google Scholar] [CrossRef] [PubMed]
- Makawita, S.; Diamandis, E.P. The bottleneck in the cancer biomarker pipeline and protein quantification through mass spectrometry-based approaches: Current strategies for candidate verification. Clin. Chem. 2010, 56, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Parekh, D.J.; Ankerst, D.P.; Troyer, D.; Srivastava, S.; Thompson, I.M. Biomarkers for prostate cancer detection. J. Urol. 2007, 178, 2252–2259. [Google Scholar] [CrossRef] [PubMed]
- Ilyin, S.E.; Belkowski, S.M.; Plata-Salamán, C.R. Biomarker discovery and validation: Technologies and integrative approaches. Trends Biotechnol. 2004, 22, 411–416. [Google Scholar] [CrossRef] [PubMed]
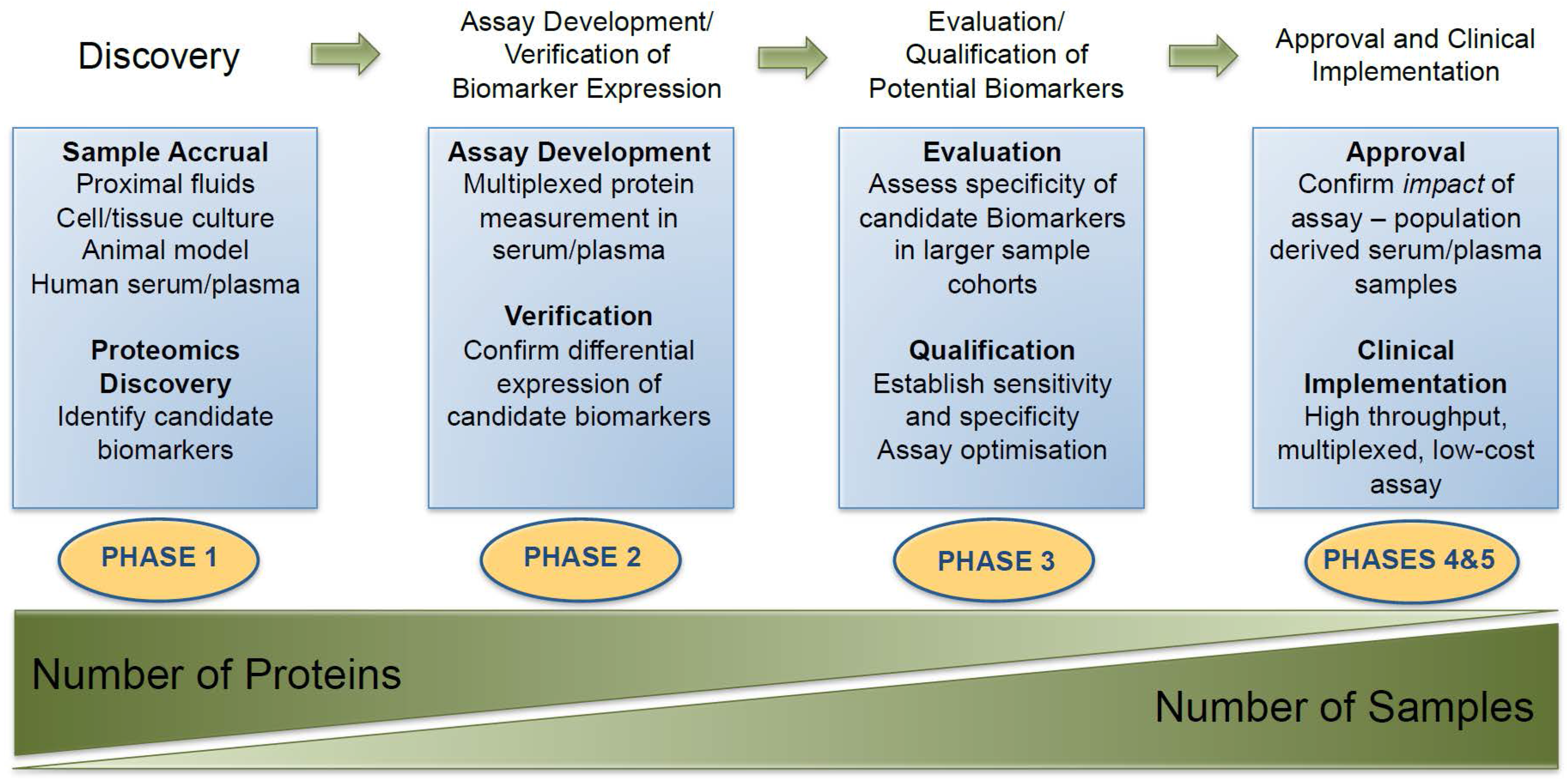
- Pepe, M.S.; Etzioni, R.; Feng, Z.; Potter, J.D.; Thompson, M.L.; Thornquist, M.; Winget, M.; Yasui, Y. Phases of biomarker development for early detection of cancer. J. Natl. Cancer Inst. 2001, 93, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Addona, T.A.; Abbatiello, S.E.; Schilling, B.; Skates, S.J.; Mani, D.R.; Bunk, D.M.; Spiegelman, C.H.; Zimmerman, L.J.; Ham, A.J.; Keshishian, H.; et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 2009, 27, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Hernández, B.; Parnell, A.; Pennington, S.R. Why have so few proteomic biomarkers “survived” validation? (Sample size and independent validation considerations). Proteomics 2014, 14, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Boyd, L.K.; Mao, X.; Lu, Y.J. The complexity of prostate cancer: Genomic alterations and heterogeneity. Nat. Rev. Urol. 2012, 9, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Fagan, A.M.; Xiong, C.; Jasielec, M.S.; Bateman, R.J.; Goate, A.M.; Benzinger, T.L.S.; Ghetti, B.; Martins, R.N.; Masters, C.L.; Mayeux, R.; et al. Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer’s disease. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Christensen, E.; Pintilie, M.; Evans, K.R.; Lenarduzzi, M.; Ménard, C.; Catton, C.N.; Diamandis, E.P.; Bristow, R.G. Longitudinal cytokine expression during IMRT for prostate cancer and acute treatment toxicity. Clin. Cancer Res. 2009, 15, 5576–5583. [Google Scholar] [CrossRef] [PubMed]
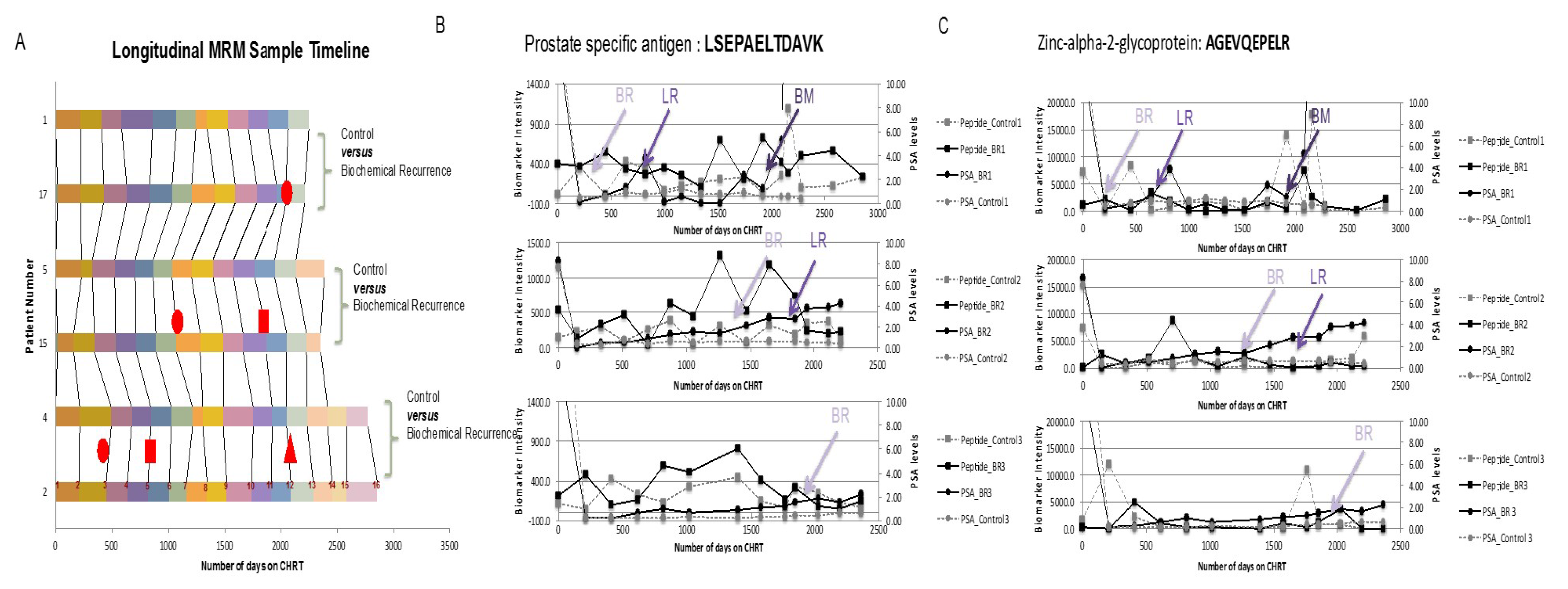
- Tonry, C.L.; Doherty, D.; O’Shea, C.; Morrissey, B.; Staunton, L.; Flatley, B.; Shannon, A.; Armstrong, J.; Pennington, S.R. Discovery and longitudinal evaluation of candidate protein biomarkers for disease recurrence in prostate cancer. J. Proteome Res. 2015, 14, 2769–2783. [Google Scholar] [CrossRef] [PubMed]
- Meo, A.D.; Diamandis, E.P.; Rodriguez, H.; Hoofnagle, A.N.; Ioannidis, J.; Lopez, M. What is wrong with clinical proteomics? Clin. Chem. 2014, 60, 1258–1266. [Google Scholar]
