
A Broad Spectrum of Liver Manifestations in Common Variable Immunodeficiency Syndrome—Two Case Reports and a Literature Overview

Abstract
1. Introduction
2. Case Reports
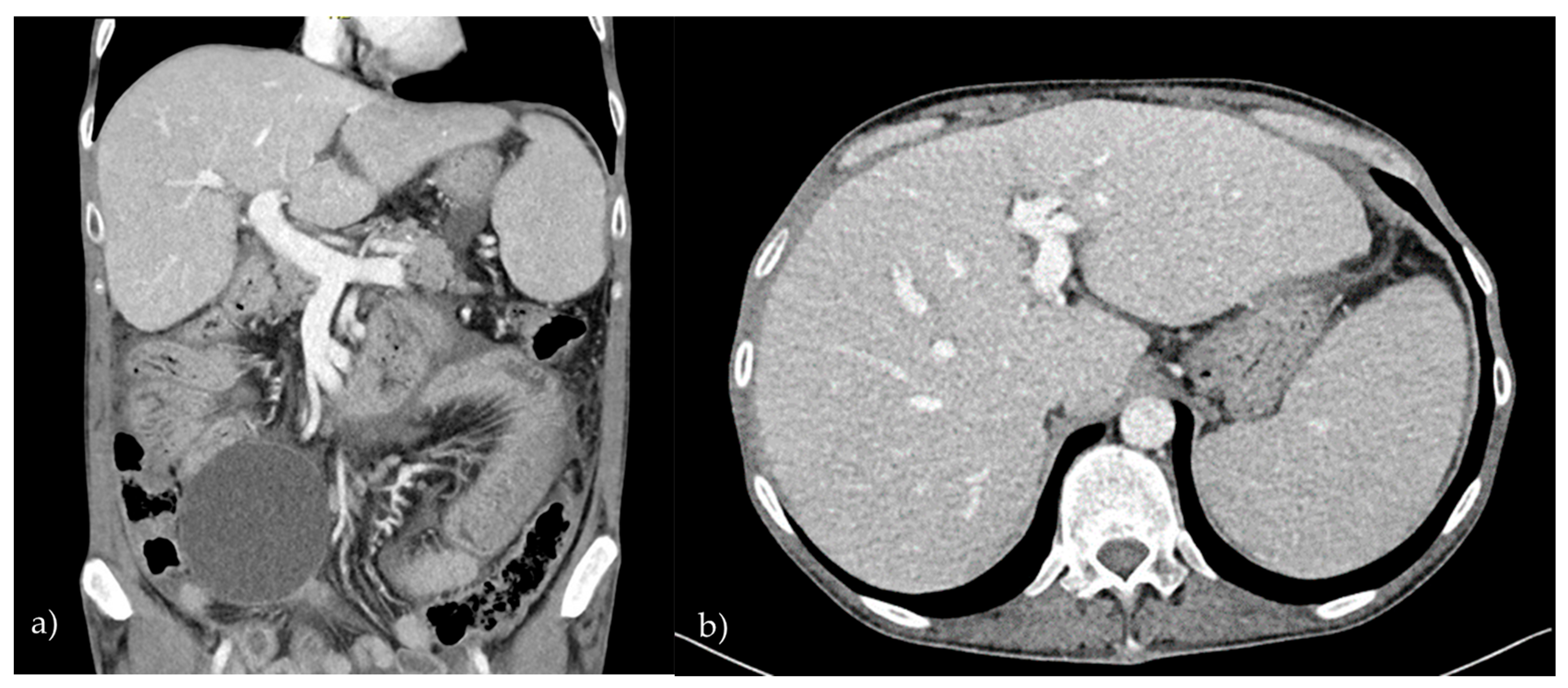
2.1. Patient 1
2.2. Patient 2
3. Discussion with Literature Overview
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIH | Autoimmune hepatitis |
| ALT | Alanine transaminase |
| ANA | Antinuclear antibodies |
| AP | Alkaline phosphatase |
| AST | Aspartate transaminase |
| CT | Contrast-enhanced computed tomography |
| CVID | Common variable immunodeficiency |
| GGT | Gamma-glutamyl transferase |
| LKM | Liver–kidney microsome |
| MRI | Magnetic resonance imaging |
| NCPH | Non-cirrhotic portal hypertension |
| NHL | Non-Hodgkin lymphoma |
| NMSC | Non-melanoma skin cancer |
| NRH | Nodular regenerative hyperplasia |
| p-ANCA | Antineutrophil cytoplasmic antibodies with a perinuclear staining pattern |
| SLA | Soluble liver antigen |
| SMAs | Smooth muscle antibodies |
References
- Bonilla, F.A.; Barlan, I.; Chapel, H.; Costa-Carvalho, B.T.; Cunningham-Rundles, C.; de la Morena, T.; Espinosa-Rosales, F.J.; Hammarström, L.; Nonoyama, S.; Quinti, I.; et al. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders. J. Allergy Clin. Immunol. Pract. 2016, 4, 38–59. [Google Scholar] [CrossRef] [PubMed]
- Ameratunga, R.; Woon, S.-T.; Gillis, D.; Koopmans, W.; Steele, R. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin. Exp. Immunol. 2013, 174, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Roskin, K.M.; Simchoni, N.; Liu, Y.; Lee, J.-Y.; Seo, K.; Hoh, R.A.; Pham, T.; Park, J.H.; Furman, D.; Dekker, C.L.; et al. IgH sequences in common variable immune deficiency reveal altered B cell development and selection. Sci. Transl. Med. 2015, 7, 302ra135. [Google Scholar] [CrossRef] [PubMed]
- Grimbacher, B.; Hutloff, A.; Schlesier, M.; Glocker, E.; Warnatz, K.; Dräger, R.; Eibel, H.; Fischer, B.; Schäffer, A.A.; Mages, H.W.; et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat. Immunol. 2003, 4, 261–268. [Google Scholar] [CrossRef]
- van Zelm, M.C.; Reisli, I.; van der Burg, M.; Castaño, D.; van Noesel, C.J.M.; van Tol, M.J.D.; Woellner, C.; Grimbacher, B.; Patiño, P.J.; van Dongen, J.J.M.; et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N. Engl. J. Med. 2006, 354, 1901–1912. [Google Scholar] [CrossRef]
- van Zelm, M.C.; Smet, J.; Adams, B.; Mascart, F.; Schandené, L.; Janssen, F.; Fester, A.; Kuo, C.-C.; Levy, S.; van Dongen, J.J.M.; et al. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J. Clin. Invest. 2010, 120, 1265–1274. [Google Scholar] [CrossRef]
- Kuijpers, T.W.; Bende, R.J.; Baars, P.A.; Grummels, A.; Derks, I.A.M.; Dolman, K.M.; Beaumont, T.; Tedder, T.F.; van Noesel, C.J.M.; Eldering, E.; et al. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J. Clin. Invest. 2010, 120, 214–222. [Google Scholar] [CrossRef]
