
Broad Electrocardiogram Syndromes Spectrum: From Common Emergencies to Particular Electrical Heart Disorders—Part II

 ,
,  ,
, 

 , , ,
, , ,  , ,
, ,  and
and
Abstract
1. Introduction
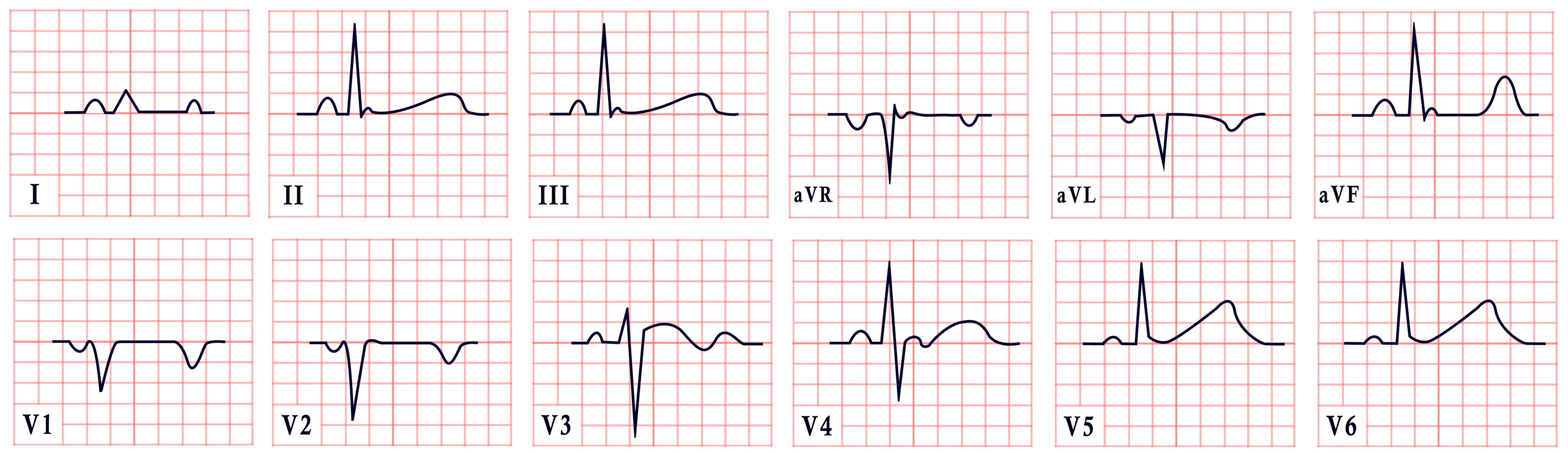
2. Long QT Syndrome
3. Jervell and Lange-Nielsen Syndrome
- Prolonged QTc > 500 ms.
- Congenital bilateral sensorineural hearing loss.
- Family history of consanguinity or SCD.
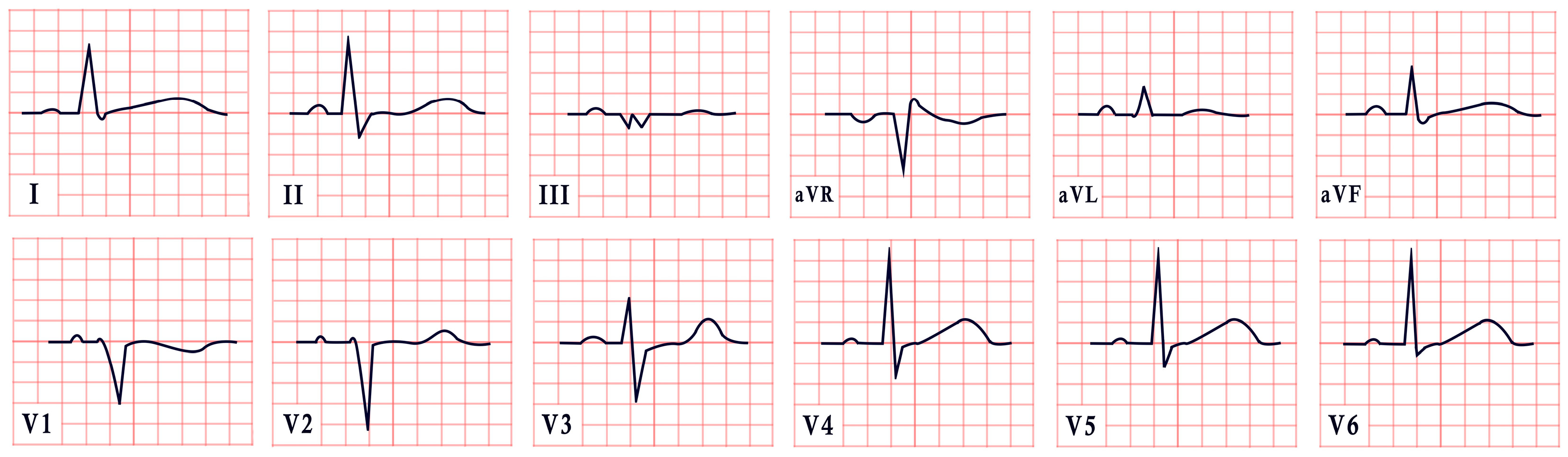
4. Romano–Ward Syndrome
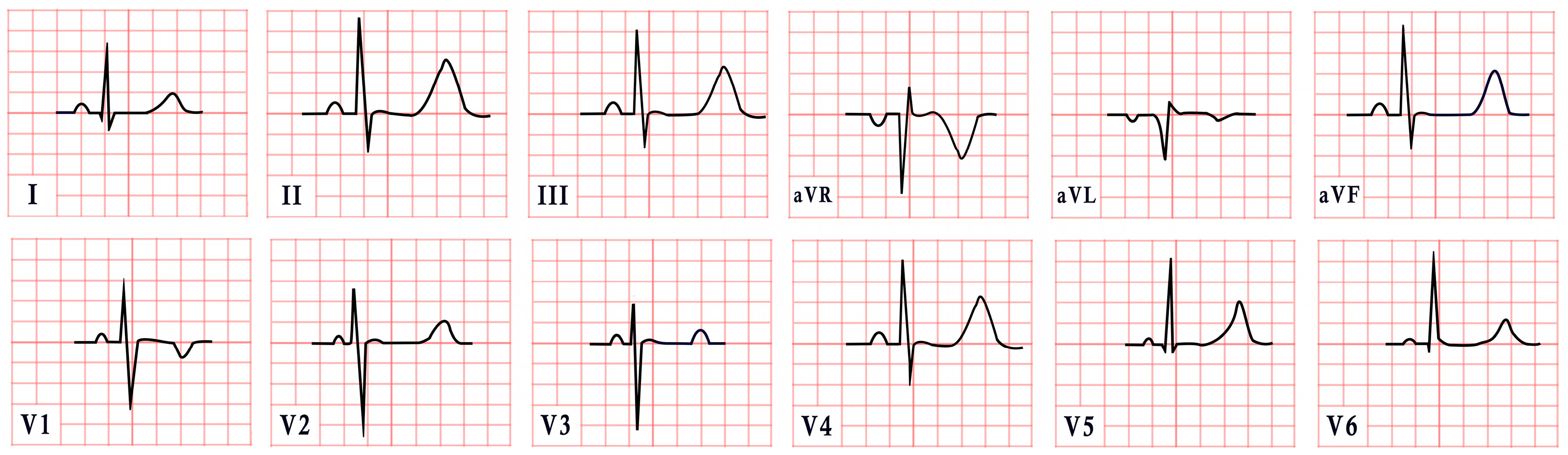
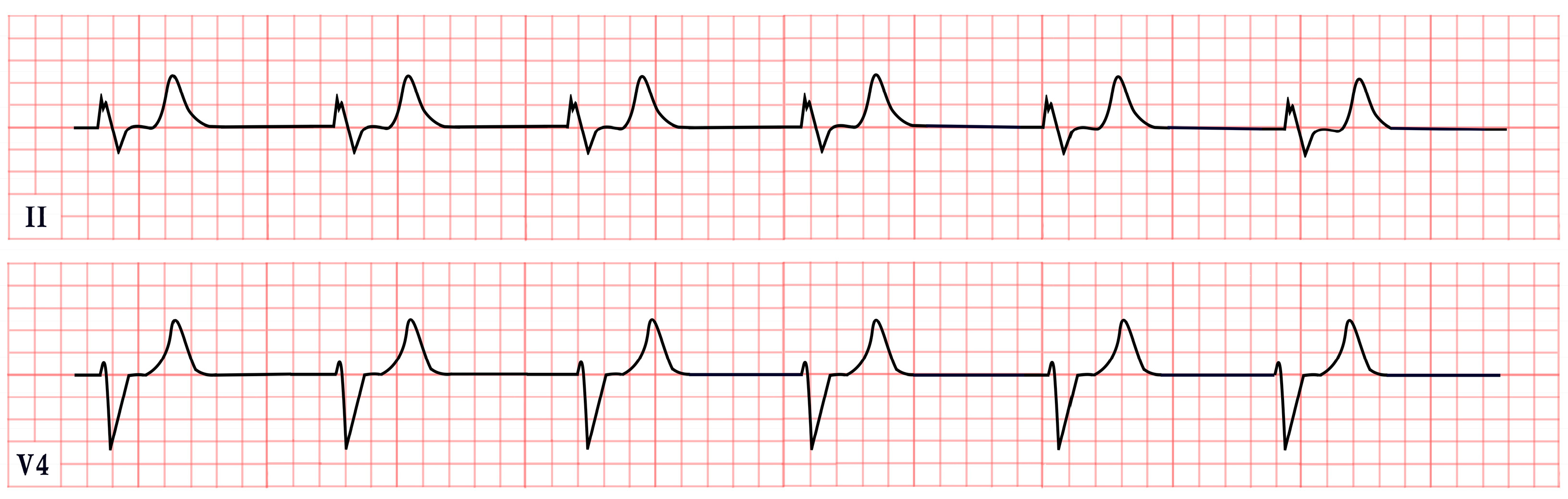
5. Andersen–Tawil Syndrome
- Periodic paralysis.
- Cardiac manifestations such as ventricular arrhythmias (e.g., frequent ventricular ectopic beats or ventricular tachycardia), a prolonged QTc, and/or a prominent U wave.
- Dysmorphic features, with at least two of the following: low-set ears, wide-set eyes, a small mandible, fifth-digit clinodactyly, or syndactyly.
- A family history of confirmed ATS [55].
6. Timothy Syndrome
- Characteristic craniofacial features (e.g., receding hairline, flat nasal bridge, thin upper lip) and, in TS1, syndactyly.
- ECG findings, particularly QTc prolongation (>480 ms in TS1, often > 600 ms in TS2), and 2:1 AV block, history of lethal ventricular arrhythmias, or cardiac arrest.
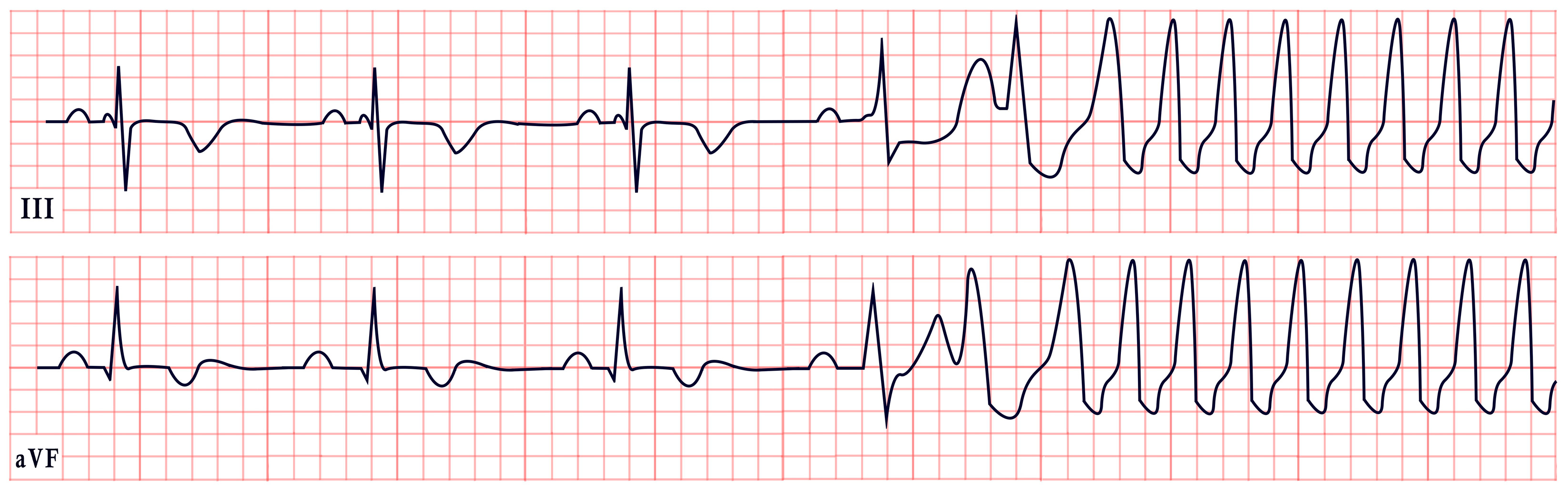
7. Short QT Syndrome
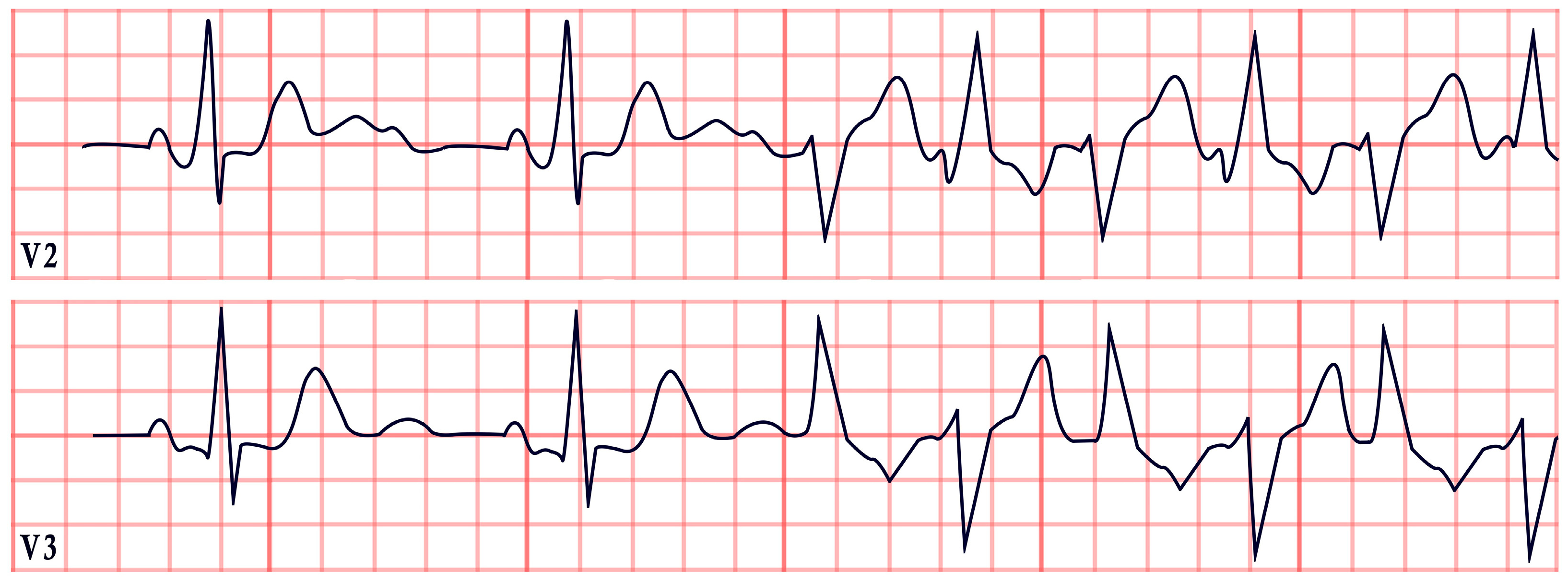
8. Twiddler’s Syndrome
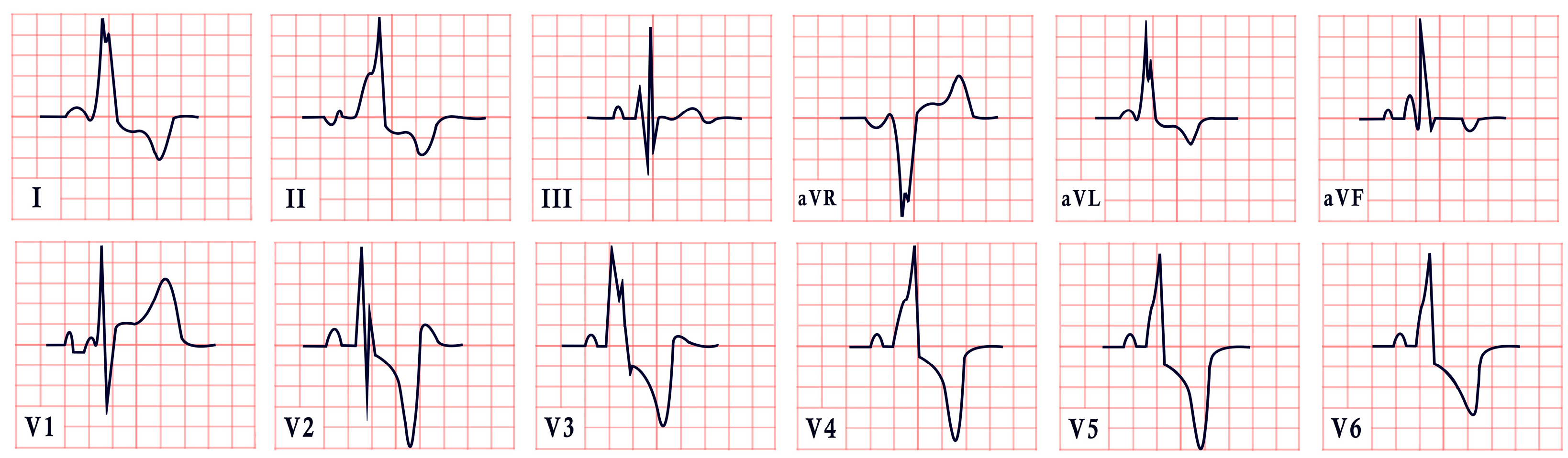
9. Noonan Syndrome
10. Barlow’s Disease
- Ballooning or hooding of the mitral leaflets between the chordae tendineae.
- Arching of the leaflets into the left atrium.
- Enlargement of the leaflets.
- Increase in the mitral valve area due to dilation of the annulus.
- Enlargement of the spongiosa and fibrosa layers.
- Deposition of fibrin and platelets.
- Changes in the atrialis layer, where the collagen fibers become compacted [Guy TS., 2012].
11. Danon Disease
- ECG abnormalities.
- Late gadolinium enhancement on CMR.
- Left ventricular hypertrophy.
- Ejection fraction < 50%.
- Muscular abnormalities, detected by EMG, or elevated biomarkers, such as liver enzymes or creatine kinase at two times the normal limit.
- Neurocognitive signs, such as attention deficit, or developmental delay [130].
12. BRASH Syndrome
13. Bundgaard Syndrome
- Concave and up-sloping ST-segment depression greater than 0.1 mV in at least four leads (V3–V6, I–III), measured 80 milliseconds after the J-point, in the absence of a secondary cause.
- ST-segment elevation greater than 0.1 mV observed in the lead aVR.
- Persistent ST-segment abnormalities, with no evidence of normalization over time.
- ST-segment depression exacerbated with exertion.
- Evidence of autosomal-dominant inheritance [139].
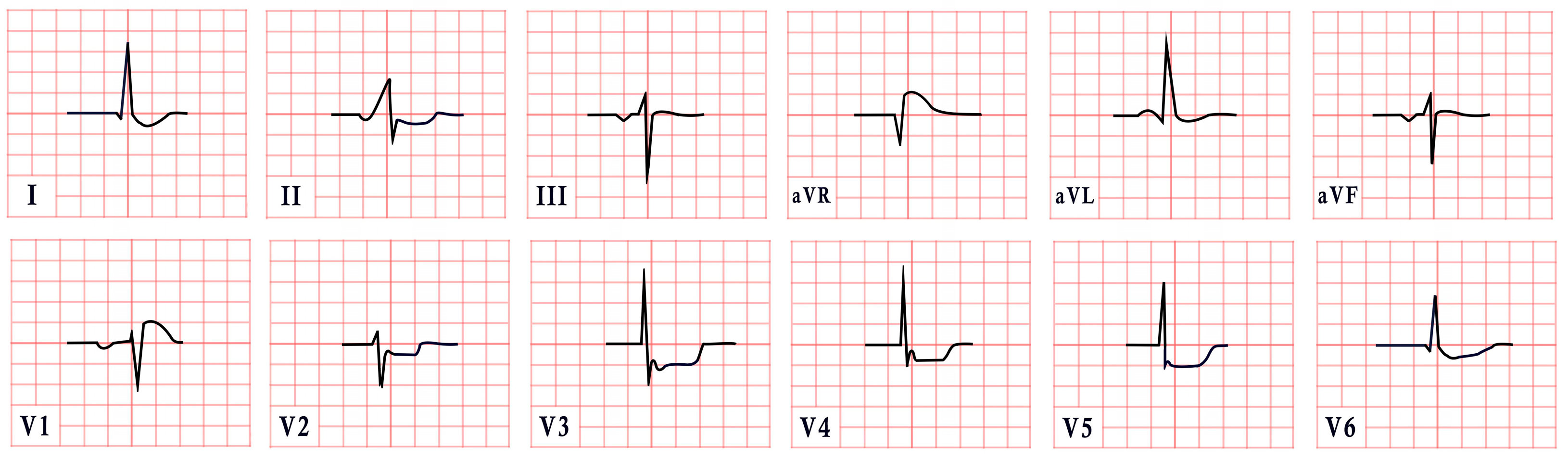
14. Naxos Disease
- Regional right ventricular kinetic abnormalities with RV systolic dysfunction or global dilation.
- Histological evidence of fibrofatty replacement in more than one myocardial sample.
- Electrocardiographic findings, including
- -
- Inverted T waves in leads V1–V3, excluding cases with complete right bundle branch block (RBBB).
- -
- ST-segment or J-point elevation in V1–V3, with the presence of epsilon waves in leads V1–V3.
- -
- Ventricular arrhythmias with a non-inferior axis, including premature ventricular beats or ventricular tachycardia (VT) (sustained or nonsustained).
- Genetic confirmation of a pathogenic variant in a patient, with either a family member meeting diagnostic criteria or postmortem confirmation on autopsy.
- Isolated regional RV kinetic abnormalities.
- Gadolinium enhancement in more than one area of the RV, confirmed in two different orthogonal views.
- ECG modifications:
- -
- Inverted T waves in leads V1–V2 (without complete right bundle branch block) in patients older than 14.
- -
- ST-segment or J-point elevation.
- -
- Inverted T waves beyond V3 with complete right bundle branch block.
- -
- Inverted T waves beyond V3 in patients younger than 14 years.
- -
- Terminal activation duration of the QRS ≥ 55 ms, measured from the nadir of the S wave to the end of the QRS in leads V1, V2, or V3.
- Ventricular arrhythmias, including
- -
- Premature ventricular beats or VT with an inferior axis.
- -
- History of aborted cardiac arrest due to VT or ventricular fibrillation.
- Genetic and family history criteria, including
15. Carvajal Syndrome
- Characteristic ring-pattern late gadolinium enhancement (LGE) involving more than three left ventricular segments in at least two orthogonal views.
- Low QRS amplitude in leads aVF, aVR, and aVL, excluding cases due to obesity, pulmonary emphysema, pericardial effusion, or cardiac amyloidosis.
- Global LV systolic dysfunction, which may occur without left ventricular dilation.
- Regional late gadolinium enhancement involving the lateral, inferior, and/or septal wall segments in at least two orthogonal views.
- T-wave inversions in leads V4–V6, excluding cases with complete left bundle branch block.
- More than 500 premature ventricular contractions per 24 h with RBBB morphology, excluding ectopies arising from the His–Purkinje system.
- Ventricular tachycardia (sustained or nonsustained) with an RBBB morphology, excluding ectopies arising from the His–Purkinje system.
- Aborted cardiac arrest due to sustained ventricular tachycardia or ventricular fibrillation [149].
16. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Friedman, P.A. The electrocardiogram at 100 years: History and future. Circulation 2024, 149, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Balta, A.; Ceasovschih, A.; Șorodoc, V.; Dimitriadis, K.; Güzel, S.; Lionte, C.; Stătescu, C.; Sascău, R.A.; Mantzouranis, E.; Sakalidis, A.; et al. Broad electrocardiogram syndromes spectrum: From common emergencies to particular electrical heart disorders. J. Pers. Med. 2022, 12, 1754. [Google Scholar] [CrossRef]
- Viskin, S. Long QT syndromes and torsade de pointes. Lancet 1999, 354, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Li, J. Long QT syndrome: Long story short. Eur. Heart J. 2022, 43, 4976–4977. [Google Scholar] [CrossRef]
- Huang, L.; Bitner-Glindzicz, M.; Tranebjærg, L.; Tinker, A. A spectrum of functional effects for disease causing mutations in the Jervell and Lange-Nielsen syndrome. Cardiovasc. Res. 2001, 51, 670–680. [Google Scholar] [CrossRef]
- Romano, C. Rare cardiac arrythmias of the pediatric age. II. Syncopal attacks due to paroxysmal ventricular fibrillation. (Presentation of 1st case in Italian pediatric literature). Clin. Pediatr. 1963, 45, 656–683. [Google Scholar]
- Vincent, G.M. The long QT syndrome. Indian Pacing Electrophysiol. J. 2002, 2, 127. [Google Scholar]
- Adler, A.; Novelli, V.; Amin, A.S.; Abiusi, E.; Care, M.; Nannenberg, E.A.; Feilotter, H.; Amenta, S.; Mazza, D.; Bikker, H. An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation 2020, 141, 418–428. [Google Scholar] [CrossRef]
- Shimizu, W. The long QT syndrome: Therapeutic implications of a genetic diagnosis. Cardiovasc. Res. 2005, 67, 347–356. [Google Scholar] [CrossRef]
- Rohatgi, R.K.; Sugrue, A.; Bos, J.M.; Cannon, B.C.; Asirvatham, S.J.; Moir, C.; Owen, H.J.; Bos, K.M.; Kruisselbrink, T.; Ackerman, M.J. Contemporary outcomes in patients with long QT syndrome. J. Am. Coll. Cardiol. 2017, 70, 453–462. [Google Scholar] [CrossRef]
- Goldenberg, I.; Moss, A.J.; Peterson, D.R.; McNitt, S.; Zareba, W.; Andrews, M.L.; Robinson, J.L.; Locati, E.H.; Ackerman, M.J.; Benhorin, J. Risk factors for aborted cardiac arrest and sudden cardiac death in children with the congenital long-QT syndrome. Circulation 2008, 117, 2184–2191. [Google Scholar] [CrossRef] [PubMed]
- Galić, E.; Bešlić, P.; Kilić, P.; Planinić, Z.; Pašalić, A.; Galić, I.; Ćubela, V.-V.; Pekić, P. Congenital long QT syndrome: A systematic review. Acta Clin. Croat. 2021, 60, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Lankaputhra, M.; Voskoboinik, A. Congenital long QT syndrome: A clinician’s guide. Intern. Med. J. 2021, 51, 1999–2011. [Google Scholar] [CrossRef] [PubMed]
- Postema, P.G.; Wilde, A.A.M. The measurement of the QT interval. Curr. Cardiol. Rev. 2014, 10, 287–294. [Google Scholar] [CrossRef]
- Mittal, S.R. QT interval–its measurement and clinical significance. J. Clin. Prev. Cardiol. 2019, 8, 71–79. [Google Scholar] [CrossRef]
- Khatib, R.; Sabir, F.R.; Omari, C.; Pepper, C.; Tayebjee, M.H. Managing drug-induced QT prolongation in clinical practice. Postgrad. Med. J. 2021, 97, 452–458. [Google Scholar] [CrossRef]
- Zeppenfeld, K.; Tfelt-Hansen, J.; De Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; De Chillou, C. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: Developed by the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC) Endorsed by the Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar]
- Lane, C.M.; Bos, J.M.; Rohatgi, R.K.; Ackerman, M.J. Beyond the length and look of repolarization: Defining the non-QTc electrocardiographic profiles of patients with congenital long QT syndrome. Heart Rhythm 2018, 15, 1413–1419. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Priori, S.G.; Spazzolini, C.; Moss, A.J.; Vincent, G.M.; Napolitano, C.; Denjoy, I.; Guicheney, P.; Breithardt, G.N.; Keating, M.T. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation 2001, 103, 89–95. [Google Scholar] [CrossRef]
- Garson, A., Jr.; Dick, M., 2nd; Fournier, A.; Gillette, P.C.; Hamilton, R.; Kugler, J.D.; Van Hare, G., 3rd; Vetter, V.; Vick, G., 3rd. The long QT syndrome in children. An international study of 287 patients. Circulation 1993, 87, 1866–1872. [Google Scholar] [CrossRef]
- Passman, R.; Kadish, A. Polymorphic ventricular tachycardia, long QT syndrome, and torsades de pointes. Med. Clin. N. Am. 2001, 85, 321–341. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 2011, 13, 1077–1109. [Google Scholar] [CrossRef] [PubMed]
- Katritsis, D.G.; Siontis, G.C.; Camm, A.J. Prognostic significance of ambulatory ECG monitoring for ventricular arrhythmias. Progress. Cardiovasc. Dis. 2013, 56, 133–142. [Google Scholar] [CrossRef]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.-E.; Huikuri, H. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace 2013, 15, 1389–1406. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Moss, A.J.; Vincent, G.M.; Crampton, R.S. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993, 88, 782–784. [Google Scholar] [CrossRef]
- Pappone, C.; Ciconte, G.; Anastasia, L.; Gaita, F.; Grant, E.; Micaglio, E.; Locati, E.T.; Calovic, Z.; Vicedomini, G.; Santinelli, V. Right ventricular epicardial arrhythmogenic substrate in long-QT syndrome patients at risk of sudden death. Europace 2023, 25, 948–955. [Google Scholar] [CrossRef]
- Pelliccia, A.; Sharma, S.; Gati, S.; Bäck, M.; Börjesson, M.; Caselli, S.; Collet, J.-P.; Corrado, D.; Drezner, J.A.; Halle, M. 2020 ESC Guidelines on sports cardiology and exercise in patients with cardiovascular disease: The Task Force on sports cardiology and exercise in patients with cardiovascular disease of the European Society of Cardiology (ESC). Eur. Heart J. 2021, 42, 17–96. [Google Scholar] [CrossRef] [PubMed]
- Ingles, J.; Yeates, L.; Hunt, L.; McGaughran, J.; Scuffham, P.A.; Atherton, J.; Semsarian, C. Health status of cardiac genetic disease patients and their at-risk relatives. Int. J. Cardiol. 2013, 165, 448–453. [Google Scholar] [CrossRef]
- Priori, S.G.; Napolitano, C. Genetics of cardiac arrhythmias and sudden cardiac death. Ann. N. Y. Acad. Sci. 2004, 1015, 96–110. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Spazzolini, C.; Crotti, L.; Bathen, J.; Amlie, J.P.; Timothy, K.; Shkolnikova, M.; Berul, C.I.; Bitner-Glindzicz, M.; Toivonen, L. The Jervell and Lange-Nielsen syndrome: Natural history, molecular basis, and clinical outcome. Circulation 2006, 113, 783–790. [Google Scholar] [CrossRef]
- Zareba, W.; Moss, A.J.; Schwartz, P.J.; Vincent, G.M.; Robinson, J.L.; Priori, S.G.; Benhorin, J.; Locati, E.H.; Towbin, J.A.; Keating, M.T. Influence of the genotype on the clinical course of the long-QT syndrome. N. Engl. J. Med. 1998, 339, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Authors/Task Force Members; Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC) Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). EP Eur. 2015, 17, 1601–1687. [Google Scholar]
- Goldenberg, I.; Zareba, W.; Moss, A.J. Long QT syndrome. Curr. Probl. Cardiol. 2008, 33, 629–694. [Google Scholar] [CrossRef] [PubMed]
- Früh, A.; Siem, G.; Holmström, H.; Døhlen, G.; Haugaa, K.H. The Jervell and Lange-Nielsen syndrome; atrial pacing combined with ß-blocker therapy, a favorable approach in young high-risk patients with long QT syndrome? Heart Rhythm 2016, 13, 2186–2192. [Google Scholar] [CrossRef]
- Green, J.D.; Schuh, M.J.; Maddern, B.R.; Haymond, J.; Helffrich, R.A. Cochlear implantation in Jervell and Lange-Nielsen syndrome. Ann. Otol. Rhinol. Laryngol. 2000, 109, 27–28. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Priori, S.G.; Dubin, A.M.; Kowey, P.; Linker, N.J.; Slotwiner, D.; Triedman, J.; Van Hare, G.F.; Gold, M.R. Beta-blocker therapy for long QT syndrome and catecholaminergic polymorphic ventricular tachycardia: Are all beta-blockers equivalent? Heart Rhythm 2016, 14, e41–e44. [Google Scholar] [CrossRef]
- Nakano, Y.; Shimizu, W. Genetics of long-QT syndrome. J. Hum. Genet. 2016, 61, 51–55. [Google Scholar] [CrossRef]
- Priori, S.G.; Schwartz, P.J.; Napolitano, C.; Bloise, R.; Ronchetti, E.; Grillo, M.; Vicentini, A.; Spazzolini, C.; Nastoli, J.; Bottelli, G. Risk stratification in the long-QT syndrome. N. Engl. J. Med. 2003, 348, 1866–1874. [Google Scholar] [CrossRef]
- Mizusawa, Y.; Horie, M.; Wilde, A.A. Genetic and clinical advances in congenital long QT syndrome. Circ. J. 2014, 78, 2827–2833. [Google Scholar] [CrossRef]
- Yap, Y.G.; Camm, A.J. Drug induced QT prolongation and torsades de pointes. Heart 2003, 89, 1363–1372. [Google Scholar] [CrossRef]
- Klein, R.; Ganelin, R.; Marks, J.; Usher, P.; Richards, C. Periodic paralysis with cardiac arrhythmia. J. Pediatr. 1963, 62, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Wójcik-Borowska, K.; Chrościńska-Krawczyk, M.; Zienkiewicz, E.; Małka, M.; Klatka, M. Andersen-Tawil Syndrome—A Case Report. Neurol. Dziecięca 2018, 27, 71–73. [Google Scholar] [CrossRef]
- Onore, M.E.; Picillo, E.; D’Ambrosio, P.; Morra, S.; Nigro, V.; Politano, L. Phenotypic Variability of Andersen–Tawil Syndrome Due to Allelic Mutation c. 652C> T in the KCNJ2 Gene—A New Family Case Report. Biomolecules 2024, 14, 507. [Google Scholar] [CrossRef]
- Andersen, E.D.; Krasilnikoff, P.A.; Overvad, H. Intermittent muscular weakness, extrasystoles, and multiple developmental anomalies: A new syndrome? Acta Paediatr. 1971, 60, 559–564. [Google Scholar] [CrossRef]
- Tawil, R.; Ptacek, L.J.; Pavlakis, S.G.; DeVivo, D.C.; Penn, A.S.; Özdemir, C.; Griggs, R.C. Andersen’s syndrome: Potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Ann. Neurol. 1994, 35, 326–330. [Google Scholar] [CrossRef]
- Donaldson, M.; Yoon, G.; Fu, Y.H.; Ptacek, L. Andersen-Tawil syndrome: A model of clinical variability, pleiotropy, and genetic heterogeneity. Ann. Med. 2004, 36, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Santana, L.F.; Cheng, E.P.; Lederer, W.J. How does the shape of the cardiac action potential control calcium signaling and contraction in the heart? J. Mol. Cell Cardiol. 2010, 49, 901. [Google Scholar] [CrossRef]
- Barrón-Díaz, D.R.; Totomoch-Serra, A.; Escobar-Cedillo, R.E.; García-Gutierrez, A.; Reyes-Quintero, Á.E.; Villegas Davirán, S.E.; Ibarra-Miramón, C.B.; Márquez, M.F. Andersen-Tawil syndrome with high risk of sudden cardiac death in four mexican patients. Cardiac and extra-cardiac phenotypes. Rev. Investig. Clin. 2021, 73, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.-L.; Pieper, G.H.; Wilders, R. Andersen–Tawil syndrome: Clinical and molecular aspects. Int. J. Cardiol. 2013, 170, 1–16. [Google Scholar] [CrossRef]
- Tristani-Firouzi, M.; Etheridge, S.P. Kir 2.1 channelopathies: The Andersen–Tawil syndrome. Pflugers Arch. 2010, 460, 289–294. [Google Scholar] [CrossRef]
- Yoon, G.; Quitania, L.; Kramer, J.; Fu, Y.; Miller, B.; Ptacek, L. Andersen–Tawil syndrome: Definition of a neurocognitive phenotype. Neurology 2006, 66, 1703–1710. [Google Scholar] [CrossRef]
- Tan, S.V.; Z’graggen, W.J.; BoËrio, D.; Rayan, D.L.R.; Howard, R.; Hanna, M.G.; Bostock, H. Membrane dysfunction in Andersen-Tawil syndrome assessed by velocity recovery cycles. Muscle Nerve 2012, 46, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Kukla, P.; K Biernacka, E.; Baranchuk, A.; Jastrzebski, M.; Jagodzinska, M. Electrocardiogram in Andersen-Tawil syndrome. New electrocardiographic criteria for diagnosis of type-1 Andersen-Tawil syndrome. Curr. Cardiol. Rev. 2014, 10, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Vivekanandam, V.; Männikkö, R.; Skorupinska, I.; Germain, L.; Gray, B.; Wedderburn, S.; Kozyra, D.; Sud, R.; James, N.; Holmes, S. Andersen–Tawil syndrome: Deep phenotyping reveals significant cardiac and neuromuscular morbidity. Brain 2022, 145, 2108–2120. [Google Scholar] [CrossRef]
- Statland, J.M.; Fontaine, B.; