Catecholamines and Neurodegeneration in Parkinson’s Disease—From Diagnostic Marker to Aggregations of α-Synuclein

Abstract
:1. Introduction
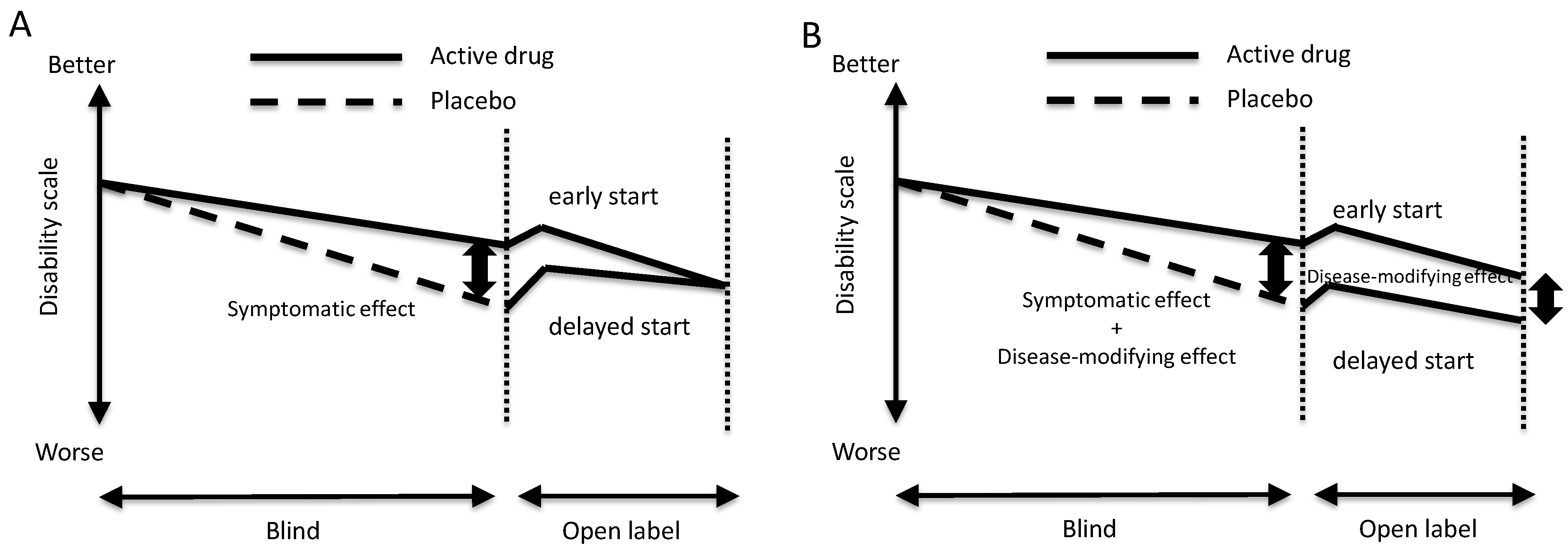
2. Possible Disease-Modifying Effects and Diagnostic Tests for Detecting Early-Stage Disease
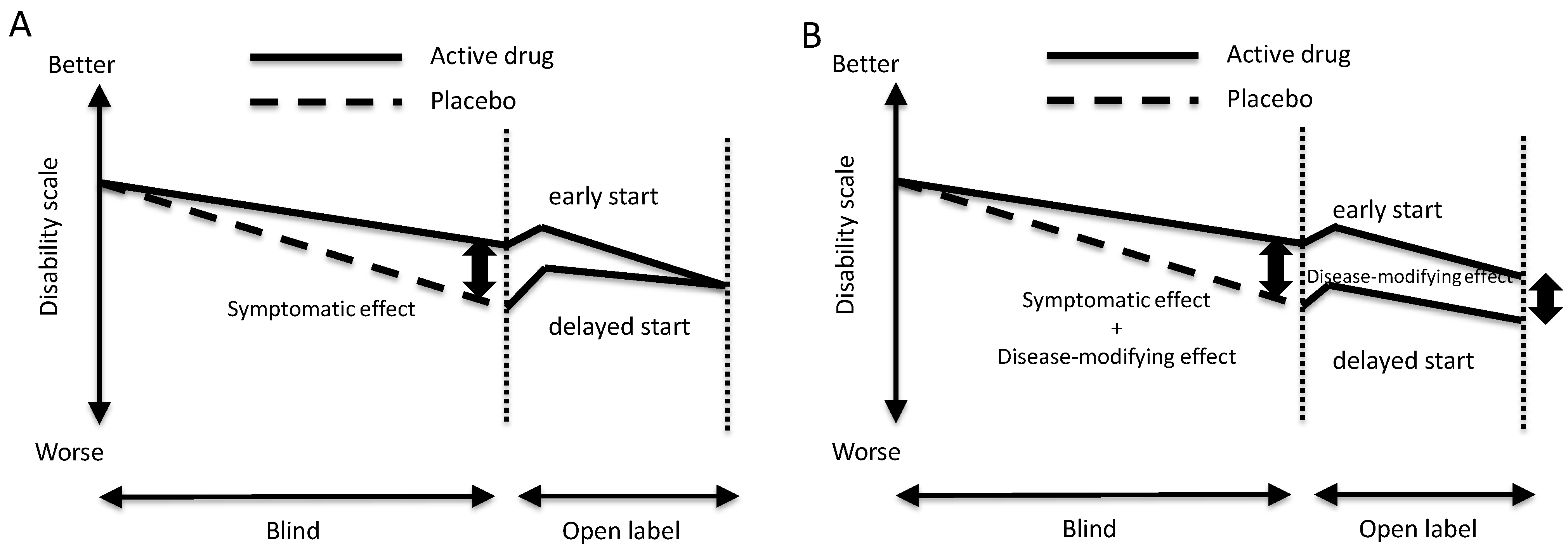
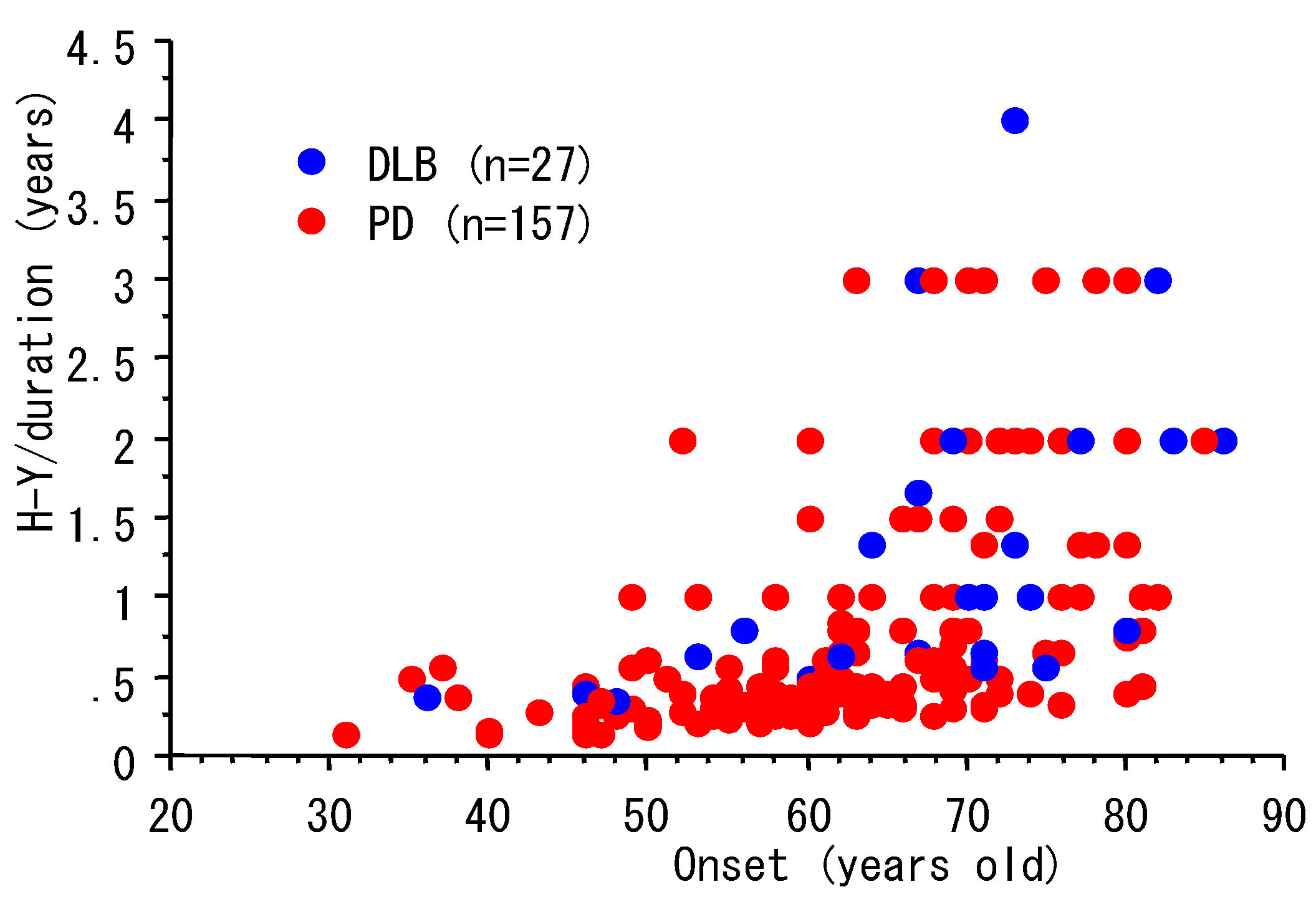
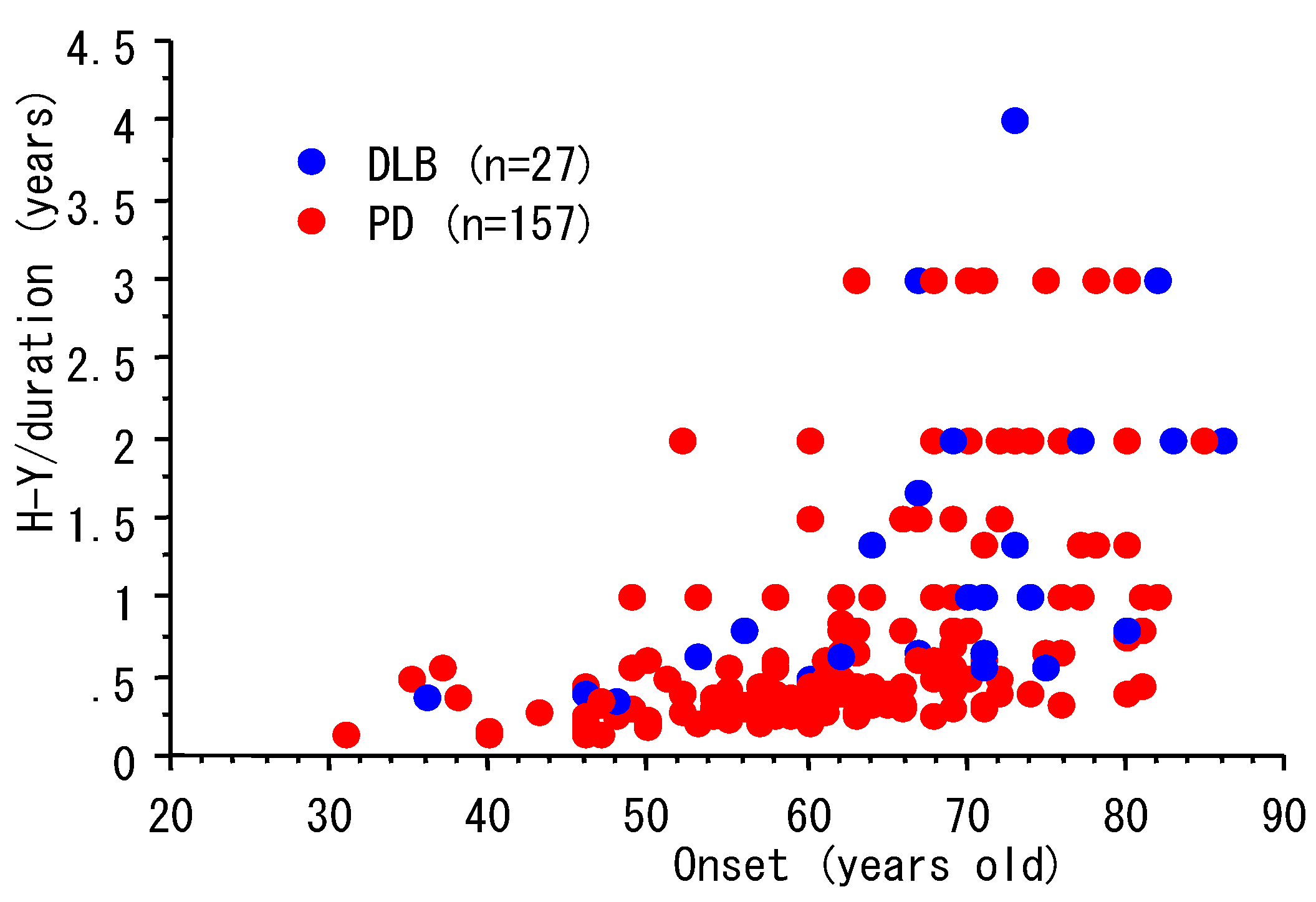
3. Progression and Age of Onset

{kind=link}
{kind=link}
{kind=link}
| n | Male, n (%) | Age, Y (mean, SD) | duration, Y (mean, SD) | |
|---|---|---|---|---|
| PD | 157 | 72 (46%) | 69.8 (8.9) | 7.4 (5.9) |
| DLB | 27 | 15 (56%) | 72.5 (9.6) | 4.8 (3.5) |
4. Olfaction and Parkinson’s Disease
5. Cardiac Denervation and Parkinson’s Disease
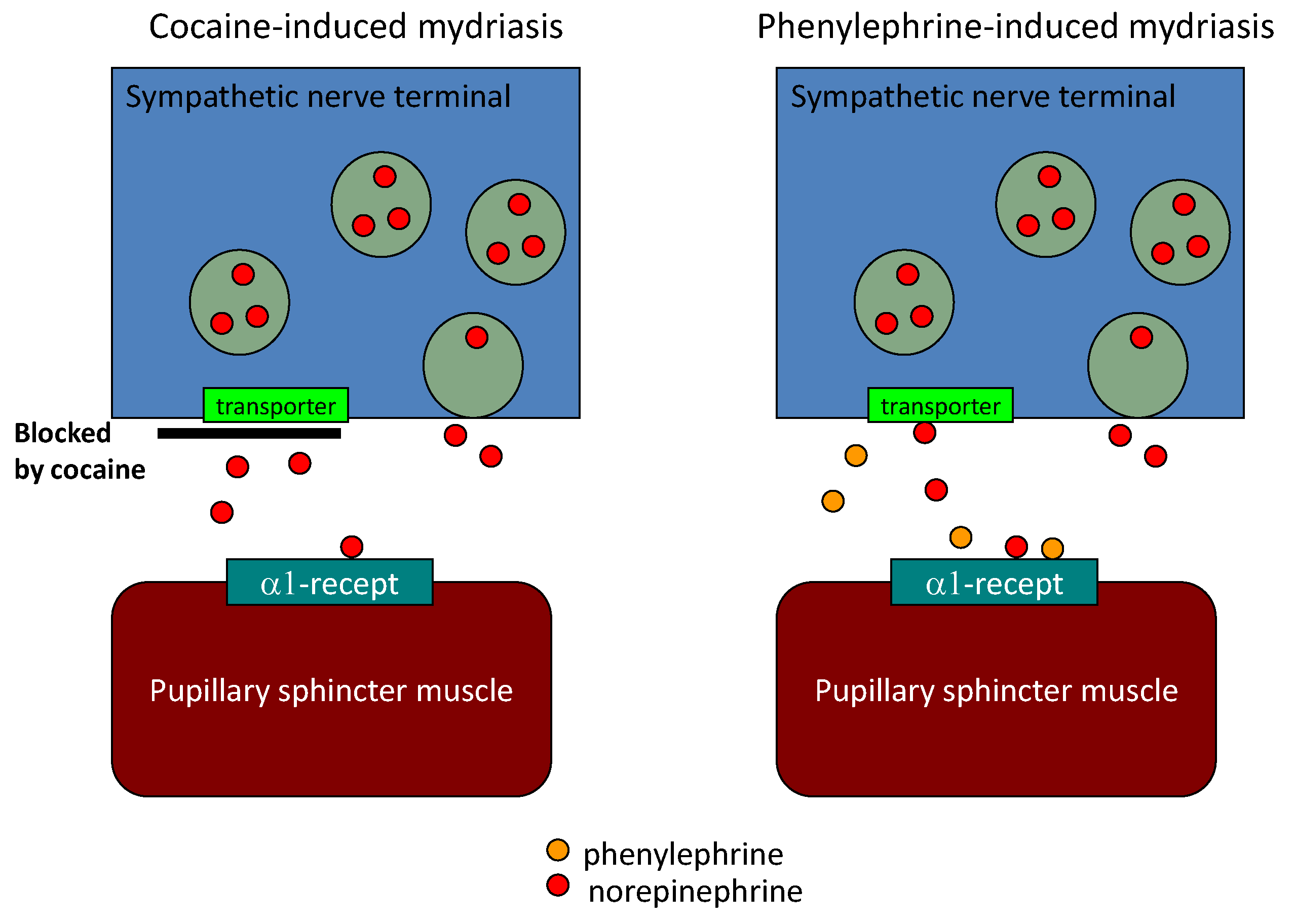
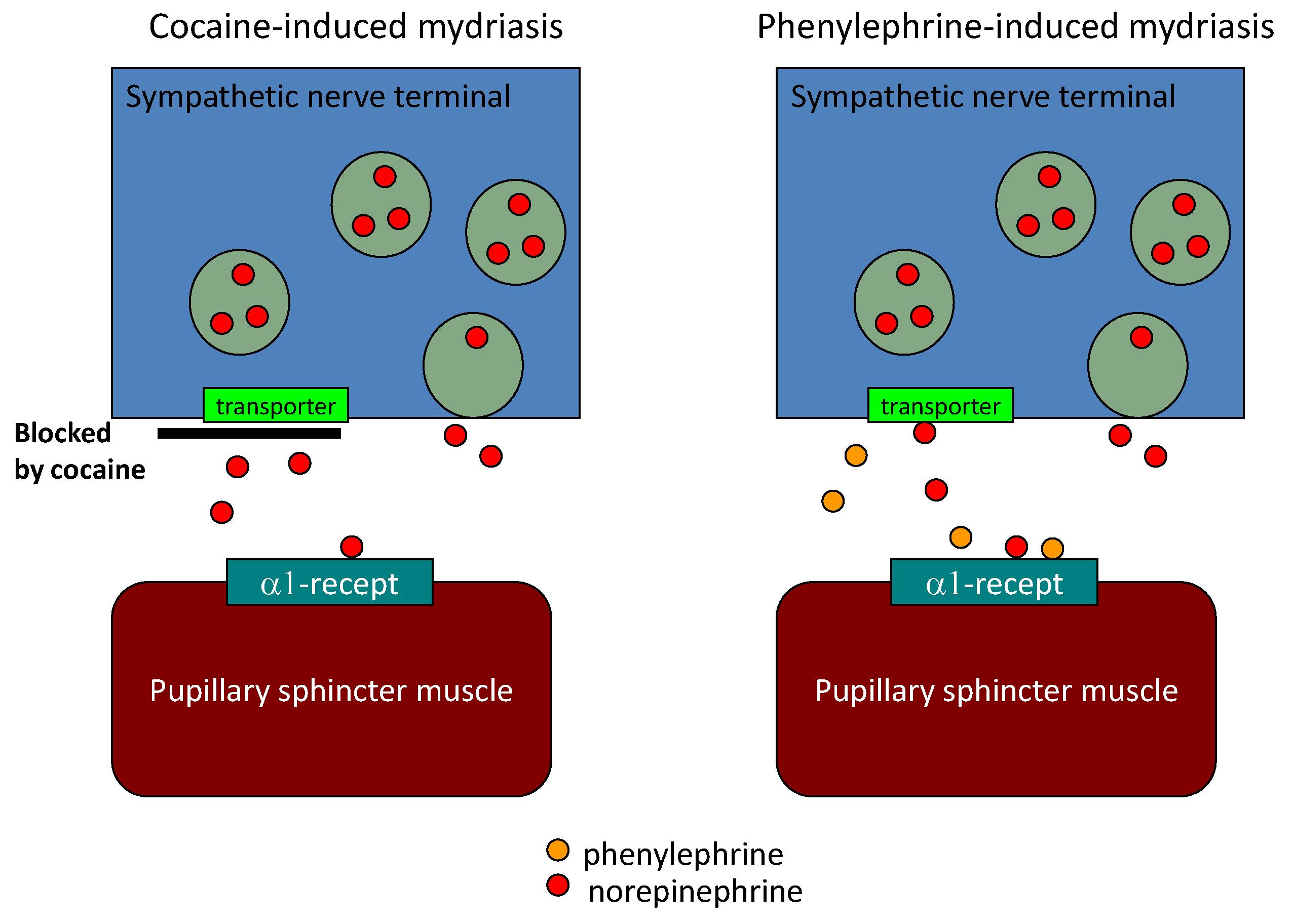
6. Diagnostic Eye Drop Test for Parkinson’s Disease
| severity | non-PD/PD | cut-off | Sn | (95% CI) | Sp | (95% CI) | AUC of ROC | (95% CI) | |
|---|---|---|---|---|---|---|---|---|---|
| Hoehn and Yahr | I | 132/12 | <2.12 | 75.0 | (42.8–94.5) | 74.2 | (65.9–81.4) | 0.80 | (0.67–0.92) |
| II | 132/45 | <1.88 | 60.0 | (44.3–74.3) | 84.1 | (76.7–89.9) | 0.81 | (0.75–0.88) | |
| III | 132/62 | <1.86 | 77.4 | (65.0–87.1) | 84.9 | (77.6–90.5) | 0.88 | (0.83–0.93) | |
| IV | 132/79 | <1.82 | 87.3 | (78.0–93.8) | 86.4 | (79.3–91.7) | 0.90 | (0.86–0.94) | |
| V | 132/15 | <1.83 | 86.7 | (59.5–98.3) | 85.6 | (78.4–91.1) | 0.93 | (0.88–0.97) | |
| Duration of illness (Y) | <3 | 132/45 | <1.81 | 64.4 | (48.8–78.1) | 86.4 | (79.3–91.7) | 0.82 | (0.75–0.89) |
| 3–5 | 132/61 | <1.86 | 71.1 | (59.2–82.9) | 84.9 | (77.6–90.4) | 0.86 | (0.81–0.92) | |
| 6–9 | 132/50 | <1.82 | 76.0 | (61.8–86.9) | 86.4 | (79.3–91.7) | 0.89 | (0.85–0.94) | |
| >9 | 132/62 | <1.81 | 88.7 | (78.1–95.3) | 86.4 | (79.3–91.7) | 0.91 | (0.87–0.95) | |
| Total | 132/253 | <1.89 | 79.8 | (74.4–84.6) | 84.1 | (76.7–89.9) | 0.88 | (0.84–0.92) |
| severity | non-PD/PD | cut-off | Sn | (95% CI) | Sp | (95% CI) | AUC of ROC | (95% CI) | |