- Abolhassani, H.; Wang, N.; Aghamohammadi, A.; Rezaei, N.; Lee, Y.N.; Frugoni, F.; Notarangelo, L.D.; Pan-Hammarström, Q.; Hammarström, L. A hypomorphic recombination-activating gene 1 (RAG1) mutation resulting in a phenotype resembling common variable immunodeficiency. J. Allergy Clin. Immunol. 2014, 134, 1375–1380. [Google Scholar] [CrossRef]
- Salzer, U.; Chapel, H.M.; Webster, A.D.B.; Pan-Hammarström, Q.; Schmitt-Graeff, A.; Schlesier, M.; Peter, H.H.; Rockstroh, J.K.; Schneider, P.; Schäffer, A.A.; et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat. Genet. 2005, 37, 820–828. [Google Scholar] [CrossRef]
- Castigli, E.; Wilson, S.A.; Garibyan, L.; Rachid, R.; Bonilla, F.; Schneider, L.; Geha, R. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat. Genet. 2005, 37, 829–834. [Google Scholar] [CrossRef]
- Martinez-Gallo, M.; Radigan, L.; Almejún, M.B.; Martínez-Pomar, N.; Matamoros, N.; Cunningham-Rundles, C. TACI mutations and impaired B-cell function in subjects with CVID and healthy heterozygotes. J. Allergy Clin. Immunol. 2013, 131, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Kutukculer, N.; Gulez, N.; Karaca, N.E.; Aksu, G.; Berdeli, A. Three different classifications, B lymphocyte subpopulations, TNFRSF13B (TACI), TNFRSF13C (BAFF-R), TNFSF13 (APRIL) gene mutations, CTLA-4 and ICOS gene polymorphisms in Turkish patients with common variable immunodeficiency. J. Clin. Immunol. 2012, 32, 1165–1179. [Google Scholar] [CrossRef]
- Azizi, G.; Rezaei, N.; Kiaee, F.; Tavakolinia, N.; Yazdani, R.; Mirshafiey, A.; Aghamohammadi, A. T-Cell Abnormalities in Common Variable Immunodeficiency. J. Investig. Allergol. Clin. Immunol. 2016, 26, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, L.; Mirshafiey, A.; Rezaei, N.; Azizi, G.; Magaji Hamid, K.; Amirzargar, A.A.; Asgardoon, M.H.; Aghamohammadi, A. The role of toll-like receptors in B-cell development and immunopathogenesis of common variable immunodeficiency. Expert Rev. Clin. Immunol. 2016, 12, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Lleo, A.; Yang, G.X.; Zhang, W.; Bowlus, C.L.; Gershwin, M.E.; Leung, P.S.C. Common variable immunodeficiency and liver involvement. Clin. Rev. Allergy Immunol. 2018, 55, 340–351. [Google Scholar] [CrossRef]
- Daza-Cajigal, V.; Segura-Guerrero, M.; López-Cueto, M.; Robles-Marhuenda, A.; Camara, C.; Gerra-Galán, T.; Gómez-de-la-Torre, R.; Avendaño-Monje, C.L.; Sánchez-Ramón, S.; Bosque-Lopez, M.J.; et al. Clinical manifestations and approach to the management of patients with common variable immunodeficiency and liver disease. Front. Immunol. 2023, 14, 1197361. [Google Scholar] [CrossRef]
- Salzer, U.; Warnatz, K.; Peter, H.H. Common variable immunodeficiency: An update. Arthritis Res. Ther. 2012, 14, 223. [Google Scholar] [CrossRef]