- Kim, K.; Kim, Y. Preparing multiple-reaction monitoring for quantitative clinical proteomics. Expert Rev. Proteom. 2009, 6, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.J.; Abbatiello, S.E.; Kim, K.; Yan, P.; Jeffrey, R.; Lin, C.; Kim, J.S.; Zhang, Y.; Wang, X.; Ivey, R.G.; et al. Demonstrating the feasibility of large-scale development of standardized assays to quantify human proteins. Nat. Methods 2014, 11, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Harlan, R.; Zhang, H. Targeted proteomics: A bridge between discovery and validation. Expert Rev. Proteom. 2014, 11, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Abbatiello, S.E.; Mani, D.R.; Schilling, B.; Maclean, B.; Zimmerman, L.J.; Feng, X.; Cusack, M.P.; Sedransk, N.; Hall, S.C.; Addona, T.; et al. Design, implementation and multisite evaluation of a system suitability protocol for the quantitative assessment of instrument performance in liquid chromatography-multiple reaction monitoring-MS (LC-MRM-MS). Mol. Cell. Proteom. 2013, 12, 2623–2639. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.; Hunter, C.L. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell. Proteom. 2006, 5, 573–588. [Google Scholar] [CrossRef] [PubMed]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.L.; et al. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C. MRM3 quantitation for highest selectivity of proteins in complex matrices. J. Biomol. Tech. 2010, 21, S34–S35. [Google Scholar]
- Niessen, J.; Jedlitschky, G.; Grube, M.; Bien, S.; Schwertz, H.; Ohtsuki, S.; Kawakami, H.; Kamiie, J.; Oswald, S.; Starke, K.; et al. Human platelets express organic anion-transporting peptide 2b1, an uptake transporter for atorvastatin. Drug Metab. Dispos. 2009, 37, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Vogeser, M.; Kyriatsoulis, A.; Huber, E.; Kobold, U. Candidate reference method for the quantification of circulating 25-hydroxyvitamin D3 by liquid chromatography-tandem mass spectrometry. Clin. Chem. 2004, 50, 1415–1417. [Google Scholar] [CrossRef] [PubMed]
- Maunsell, Z.; Wright, D.J.; Rainbow, S.J. Routine isotope-dilution liquid chromatography-tandem mass spectrometry assay for simultaneous measurement of the 25-hydroxy metabolites of vitamins D2 and D3. Clin. Chem. 2005, 51, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- Lacey, J.M.; Bergen, H.R.; Magera, M.J.; Naylor, S.; O’Brien, J.F. Rapid determination of transferrin isoforms by immunoaffinity liquid chromatography and electrospray mass spectrometry. Clin. Chem. 2001, 47, 513–518. [Google Scholar] [PubMed]
- Trenchevska, O.; Nedelkov, D. Targeted quantitative mass spectrometric immunoassay for human protein variants. Proteome Sci. 2011, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Trenchevska, O.; Kamcheva, E.; Nedelkov, D. Mass spectrometric immunoassay for quantitative determination of protein biomarker isoforms. J. Proteome Res. 2010, 9, 5969–5973. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, U.A.; Phillips, D.A.; Trenchevska, O.; Nedelkov, D. Quantitative mass spectrometry evaluation of human retinol binding protein 4 and related variants. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Bystrom, C.; Sheng, S.; Zhang, K.; Caulfield, M.; Clarke, N.J.; Reitz, R. Clinical utility of insulin-like growth factor 1 and 2; determination by high resolution mass spectrometry. PLoS ONE 2012, 7, e43457. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Caulfield, M.P.; McPhaul, M.J.; Reitz, R.E.; Taylor, S.W.; Clarke, N.J. Quantitative insulin analysis using liquid chromatography-tandem mass spectrometry in a high-throughput clinical laboratory. Clin. Chem. 2013, 59, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Peterman, S.; Niederkofler, E.E.; Phillips, D.A.; Krastins, B.; Kiernan, U.A.; Tubbs, K.A.; Nedelkov, D.; Prakash, A.; Vogelsang, M.S.; Schoeder, T.; et al. An automated, high-throughput method for targeted quantification of intact insulin and its therapeutic analogs in human serum or plasma coupling mass spectrometric immunoassay with high resolution and accurate mass detection (MSIA-HR/AM). Proteomics 2014, 14, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- Bystrom, C.E.; Salameh, W.; Reitz, R.; Clarke, N.J. Plasma renin activity by LC-MS/MS: Development of a prototypical clinical assay reveals a subpopulation of human plasma samples with substantial peptidase activity. Clin. Chem. 2010, 56, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, F.; Solomon, B.; Gregorc, V.; Roder, H.; Gray, R.; Kasahara, K.; Nishio, M.; Brahmer, J.; Spreafico, A.; Ludovini, V.; et al. Mass spectrometry to classify non-small-cell lung cancer patients for clinical outcome after treatment with epidermal growth factor receptor tyrosine kinase inhibitors: A multicohort cross-institutional study. J. Natl. Cancer Inst. 2007, 99, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Gregorc, V.; Novello, S.; Lazzari, C.; Barni, S.; Aieta, M.; Mencoboni, M.; Grossi, F.; De Pas, T.; de Marinis, F.; Bearz, A.; et al. Predictive value of a proteomic signature in patients with non-small-cell lung cancer treated with second-line erlotinib or chemotherapy (PROSE): A biomarker-stratified, randomised phase 3 trial. Lancet Oncol. 2014, 15, 713–721. [Google Scholar] [CrossRef]
- Butts, C.A. VeriStrat validated in patients with non-small-cell lung cancer. Lancet Oncol. 2014, 15, 671–672. [Google Scholar] [CrossRef]
- Vachani, A.; Pass, H.I.; Rom, W.N.; Midthun, D.E.; Edell, E.S.; Laviolette, M.; Li, X.J.; Fong, P.Y.; Hunsucker, S.W.; Hayward, C.; et al. Validation of a multiprotein plasma classifier to identify benign lung nodules. J. Thorac. Oncol. 2015, 10, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.E.; Borchers, C.H. Mass spectrometry based biomarker discovery, verification, and validation—Quality assurance and control of protein biomarker assays. Mol. Oncol. 2014, 8, 840–858. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.S.; Liu, T.; Petyuk, V.A.; Burnum-Johnson, K.E.; Ibrahim, Y.M.; Anderson, G.A.; Smith, R.D. Mass spectrometry for translational proteomics: Progress and clinical implications. Genome Med. 2012, 4, 63. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L. Within sight of a rational pipeline for development of protein diagnostics. Clin. Chem. 2012, 58, 28–30. [Google Scholar] [CrossRef] [PubMed]
- Boja, E.; Rivers, R.; Kinsinger, C.; Mesri, M.; Hiltke, T.; Rahbar, A.; Rodriguez, H. Restructuring proteomics through verification. Biomark. Med. 2010, 4, 799–803. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
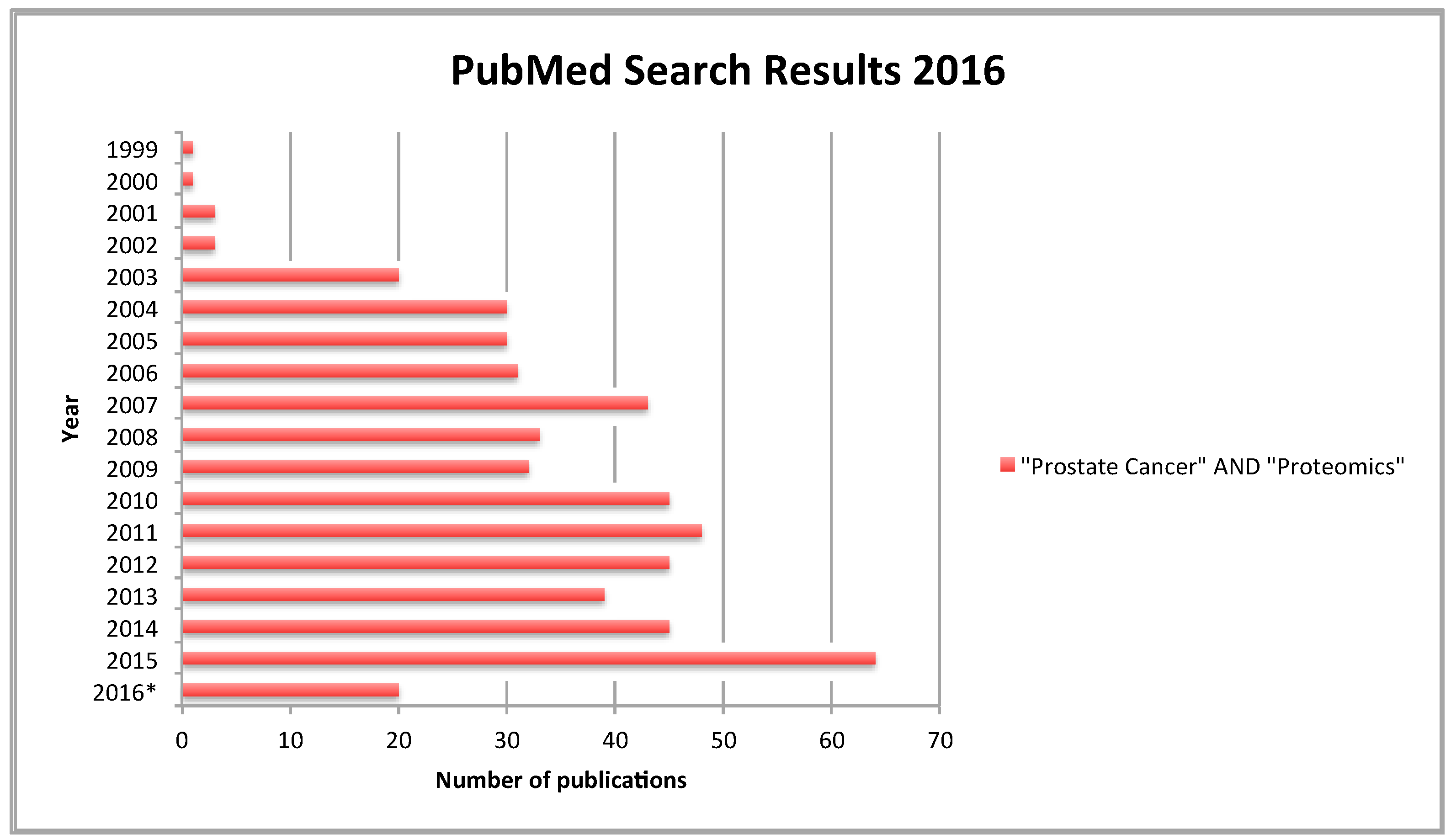{kind=link}
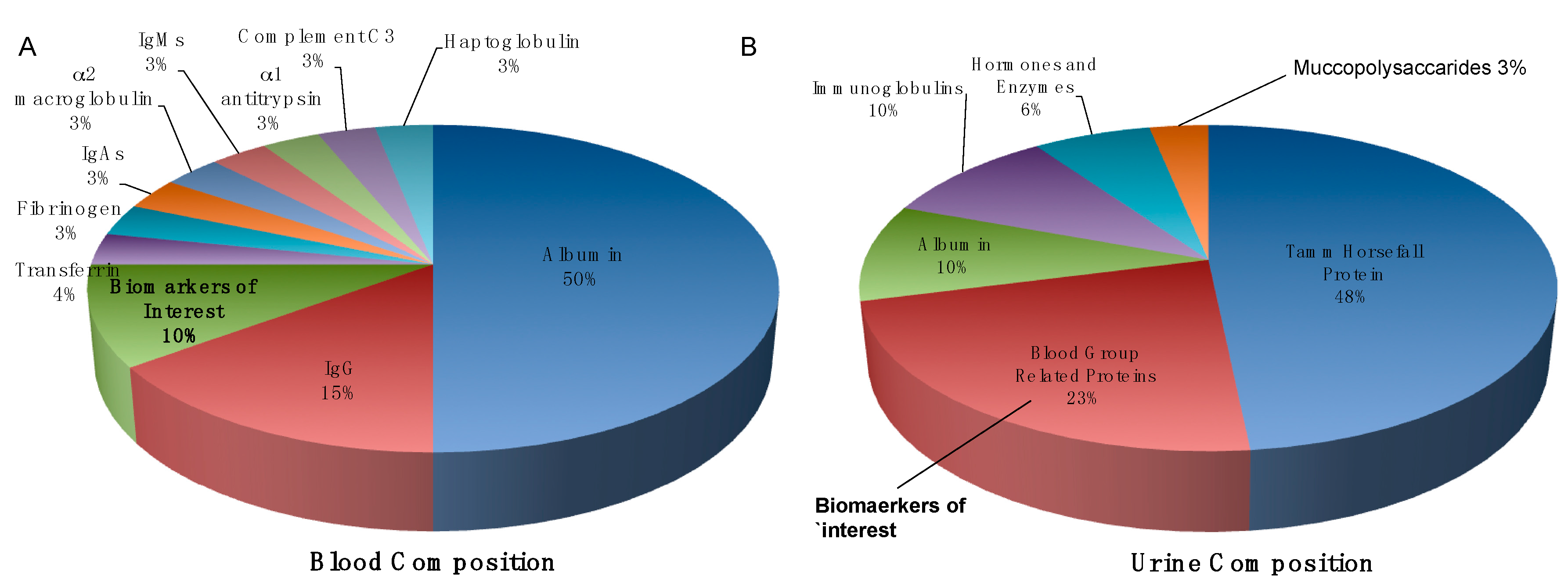{kind=link}
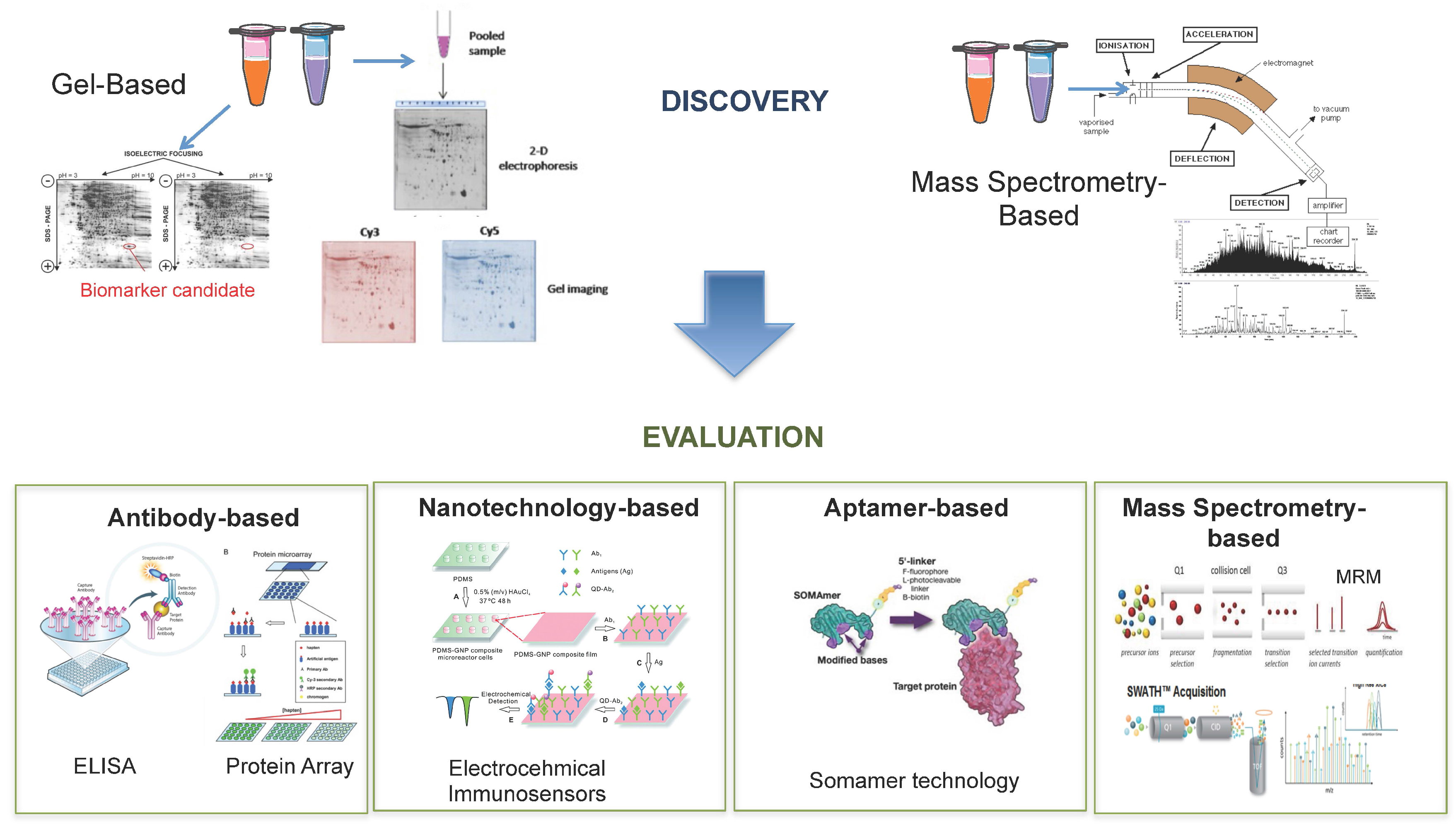{kind=link}
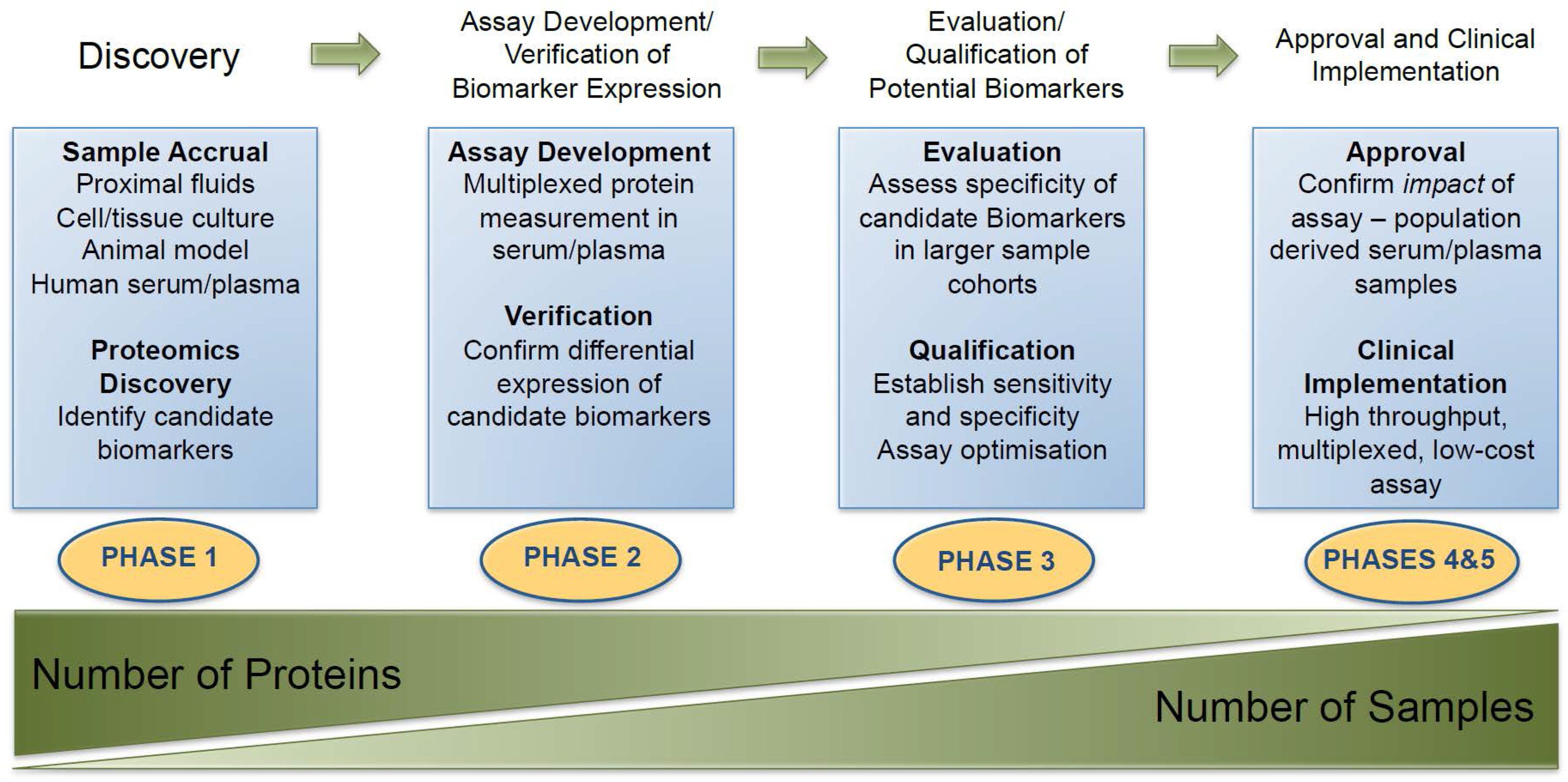{kind=link}
{kind=link}
| Category | Screening and Diagnosis |
|---|---|
| Epidemiology | 214 cases per 1000 men |
| Risk Factors | Increasing age, ethnic origin and heredity |
| Classifications | Union Internationale Contre le Cancer 2010 TNM |
| Gleason scoring recommended for grading | |
| Prostate Cancer Screening | 1. Routine screening not recommended for men ages 40–54 years |
| 2. Recommended shared decision making for men aged 55–69 years | |
| 3. Routine screening interval of ≥2 years in men who decide on screening | |
| 4. Routine screening not recommended for men ≥70 years or with life expectancy <10–15 years | |
| Diagnosis and Staging | 1. Abnormal DRE/elevated PSA (cut-off level for normal PSA not yet determined) |
| 2. Diagnosis depends on histopathologic confirmation | |
| 3. TRUS-guided systemic biopsy with ≥10 systemic, laterally directed cores | |
| 4. One set of repeat biopsies recommended in cases with persistent indication for prostate biopsy (abnormal DRE, elevated PSA, ASAP, multifocal PIN) | |
| 5. MRI to investigate anteriorly located PCa if biopsy negative and clinical indications of PCa persist | |
| Primary Local Treatment | |
| Active Surveillance | 1. >10 years life expectancy |
| 2. Stage T1–T2 | |
| 3. PSA ≤ 10 ng/mL | |
| 4. Biopsy Gleason score <6 | |
| 5. ≤2 positive biopsies | |
| 6. ≤50% cancer per biopsy | |
| Radical Prostatectomy | 1. Patients with life expectancy >10 years |
| 2. In patients with high-risk localised PCa, life expectancy >10 years, offered in multimodality setting | |
| 3. In patients with high risk locally advanced with life expectancy >10, may be offered in multimodality setting | |
| Radiation therapy (low risk) | Dose of 74–78 Gy |
| Radiation Therapy (intermediate risk) | EBRT dose of 76–78 Gy in combination with short-term (4–6 months) ADT |
| Radiation therapy (high risk, localised) | EBRT dose 76–78 Gy in combination with long-term (2–3 years) ADT |
| Transperineal brachytherapy as monotherapy | 1. Stage cT1c-T2a, NOMO 1 |
| 2. Gleason score ≤7 on at least 12 random biopsies | |
| 3. Initial PSA ≤10 ng/mL | |
| 3. ≤50% biopsy cores involved with cancer | |
| 4. A prostate volume of <50 mL | |
| 5. A good International Prostate Symptom Score (≤17) | |
| 6. No previous transurethral resection of the prostate |
| Assay | Marker Description | Assay Type | Biomaterial | FDA Approved |
|---|---|---|---|---|
| Tissue-Based | ||||
| Oncotype DX | 17 genes | RT-PCR | Fixed paraffin embedded needle core biopsy | No |
| Prolaris | 46 genes | RNA expression | Formalin-fixed paraffin embedded needle core biopsy | No |
| ProMark | 8 proteins | Immunofluorescent imaging | Formalin-fixed paraffin embedded needle core biopsy | No |
| Decipher | 22 coding and non-coding RNAs | Whole-transcriptome microarray | Formalin-fixed paraffin embedded needle core biopsy | No |
| Confirm MDx | 3 genes | Quantitative methylation-specific PCR | Prostate needle core biopsy | No |
| PCMT | mtDNA deletions | Quantitative PCR (specific for mtDNA) | Prostate needle core biopsy | No |
| Fluid-Based | ||||
| phi | PSA, fPSA, p2PSA | Multi-analyte Immunoassay | Serum | No |
| 4K score | total PSA, fPSA, intact PSA, hK2 | Multi-analyte Immunoassay | Plasma | No |
| Progensa (PCA3) | PSA and PCA3 mRNA | in vitro RNA TMA assay | Post-DRE first void urine | Only when repeat biopsy considered |
| SelectMDx | HOXC6, DLX1, KLK3 | Reverse Transcription PCR (RT-PCR) | Post-DRE first void urine | No |
| MiPS | PSA,PCA3 and TMPRSS2:ERG mRNAs | in vitro RNA TMA and Hybrid Protection Assay (HPA) | Post-DRE first void urine | No |
| Prostarix | 4 amino acids: sarcosine, alanine, glycine and glutamate | Liquid chromatography and mass spectrometry | Post-DRE urine | No |
| ExoDx Prostate (IntelliScore) | Exosomal RNA (ERG, PCA3, SPDEF) | RT-PCR | Urine | No |
| Reference | Title | Marker(s) |
|---|---|---|
| Webber, JP et al. Oncotarget 2016 [117] | Prostate stromal cell proteomics analysis discriminates normal from tumour reactive stromal phenotypes | Proteins including TAGLN, VDAC1, VDAC2, ALDH1A1 |
| Adeola, HA et al. Oncotarget 2016 [118] | Novel potential serological prostate cancer biomarkers using CT100+ cancer antigen microarray platform in a multi-cultural South African cohort. | 41 antigen biomarkers including GAGE1, ROPN1, SPANXA1, PRKCZ, MAGEB1, p53, S15A, S46A, FGFR2, COL6A1, CALM1 |
| Li, Q et al. Int. J. Oncol. 2016 [119] | Quantitative proteomic study of human prostate cancer cells with different metastatic potentials | SETDB1 |
| Ino, Y et al. Proteomics 2016 [120] | Phosphoproteome analysis demonstrates the potential role of THRAP3 phosphorylation in androgen-independent prostate cancer cell growth. | THRAP3 |
| Kazuno, S et al. Cancer Med. 2016 [121] | Glycosylation status of serum immunoglobulin G in patients with prostate diseases | Glycosylation changes in IgG |
| Stone, L. Nat. Rev. Urol. 2016 [122] | Prostate cancer: Proteomics provides a prognostic marker. | - |
| Davalieva, K et al. Prostate 2015 [123] | Proteomics analysis of malignant and benign prostate tissue by 2D DIGE/MS reveals new insights into proteins involved in prostate cancer | 9 proteins (CSNK1A1, ARID5B, LYPLA1, PSMB6, RABEP1, TALDO1, UBE2N, PPP1CB, and SERPINB1) |
| Arner, P et al. PLoS ONE 2015 [124] | Circulating carnosine dipeptidase 1 associates with weight loss and poor prognosis in gastrointestinal cancer | CNDP1 |
| Shipitsin, M et al. Br. J. Cancer 2014 [90] | Identification of proteomic biomarkers predicting prostate cancer aggressiveness and lethality despite biopsy-sampling error | 12 proteins (ACTN1, CUL2, DERL1, FUS, HSPA9, PDSS2, PLAG1, pS6, SMAD2, SMAD4, VDAC1, YBX1) |
| Bergamini, S et al. Proteome Sci. 2014 [125] | Inflammation: an important parameter in the search of prostate cancer biomarkers. | 9 Proteins (F2, C4B, C3, AZGP1, HPX, SERPINC1, SERPINF1, HP, SAA1) |
| Pallua, JD et al. J. Proteomics 2013 [126] | MALDI-MS tissue imaging identification of biliverdin reductase B overexpression in prostate cancer | BLVRB |
| Leymarie, N et al. Mol. Cell Proteom. 2013 [127] | Interlaboratory study on differential analysis of protein glycosylation by mass spectrometry: the ABRF glycoprotein research multi-institutional study 2012 | Glycoforms of PSA |
| Jiang, FN et al. PLoS ONE 2013 [128] | An integrative proteomics and interaction network-based classifier for prostate cancer diagnosis | 3 proteins (PTEN, SFPQ, HDAC1) |
| Han, ZD et al. Med. Oncol. 2012 [129] | Identification of novel serological tumor markers for human prostate cancer using integrative transcriptome and proteome analysis | IMPDH2 |