|---|---|---|---|---|---|---|---|---|---|
| Hoehn and Yahr | I | 132/12 | <2.05 | 66.7 | (34.9–90.1) | 72.7 | (64.3–80.1) | 0.74 | (0.60–0.89) |
| II | 132/45 | <2.06 | 80.0 | (65.4–90.4) | 72.7 | (64.3–80.1) | 0.83 | (0.76–0.89) | |
| III | 132/62 | <1.78 | 83.9 | (72.3–92.0) | 86.6 | (78.4–91.1) | 0.88 | (0.83–0.93) | |
| IV | 132/79 | <1.67 | 94.9 | (87.5–98.4) | 87.1 | (80.2–92.3) | 0.91 | (0.87–0.95) | |
| V | 132/15 | <1.56 | 93.3 | (68.1–99.8) | 88.6 | (82.0–93.5) | 0.93 | (0.88–0.97) | |
| Duration of illness (Y) | <3 | 132/45 | <1.84 | 73.3 | (58.1–85.4) | 84.1 | (76.7–89.9) | 0.80 | (0.73–0.88) |
| 3–5 | 132/61 | <1.78 | 80.3 | (68.2–89.4) | 85.6 | (78.4–91.1) | 0.87 | (0.82–0.92) | |
| 6–9 | 132/50 | <1.85 | 90.0 | (78.2–96.7) | 84.1 | (76.7–89.9) | 0.90 | (0.85–0.94) | |
| >9 | 132/62 | <1.66 | 96.8 | (88.8–99.6) | 87.1 | (80.2–92.3) | 0.92 | (0.88–0.96) | |
| Total | 132/253 | <1.79 | 86.6 | (81.7–90.5) | 85.6 | (78.4–91.1) | 0.92 | (0.88–0.92) |
| Authors | Test modality | Method | LR | Sn (%) | Sp (%) |
|---|---|---|---|---|---|
| Doty et al. [38] | Olfaction (<59 y.o., male) | UPSIT | 6.1 | 91 | 85 |
| (<59 y.o., female) | UPSIT | 5.2 | 79 | 85 | |
| (60-70 y.o., male) | UPSIT | 4.4 | 81 | 82 | |
| (60-70 y.o., female) | UPSIT | 6.9 | 80 | 88 | |
| (≥71 y.o., male) | UPSIT | 6.3 | 76 | 88 | |
| (≥71 y.o., female) | UPSIT | 4.0 | 74 | 82 | |
| Double et al. [39] | Olfaction | B-SIT | 3.5 | 82 | 77 |
| Sawada et al. [15] | Pupillary sympathetic Denervation | Eye drop test | 3.9 | 80 | 79 |
| Sawada et al. [14] | Cardiac sympathetic denervation | MIBG early H/M | 5.4 | 81 | 85 |
| MIBG late H/M | 8.0 | 84 | 90 |
| Organ | Sympathetic Nerve Origin | Sympathetic Nerve Terminals |
|---|---|---|
| Heart (epicardium) | Paraspinal sympathetic ganglion | Denervated |
| Pupil (pupilary sphincter) | ||
| Thyroid gland | ||
| Liver | Celiac ganglion | Preserved |
| kidney |
7. Catecholamines and Dopaminergic Neuronal Death
8. Summary
Acknowledgements
References
- Lees, A.J.; Hardy, J.; Reves, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.; Lees, A.; Stern, M. Milestones in Parkinson’s disease—Clinical and pathologic features. Mov. Disord. 2011, 26, 1015–1021. [Google Scholar] [CrossRef]
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef]
- De Rijk, M.C.; Tzourio, C.; Breteler, M.M.; Dartigues, J.F.; Amaducci, L.; Lopez-Pousa, S.; Manubens-Bertran, J.M.; Alperovitch, A.; Rocca, W.A. Prevalence of parkinsonism and Parkinson’s disease in Europe: the Europarkinson collaborative study. European Community concerted action on the epidemiology of Parkinson’s disease. J. Neurol. Neurosurg. Psychiat. 1997, 62, 10–15. [Google Scholar] [CrossRef]
- Daniel, S.E.; Lees, A.J. Parkinson’s disease society brain bank, London: Overview and research. J. Neural Transm. 1993, 39, 165–172. [Google Scholar]
- Chaudhuri, K.R.; Martinez-Martin, P. Quatitation of non-motor symptoms in Parkinson’s disease. E. J. Neurol. 2008, 15 (Suppl. 2), 2–8. [Google Scholar] [CrossRef]
- Tan, A.; Salqado, M.; Fahn, S. Rapid eye movement sleep behavior disorder preceding Parkinson’s disease with therapeutic response to levodopa. Mov. Disord. 1996, 11, 214–216. [Google Scholar] [CrossRef]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; Tanner, C.M.; White, L.R. Pre-motor features of Parkinson’s disease: tHe Honolulu-Asia aging study experience. Parkinsonism Relat. Disord. 2012, S1, S199–S202. [Google Scholar]
- Savica, R.; Rocca, W.A.; Ahlskog, J.E. When does Parkinson disease start? Arch. Neurol. 2010, 67, 798–801. [Google Scholar] [CrossRef]
- Parkinson Study Group. A controlled, randomized, delayed-start study of rasagiline in early Parkinson disease. Arch. Neurol. 2004, 61, 561–566. [CrossRef]
- Olanow, C.W.; Rascol, O.; Hauser, R.; Feigin, P.D.; Jankovic, J.; Lang, A.; Langston, W.; Melamed, E.; Poewe, W.; Stocchi, F.; et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1268–1278. [Google Scholar] [CrossRef]
- Ponsen, M.M.; Stoffers, D.; Booij, J.; van Eck-Smit, B.L.; Wolters, E.; Berendse, H.W. Idiopathic hyposmia as a preclinical sign of Parkinson’s disease. Ann. Neurol. 2004, 56, 173–181. [Google Scholar] [CrossRef]
- Ponsen, M.M.; Stoffers, D.; Twisk, J.W.; Wolters, E.; Berendse, H.W. Hyposmia and executive dysfunction as predictors of future Parkinson’s disease: A prospective study. Mov. Disord. 2009, 24, 1060–1065. [Google Scholar] [CrossRef]
- Sawada, H.; Oeda, T.; Yamamoto, K.; Kitagawa, N.; Mizuta, E.; Hosokawa, R.; Ohba, M.; Nishio, R.; Yamakawa, K.; Takeuchi, H.; et al. Diagnostic accuracy of cardiac metaiodobenzylguanidine scintigraphy in Parkinson disease. Eur. J. Neurol. 2009, 16, 174–182. [Google Scholar] [CrossRef]
- Sawada, H.; Yamakawa, K.; Yamakado, H.; Hosokawa, R.; Ohba, M.; Miyamoto, K.; Kawamura, T.; Shimohama, S. Cocaine and phenylephrine eye drop test for Parkinson disease. JAMA 2005, 293, 932–934. [Google Scholar] [PubMed]
- Nagatsu, T.; Sawada, M. Molecular mechanism of the relation of monoamine oxidase B and its inhibitors to Parkinson’s disease: Possible implications of glial cells. J. Neural Trasm. 2006, Suppl 71, 53–65. [Google Scholar]
- Weintraub, O.; Amit, T.; Bar-Am, O.; Youdim, M.B. Rasagiline: A novel anti-Parkinsonian monoamine oxidase-B inihibitor with neuroprotective activity. Prog. Neurobiol. 2010, 92, 330–344. [Google Scholar] [CrossRef]
- Parkinson Study Group. A controlled trial of rasagiline in early Parkinson disease: The TEMPO study. Arch. Neurol. 2002, 59, 1937–1943. [CrossRef]
- Horimoto, Y.; Matsumoto, M.; Nakazawa, H.; Yuasa, H.; Morishita, M.; Akatsu, H.; Ikari, H.; Yamamoto, T.; Kosaka, K. Cognitive conditions of pathologically confirmed dementia with Lewy bodies and Parkinson’s disease with dementia. J. Neurol. Sci. 2003, 216, 105–108. [Google Scholar] [CrossRef]
- Koss, E.; Weiffenbach, J.M.; Haxby, J.V.; Friedland, R.P. Olfactory detection and identification performance are dissociated in early Alzheimer’s disease. Neurology 1988, 38, 1228–1232. [Google Scholar] [CrossRef]
- Serby, M.; Larson, P.; Kallstein, D. The nature and course of olfactory deficits in Alzheimer’s disease. Am. J. Psychiat. 1991, 148, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Doty, R.L.; Deems, D.A.; Stellar, S. Olfactory dysfunction in parkinsonism: A general deficit unrelated to neurologic signs, disease stage, or disease duration. Neurology 1988, 38, 1237–1244. [Google Scholar] [CrossRef]
- Müller, A.; Reichmann, H.; Livermore, A.; Hummel, T. Olfactory function in idiopathic Parkinson’s disease (IPD): Results from cross-sectional studies in IPD patients and long-term follow-up of de-novo IPD patients. J. Neural Transm. 2002, 109, 805–811. [Google Scholar] [CrossRef]
- Braak, H.; Ghebremedhin, E.; Rub, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef]
- Haehner, A.; Hummel, T.; Reichman, H. Olfactory loss in Parkinson’s disease. Parkinson’s Dis. 2011, 2011. [Google Scholar] [CrossRef]
- Doty, R.L. Olfactory dysfunction in Parkinson disease. Nat. Rev. Neurol. 2012, 8, 329–339. [Google Scholar] [CrossRef]
- Baba, T.; Kikuchi, A.; Hirayama, K.; Nishio, Y.; Hosokai, Y.; Kanno, S.; Hasegawa, T.; Sugeno, N.; Konno, M.; Suzuki, K.; et al. Severe olfactory dysfunction is a prodromal symptom of dementia associated with Parkinson’s disease: A 3 year longitudinal study. Brain 2012, 135, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Perl, D.P.; DeMartino, G.N.; McNaught, K.S. Lewy-body formation is aggresome-related process: A hypothesis. Lancet Neurol. 2004, 3, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef]
- Iwanaga, K.; Wakabayashi, K.; Yoshimoto, M.; Tomita, I.; Satoh, H.; Takashima, H.; Satoh, A.; Seto, M.; Tsujihata, M.; Takahashi, H. Lewy body-type degeneration in cardiac plexus in Parkinson’s and incidental Lewy body diseases. Neurology 1999, 52, 1269–1271. [Google Scholar] [CrossRef]
- Orimo, S.; Ozawa, E.; Oka, T.; Nakade, S.; Tsuchiya, K.; Yoshimoto, M.; Wakabayashi, K.; Takahashi, H. Different histopathology accounting for a decrease in myocardial MIBG uptake in PD and MSA. Neurology 2001, 57, 1140–1141. [Google Scholar] [CrossRef]
- Orimo, S.; Takahashi, A.; Uchihara, T.; Mori, F.; Kakita, A.; Wakabayashi, K.; Takahashi, H. Degeneration of cardiac sympathetic nerve begins in the early disase process of Parkinson’s disease. Brain Pathol. 2007, 17, 24–30. [Google Scholar] [CrossRef]
- Golstein, D.S.; Holmes, C.; Li, S.T.; Bruce, S.; Metman, L.V.; Cannon, R.O., III. Cardiac sympathetic denervation in Parkinson’s disease. Ann. Intern. Med. 2000, 133, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Braune, S.; Reinhardt, M.; Schnitzer, R.; Riedel, A.; Lücking, C.H. Cardiac uptake of [123I]MIBG separates Parkinson’s disease from multiple system atrophy. Neurology 1999, 53, 1020–1025. [Google Scholar] [CrossRef]
- Druschky, A.; Hilz, M.J.; Platsch, G.; Radespiel-Tröger, M.; Druschky, K.; Kuwert, T.; Neundörfer, B. Differentiation of Parkinson’s disease and multiple system atrophy in early disease stages by means of I-123-MIBG-SPECT. J. Neurol. Sci. 2000, 175, 3–12. [Google Scholar] [CrossRef]
- Takatsu, H.; Nagashima, K.; Murase, M.; Fujiwara, H.; Nishida, H.; Matsuo, H.; Watanabe, S.; Satomi, K. Differentiating Parkinson disease from multiple-system atrophy by measuring cardiac iodine-123 metaiodobenzylguanidine accumulation. JAMA 2000, 284, 44–45. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, A.; Baba, T.; Hasegawa, T.; Sugeno, N.; Konno, M.; Takeda, A. Differentiating Parkinson’s disease from multiple system atrophy by [123I] meta-iodobenzylguanidine myocardial scintigraphy and olfactory test. Parkinsonism Relat. Disord. 2011, 17, 698–700. [Google Scholar] [CrossRef]
- Doty, R.L.; Bromley, S.M.; Stern, M.B. Olfactory testing as an aid in the diagnosis of Parkinson’s disease: Development of optimal discrimination criteria. Neurodegeneration 1995, 4, 93–97. [Google Scholar] [CrossRef]
- Double, K.L.; Rowe, D.B.; Hayes, M.; Chan, D.K.; Blackie, J.; Corbett, A.; Joffe, R.; Fung, V.S.; Morris, J.; Halliday, G.M. Identifying the pattern of olfactory deficits in Parkinson disease using the brief smell identification test. Arch. Neurol. 2003, 60, 545–549. [Google Scholar] [CrossRef]
- Tipre, D.N.; Goldstein, D.S. Cardiac and extracardiac sympathetic denervation in Parkinson’s disease with orthostatic hypotension and in pure autonomic failure. J. Nucl. Med. 2005, 46, 1775–1781. [Google Scholar] [PubMed]
- Matsui, H.; Udaka, F.; Oda, M.; Tamura, A.; Kubori, T.; Nishinaka, K.; Kameyama, M. Metaiodobenzylguanidine (MIBG) uptake in Parkinson’s disease also decreases at thyroid. Ann. Nucl. Med. 2005, 19, 225–229. [Google Scholar] [CrossRef]
- Nunomura, A.; Moreira, P.I.; Lee, H.G.; Zhu, X.; Castellani, R.J.; Smith, M.A.; Perry, G. Neuronal death and survival under oxidative stress in Alzheimer and Parkinson diseases. CNS Neurol. Disord. Drug Targets 2007, 6, 411–423. [Google Scholar] [CrossRef]
- Yamamoto, N.; Sawada, H.; Izumi, Y.; Kume, T.; Katsuki, H.; Shimohama, S.; Akaike, A. Proteasome inhibition induces glutathione synthesis and protects cells from oxidative stress: Relevance to Parkinson disease. J. Biol. Chem. 2007, 282, 4364–4372. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.; Smith, M.A.; Richey, P.L.; Perry, G. Glycoxidation and oxidative stress in Parkinson disease and diffuse Lewy body disease. Brain Res. 1996, 737, 195–200. [Google Scholar] [CrossRef]
- Sakka, N.; Sawada, H.; Izumi, Y.; Kume, T.; Katsuki, H.; Kaneko, S.; Shimohama, S.; Akaike, A. Dopamine is involved in selectivity of dopaminergic neuronal death by rotenone. Neuroreport 2003, 14, 2425–2428. [Google Scholar] [CrossRef] [PubMed]
- Bisaglia, M.; Tosatto, L.; Munari, F.; Tessari, I.; de Laureto, P.P.; Mammi, S.; Bubacco, L. Dopamine quinones interact with α-synuclein to form unstructured adducts. Biochem. Biophys. Res. Commun. 2010, 394, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, K.; Izumi, Y.; Takeuchi, H.; Yamamoto, N.; Kume, T.; Akaike, A.; Takahashi, R.; Shimohama, S.; Sawada, H. Dopamine facilitates alpha-synuclein oligomerization in human neuroblastoma SH-SY5Y cells. Biochem. Biophys. Res. Commun. 2010, 391, 129–134. [Google Scholar] [CrossRef]
- Lee, H.J.; Baek, S.M.; Ho, D.H.; Suk, J.E.; Cho, E.D.; Lee, S.J. Dopamine promotes formation and secretion of non-fibrillar alpha-synuclein oligomers. Exp. Mol. Med. 2011, 43, 216–222. [Google Scholar] [CrossRef]
- Leong, S.L.; Cappai, R.; Barnham, K.J.; Pham, C.L. Modulation of alpha-synuclein aggregation by dopamine: A review. Neurochem. Res. 2009, 34, 1838–1846. [Google Scholar] [CrossRef]
- Rekas, A.; Knott, R.B.; Sokolova, A.; Barnham, K.J.; Perez, K.A.; Masters, C.L.; Drew, S.C.; Cappai, R.; Curtain, C.C.; Pham, C.L. The structure of dopamine induced alpha-synuclein oligomers. Eur. Biophys. J. 2010, 39, 1407–1419. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sawada, H.; Oeda, T.; Yamamoto, K. Catecholamines and Neurodegeneration in Parkinson’s Disease—From Diagnostic Marker to Aggregations of α-Synuclein. Diagnostics 2013, 3, 210-221. https://doi.org/10.3390/diagnostics3020210
Sawada H, Oeda T, Yamamoto K. Catecholamines and Neurodegeneration in Parkinson’s Disease—From Diagnostic Marker to Aggregations of α-Synuclein. Diagnostics. 2013; 3(2):210-221. https://doi.org/10.3390/diagnostics3020210
Chicago/Turabian StyleSawada, Hideyuki, Tomoko Oeda, and Kenji Yamamoto. 2013. "Catecholamines and Neurodegeneration in Parkinson’s Disease—From Diagnostic Marker to Aggregations of α-Synuclein" Diagnostics 3, no. 2: 210-221. https://doi.org/10.3390/diagnostics3020210
APA StyleSawada, H., Oeda, T., & Yamamoto, K. (2013). Catecholamines and Neurodegeneration in Parkinson’s Disease—From Diagnostic Marker to Aggregations of α-Synuclein. Diagnostics, 3(2), 210-221. https://doi.org/10.3390/diagnostics3020210