- Oksenhendler, E.; Gérard, L.; Fieschi, C.; Malphettes, M.; Mouillot, G.; Jaussaud, R.; Viallard, J.-F.; Gardembas, M.; Galicier, L.; Schleinitz, N.; et al. Infections in 252 patients with common variable immunodeficiency. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2008, 46, 1547–1554. [Google Scholar] [CrossRef]
- Chapel, H.; Lucas, M.; Lee, M.; Bjorkander, J.; Webster, D.; Grimbacher, B.; Fieschi, C.; Thon, V.; Abedi, M.R.; Hammarstrom, L. Common variable immunodeficiency disorders: Division into distinct clinical phenotypes. Blood. 2008, 112, 277–286. [Google Scholar] [CrossRef]
- Cunningham-Rundles, C.; Bodian, C. Common variable immunodeficiency: Clinical and immunological features of 248 patients. Clin. Immunol. 1999, 92, 34–48. [Google Scholar] [CrossRef]
- Azizi, G.; Abolhassani, H.; Asgardoon, M.H.; Alinia, T.; Yazdani, R.; Mohammadi, J.; Rezaei, N.; Ochs, H.D.; Aghamohammadi, A. Autoimmunity in common variable immunodeficiency: Epidemiology, pathophysiology and management. Expert Rev. Clin. Immunol. 2017, 13, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Baumert, L.S.; Shih, A.; Chung, R.T. Management of liver disease and portal hypertension in common variable immunodeficiency (CVID). JHEP Rep. 2023, 5, 100882. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, A.; Crescenzi, L.; Varricchi, G.; Marone, G.; Spadaro, G. Heterogeneity of Liver Disease in Common Variable Immunodeficiency Disorders. Front. Immunol. 2020, 11, 338. [Google Scholar] [CrossRef] [PubMed]
- Gathmann, B.; Mahlaoui, N.; Gérard, L.; Oksenhendler, E.; Warnatz, K.; Schulze, I.; Kindle, G.; Kuijpers, T.W.; van Beem, R.T.; Guzman, D.; et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J. Allergy Clin. Immunol. 2014, 134, 116–126. [Google Scholar] [CrossRef]
- Daniels, J.A.; Torbenson, M.; Vivekanandan, P.; Anders, R.A.; Boitnott, J.K. Hepatitis in common variable immunodeficiency. Hum. Pathol. 2009, 40, 484–488. [Google Scholar] [CrossRef]
- Resnick, E.S.; Moshier, E.L.; Godbold, J.H.; Cunningham-Rundles, C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012, 119, 1650–1657. [Google Scholar] [CrossRef]
- Azzu, V.; Elias, J.E.; Duckworth, A.; Davies, S.; Brais, R.; Kumararatne, D.S.; Gimson, A.E.S.; Griffiths, W.J.H. Liver transplantation in adults with liver disease due to common variable immunodeficiency leads to early recurrent disease and poor outcome. Liver Transplant Off. Publ. Am. Assoc. Study Liver Dis. Int. Liver Transplant Soc. 2018, 24, 171–181. [Google Scholar] [CrossRef]
- Ward, C.; Lucas, M.; Piris, J.; Collier, J.; Chapel, H. Abnormal liver function in common variable immunodeficiency disorders due to nodular regenerative hyperplasia. Clin. Exp. Immunol. 2008, 153, 331–337. [Google Scholar] [CrossRef]
- Malamut, G.; Ziol, M.; Suarez, F.; Beaugrand, M.; Viallard, J.F.; Lascaux, A.S.; Verkarre, V.; Bechade, D.; Poynard, T.; Hermine, O.; et al. Nodular regenerative hyperplasia: The main liver disease in patients with primary hypogammaglobulinemia and hepatic abnormalities. J. Hepatol. 2008, 48, 74–82. [Google Scholar] [CrossRef]
- Fuss, I.J.; Friend, J.; Yang, Z.; He, P.; Hooda, L.; Boyer, J.; Xi, L.; Kleiner, M.; Raffeld, D.E.; Heller, T.; et al. Nodular Regenerative Hyperplasia in Common Variable Immunodeficiency. J. Clin. Immunol. 2013, 33, 748–758. [Google Scholar] [CrossRef]
- Wanless, I.R. Micronodular Transpormation (Nodular Regenerative Hyperplasia) of the Liver: A Report of 64 Cases Among 2,500 Autopsies and A New Classification of Benign Hepatocellular Nodules. Hepatology. 1990, 11, 787. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, N.; Gulati, N.; Rastogi, A.; Chougule, A.; Bihari, C.; Jindal, A. Nodular regenerative hyperplasia—An under-recognized vascular disorder of liver. Pathol. Res. Pract. 2020, 216, 152833. [Google Scholar] [CrossRef] [PubMed]
- Lima, F.M.S.; Toledo-Barros, M.; Alves, V.A.F.; Duarte, M.I.S.; Takakura, C.; Bernardes-Silva, C.F.; Marinho, A.K.B.; Grecco, O.; Kalil, J.; Kokron, C.M. Liver disease accompanied by enteropathy in common variable immunodeficiency: Common pathophysiological mechanisms. Front. Immunol. 2022, 13, 933463. [Google Scholar] [CrossRef] [PubMed]
- Ziol, M.; Poirel, H.; Kountchou, G.N.; Boyer, O.; Mohand, D.; Mouthon, L.; Tepper, M.; Guillet, J.G.; Guettier, C.; Raphael, M.; et al. Intrasinusoidal cytotoxic CD8+ T cells in nodular regenerative hyperplasia of the liver. Hum. Pathol. 2004, 35, 1241–1251. [Google Scholar] [CrossRef]
- Perreau, M.; Vigano, S.; Bellanger, F.; Pellaton, C.; Buss, G.; Comte, D.; Roger, T.; Lacabaratz, C.; Bart, P.A.; Levy, Y.; et al. Exhaustion of bacteria-specific CD4 T cells and microbial translocation in common variable immunodeficiency disorders. J. Exp. Med. 2014, 211, 2033. [Google Scholar] [CrossRef]
- Ho, H.; Radigan, L.; Bongers, G.; El-Shamy, A.; Cunningham-Rundles, C. Circulating bioactive bacterial DNA is associated with immune activation and complications in common variable immunodeficiency. JCI Insight 2021, 6, e144777. [Google Scholar] [CrossRef]
- Hartleb, M.; Gutkowski, K.; Milkiewicz, P. Nodular regenerative hyperplasia: Evolving concepts on underdiagnosed cause of portal hypertension. World J. Gastroenterol. WJG 2011, 17, 1400. [Google Scholar] [CrossRef]
- Reshamwala, P.A.; Kleiner, D.E.; Heller, T. Nodular regenerative hyperplasia: Not all nodules are created equal. Hepatology 2006, 44, 7–14. [Google Scholar] [CrossRef]
- Jolles, S. The variable in common variable immunodeficiency: A disease of complex phenotypes. J. Allergy Clin. Immunol. Pract. 2013, 1, 545–556, quiz 557. [Google Scholar] [CrossRef]
- Ardeniz, Ö.; Cunningham-Rundles, C. Granulomatous disease in common variable immunodeficiency. Clin. Immunol. 2009, 133, 198–207. [Google Scholar] [CrossRef]
- Fasano, M.B.; Sullivan, K.E.; Sarpong, S.B.; Wood, R.A.; Jones, S.M.; Johns, C.J.; Lederman, H.M.; Bykowsky, M.J.; Greene, J.M.; Winkelstein, J.A. Sarcoidosis and common variable immunodeficiency. Report of 8 cases and review of the literature. Medicine 1996, 75, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Uzzan, M.; Ko, H.M.; Mehandru, S.; Cunningham-Rundles, C. Gastrointestinal Disorders Associated with Common Variable Immune Deficiency (CVID) and Chronic Granulomatous Disease (CGD). Curr. Gastroenterol. Rep. 2016, 18, 17. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, A.; Nappi, L.; Crescenzi, L.; D’Armiento, F.P.; Genovese, A.; Spadaro, G. Chronic Diarrhea in Common Variable Immunodeficiency: A Case Series and Review of the Literature. J. Clin. Immunol. 2018, 38, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Malamut, G.; Verkarre, V.; Suarez, F.; Viallard, J.F.; Lascaux, A.S.; Cosnes, J.; Bouhnik, Y.; Lambotte, O.; Béchade, D.; Ziol, M.; et al. The enteropathy associated with common variable immunodeficiency: The delineated frontiers with celiac disease. Am. J. Gastroenterol. 2010, 105, 2262–2275. [Google Scholar] [CrossRef]
- Mannon, P.J.; Fuss, I.J.; Dill, S.; Friend, J.; Groden, C.; Hornung, R.; Yang, Z.; Yi, C.; Quezado, M.; Brown, M.; et al. Excess IL-12 but not IL-23 accompanies the inflammatory bowel disease associated with common variable immunodeficiency. Gastroenterolog. 2006, 131, 748–756. [Google Scholar] [CrossRef]
- Agarwal, S.; Cunningham-Rundles, C. Gastrointestinal Manifestations and Complications of Primary Immunodeficiency Disorders. Immunol. Allergy Clin. N. Am. 2019, 39, 81–94. [Google Scholar] [CrossRef]
- Bruns, L.; Panagiota, V.; von Hardenberg, S.; Schmidt, G.; Adriawan, I.R.; Sogka, E.; Hirsch, S.; Ahrenstorf, G.; Witte, T.; Schmidt, R.E.; et al. Common Variable Immunodeficiency-Associated Cancers: The Role of Clinical Phenotypes, Immunological and Genetic Factors. Front. Immunol. 2022, 13, 742530. [Google Scholar] [CrossRef]
- Pulvirenti, F.; Pecoraro, A.; Cinetto, F.; Milito, C.; Valente, M.; Santangeli, E.; Crescenzi, L.; Rizzo, F.; Tabolli, S.; Spadaro, G.; et al. Gastric Cancer Is the Leading Cause of Death in Italian Adult Patients with Common Variable Immunodeficiency. Front. Immunol. 2018, 9, 2546. [Google Scholar] [CrossRef]
- Kiaee, F.; Azizi, G.; Rafiemanesh, H.; Zainaldain, H.; Sadaat Rizvi, F.; Alizadeh, M.; Jamee, M.; Mohammadi, S.; Habibi, S.; Sharifi, L.; et al. Malignancy in common variable immunodeficiency: A systematic review and meta-analysis. Expert Rev. Clin. Immunol. 2019, 15, 1105–1113. [Google Scholar] [CrossRef]
- Cunningham-Rundles, C.; Cooper, D.L.; Duffy, T.P.; Strauchen, J. Lymphomas of mucosal-associated lymphoid tissue in common variable immunodeficiency. Am. J. Hematol. 2002, 69, 171–178. [Google Scholar] [CrossRef]
- Tak Manesh, A.; Azizi, G.; Heydari, A.; Kiaee, F.; Shaghaghi, M.; Hossein-Khannazer, N.; Yazdani, R.; Abolhassani, H.; Aghamohammadi, A. Epidemiology and pathophysiology of malignancy in common variable immunodeficiency? Allergol. Immunopathol. 2017, 45, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Tabarsi, P.; Mansouri, D.; Khosravi, A.; Garssen, J.; Velayati, A.; Adcock, I.M. Cancers Related to Immunodeficiencies: Update and Perspectives. Front. Immunol. 2016, 7, 365. [Google Scholar] [CrossRef] [PubMed]
- De Petris, G.; Dhungel, B.M.; Chen, L.; Chang, Y.-H.H. Gastric adenocarcinoma in common variable immunodeficiency: Features of cancer and associated gastritis may be characteristic of the condition. Int. J. Surg. Pathol. 2014, 22, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Sumazaki, R.; Matsubara, T.; Aoki, T.; Nagai, Y.; Shibasaki, M.; Takita. Rapidly progressive hepatitis C in a patient with common variable immunodeficiency. Eur. J. Pediatr. 1996, 155, 532–534. [Google Scholar] [CrossRef]
- Gandhi, K.; Parikh, P.; Aronow, W.S.; Desai, H.; Amin, H.; Sharma, M.; Rubinstein, A. A Case of Explosive Progression of Hepatocellular Carcinoma in a Patient with Common Variable Immunodeficiency (CVID). J. Gastrointest. Cancer. 2010, 41, 281–284. [Google Scholar] [CrossRef]
- Mahdavinia, M.; Mirsaeidi, M.; Bishehsari, F.; McGrath, K. Primary sclerosing cholangitis in common variable immune deficiency. Allergol. Int. Off. J. Jpn. Soc. Allergol. 2015, 64, 187. [Google Scholar] [CrossRef]
- Macura-Biegun, A.; Kowalczyk, D. Common variable immunodeficiency concomitant with liver cirrhosis—case report. Przegl. Lek. 2002, 59, 472–473. [Google Scholar]
- Tinazzi, E.; Osti, N.; Beri, R.; Argentino, G.; Veneri, D.; Dima, F.; Bason, C.; Jadav, G.; Dolcino, M.; Puccetti, A.; et al. Pathogenesis of immune thrombocytopenia in common variable immunodeficiency. Autoimmun. Rev. 2020, 19, 102616. [Google Scholar] [CrossRef]
- Alvaro, D.; Caporaso, N.; Giannini, E.G.; Iacobellis, A.; Morelli, M.; Toniutto, P.; Violi, F. Procedure-related bleeding risk in patients with cirrhosis and severe thrombocytopenia. Eur. J. Clin. Investig. 2021, 51, e13508. [Google Scholar] [CrossRef]
- Jones, T.P.W.; Buckland, M.; Breuer, J.; Lowe, D.M. Viral infection in primary antibody deficiency syndromes. Rev. Med. Virol. 2019, 29, e2049. [Google Scholar] [CrossRef]
- Bjøro, K.; Frøland, S.S.; Yun, Z.; Samdal, H.H.; Haaland, T. Hepatitis C infection in patients with primary hypogammaglobulinemia after treatment with contaminated immune globulin. N. Engl. J. Med. 1994, 331, 1607–1611. [Google Scholar] [CrossRef] [PubMed]
- Razvi, S.; Schneider, L.; Jonas, M.M.; Cunningham-Rundles, C. Outcome of intravenous immunoglobulin-transmitted hepatitis C virus infection in primary immunodeficiency. Clin. Immunol. 2001, 101, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Myneedu, K.; Chavez, L.O.; Sussman, N.L.; Michael, M.; Padilla, A.; Zuckerman, M.J. Autoimmune Hepatitis in a Patient with Common Variable Immunodeficiency. ACG Case Rep. J. 2021, 8, e00547. [Google Scholar] [CrossRef] [PubMed]
- de Franchis, R.; Bosch, J.; Garcia-Tsao, G.; Reiberger, T.; Ripoll, C.; Baveno VII Faculty. Corrigendum to ‘Baveno VII—Renewing consensus in portal hypertension’. J. Hepatol. 2022, 77, 959–974. [Google Scholar] [CrossRef]
- Boike, J.R.; Thornburg, B.G.; Asrani, S.K.; Fallon, M.B.; Fortune, B.E.; Izzy, M.J.; Verna, E.C.; Abraldes, J.G.; Allegretti, A.S.; Bajaj, J.S.; et al. North American Practice-Based Recommendations for Transjugular Intrahepatic Portosystemic Shunts in Portal Hypertension. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2022, 20, 1636–1662.e36. [Google Scholar] [CrossRef]
- Vizzutti, F.; Schepis, F.; Arena, U.; Fanelli, F.; Gitto, S.; Aspite, S.; Turco, L.; Dragoni, G.; Laffi, G.; Marra, F. Transjugular intrahepatic portosystemic shunt (TIPS): Current indications and strategies to improve the outcomes. Intern. Emerg. Med. 2020, 15, 37–48. [Google Scholar] [CrossRef]
- García-Pagán, J.C.; Saffo, S.; Mandorfer, M.; Garcia-Tsao, G. Where does TIPS fit in the management of patients with cirrhosis? JHEP Rep. Innov. Hepatol. 2020, 2, 100122. [Google Scholar] [CrossRef]
- Smith, M.S.; Webster, A.D.; Dhillon, A.P.; Dusheiko, G.; Boulton, R.; Savage, K.; Rolles, K.; Burroughs, A.K. Orthotopic liver transplantation for chronic hepatitis in two patients with common variable immunodeficiency. Gastroenterolog 1995, 108, 879–884. [Google Scholar] [CrossRef]
- Montalti, R.; Mocchegiani, F.; Vincenzi, P.; Svegliati Baroni, G.; Nicolini, D.; Vivarelli, M. Liver transplantation in patients with common variable immunodeficiency: A report of two cases. Ann. Transplant. 2014, 19, 541–544. [Google Scholar] [CrossRef]
- Tranah, T.H.; Cargill, Z.; Tavabie, O.; Mufti, G.; Aluvihare, V.; Sanchez-Fueyo, A.; Heaton, N.; Miquel, R.; Suddle, A. Challenges in liver transplantation for common variable immunodeficiency-related liver disease: A case series and systematic review. J. Liver Transplant 2021, 4, 100038. [Google Scholar] [CrossRef]
- Jørgensen, S.F.; Macpherson, M.E.; Bjøro, K.; Karlsen, T.H.; Reims, H.M.; Grzyb, K.; Fosby, B.; Fevang, B.; Aukrust, P.; Nordøy, I. Liver transplantation in patients with primary antibody deficiency. J. Allergy Clin. Immunol. 2017, 139, 1708–1710.e2. [Google Scholar] [CrossRef]
- Murakawa, Y.; Miyagawa-Hayashino, A.; Ogura, Y.; Egawa, H.; Okamoto, S.; Soejima, Y.; Kurosawa, M.; Sumiyoshi, S.; Uemoto, S.; Haga, H. Liver transplantation for severe hepatitis in patients with common variable immunodeficiency. Pediatr. Transplant. 2012, 16, E210–E216. [Google Scholar] [CrossRef]

{kind=link}
| Liver Manifestation | Pathology | Clinical Features | Diagnostic Approach |
|---|---|---|---|
| Nodular Regenerative Hyperplasia (NRH) | No fibrosis, small parenchymal nodules, impaired blood flow (microvascular thrombosis). | Hepatomegaly; Ascites; Non-cirrhotic portal hypertension (in advanced cases); Splenomegaly. | Elevated liver enzymes (AP, GGT). Imaging: Abdominal ultrasound (poor detection of small nodules), CT/MRI, liver biopsy. |
| Autoimmune Hepatitis (AIH) | Interface hepatitis, periportal fibrosis, bridging fibrosis in severe cases. | Elevated AST, ALT, GGT; Fatigue; Jaundice. | Immunoserology: Autoantibodies ANA, Anti-SMAs, Anti-LKM, Anti-SLA, p-ANCA. Liver biopsy: Interface hepatitis, lymphocytic infiltration. Exclude viral hepatitis and other causes of hepatitis. |
| Granulomatous Hepatitis | Non-caseating granulomas associated with T-cell dysregulation. | Chronic inflammation; Fatigue; Weight loss; Jaundice; Hepatomegaly; Splenomegaly. | Liver biopsy: Granulomas, often sarcoid like. Imaging: Transabdominal ultrasound, elastography, CT/MRI to detect hepatomegaly and fibrosis. |
| Portal Hypertension (Secondary to NRH) | Compression of portal veins by regenerative nodules, altered blood flow, increased resistance. | Splenomegaly; Neutropenia; Thrombocytopenia; Ascites; Varices (esophageal, gastric). | Imaging: Doppler ultrasound, CT, MRI for portal system evaluation. Biopsy for confirmation of underlying NRH or AIH. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Supovec, E.; Drnovšek, J. A Broad Spectrum of Liver Manifestations in Common Variable Immunodeficiency Syndrome—Two Case Reports and a Literature Overview. Diagnostics 2025, 15, 1659. https://doi.org/10.3390/diagnostics15131659
Supovec E, Drnovšek J. A Broad Spectrum of Liver Manifestations in Common Variable Immunodeficiency Syndrome—Two Case Reports and a Literature Overview. Diagnostics. 2025; 15(13):1659. https://doi.org/10.3390/diagnostics15131659
Chicago/Turabian StyleSupovec, Eva, and Jan Drnovšek. 2025. "A Broad Spectrum of Liver Manifestations in Common Variable Immunodeficiency Syndrome—Two Case Reports and a Literature Overview" Diagnostics 15, no. 13: 1659. https://doi.org/10.3390/diagnostics15131659
APA StyleSupovec, E., & Drnovšek, J. (2025). A Broad Spectrum of Liver Manifestations in Common Variable Immunodeficiency Syndrome—Two Case Reports and a Literature Overview. Diagnostics, 15(13), 1659. https://doi.org/10.3390/diagnostics15131659