| Endoh, K et al. Prostate 2012 [130] | Identification of phosphorylated proteins involved in the oncogenesis of prostate cancer via Pin1-proteomic analysis | TFG |
| Cheng, HL et al. Proteom. Clin. Appl. 2011 [131] | Urinary CD14 as a potential biomarker for benign prostatic hyperplasia—discovery by combining MALDI-TOF-based biostatistics and ESI-MS/MS-based stable-isotope labeling | CD14 |
| Alaiya, AA et al. Int. J. Oncol. 2011 [132] | Proteomics-based signature for human benign prostate hyperplasia and prostate adenocarcinoma | 15 proteins (TPM1, PHB, KRT8, TUBB2, DES, Glycerol 3 phosphate, P4HB, EHHADH, HSPA5, KRT18, SERPINA1, CKB, HSPA8, ATP5B, ANXA4 |
| True, LD et al. Mod. Pathol. 2010 [133] | CD90/THY1 is overexpressed in prostate cancer-associated fibroblasts and could serve as a cancer biomarker | CD90/THY1 |
| Valmu, L et al. Methods Mol. Biol. 2010 [134] | Proteomic analysis of pancreatic secretory trypsin inhibitor/tumor-associated trypsin inhibitor from urine of patients with pancreatitis or prostate cancer | PSTI |
| Thoenes, L et al. J. Proteom. 2010 [135] | In vivo chemoresistance of prostate cancer in metronomic cyclophosphamide therapy | 3 proteins (TXN, CTSB, ANXA3) |
| Van der Deen, M et al. J. Cell Biochem. 2010 [136] | The cancer-related Runx2 protein enhances cell growth and responses to androgen and TGF-beta in prostate cancer cells | Runx2 |
| Sardana, G et al. J Proteome Res. 2008 [137] | Proteomic analysis of conditioned media from the PC3, LNCaP, and 22Rv1 prostate cancer cell lines: discovery and validation of candidate prostate cancer biomarkers | 4 proteins (FST, CXCL16, PTX3, SPON2) |
| Saito, S et al. Int. J. Cancer 2008 [138] | Haptoglobin-beta chain defined by monoclonal antibody RM2 as a novel serum marker for prostate cancer | RM2 |
| Ummanni, R et al. Cancer Lett. 2008 [139] | Prohibitin identified by proteomic analysis of prostate biopsies distinguishes hyperplasia and cancer | PHB |
| Huang, D et al. Cancer Epidemiol. Biomark. Prev. 2007 [140] | Quantitative fluorescence imaging analysis for cancer biomarker discovery: application to beta-catenin in archived prostate specimens | CTNNB1 |
| Ruan, W et al. Mol. Cell Proteom. 2006 [141] | Identification of clinically significant tumor antigens by selecting phage antibody library on tumor cells in situ using laser capture microdissection | ALCAM, MEMD, CD166 |
| Johansson, B et al. Prostate 2006 [142] | Proteomic comparison of prostate cancer cell lines LNCaP-FGC and LNCaP-r reveals heatshock protein 60 as a marker for prostate malignancy | HSP60 |
| Lam YW et al. Proteomics 2005 [143] | Mass profiling-directed isolation and identification of a stage-specific serologic protein biomarker of advanced prostate cancer | PF4 |
| Tissue | Body Fluids | ||||
|---|---|---|---|---|---|
| Biopsy | Needle Biopsy | Serum & Plasma | Urine | Prostatic Fluid and Seminal Plasma | |
| Advantages | Direct analysis of tumor protein expression/activation | Non-invasive collection | Non-invasive collection | Minimally invasive collection | |
| Diagnostic markers | Fast and low-cost sample preparation | High volume | Rich in prostate-derived proteins | ||
| Prognostic markers | Diagnostic markers | Rich in prostate-derived proteins | Fast and low-cost sample preparation | ||
| Most useful for patient stratification in terms of response to therapy | Prognostic markers | Fast and low-cost sample preparation | Diagnostic markers | ||
| - | - | Diagnostic markers | Prognostic markers | ||
| - | - | Prognostic markers | |||
| Limitations | Invasive collection | Low abundance of potential biomarkers | Low abundance of potential biomarkers | Low abundance of potential biomarkers | |
| Limited quantity | Dynamic concentration range | Dynamic concentration range | Dynamic concentration range | ||
| Must be snap-frozen within 30 minutes from collection | Intra and inter-patient variability in composition | Intra and inter-patient variability in composition | Intra and inter-patient variability in composition | ||
| Complicated sample preparation | - | Variability in sample collection | - | ||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tonry, C.L.; Leacy, E.; Raso, C.; Finn, S.P.; Armstrong, J.; Pennington, S.R. The Role of Proteomics in Biomarker Development for Improved Patient Diagnosis and Clinical Decision Making in Prostate Cancer. Diagnostics 2016, 6, 27. https://doi.org/10.3390/diagnostics6030027
Tonry CL, Leacy E, Raso C, Finn SP, Armstrong J, Pennington SR. The Role of Proteomics in Biomarker Development for Improved Patient Diagnosis and Clinical Decision Making in Prostate Cancer. Diagnostics. 2016; 6(3):27. https://doi.org/10.3390/diagnostics6030027
Chicago/Turabian StyleTonry, Claire L., Emma Leacy, Cinzia Raso, Stephen P. Finn, John Armstrong, and Stephen R. Pennington. 2016. "The Role of Proteomics in Biomarker Development for Improved Patient Diagnosis and Clinical Decision Making in Prostate Cancer" Diagnostics 6, no. 3: 27. https://doi.org/10.3390/diagnostics6030027
APA StyleTonry, C. L., Leacy, E., Raso, C., Finn, S. P., Armstrong, J., & Pennington, S. R. (2016). The Role of Proteomics in Biomarker Development for Improved Patient Diagnosis and Clinical Decision Making in Prostate Cancer. Diagnostics, 6(3), 27. https://doi.org/10.3390/diagnostics6030027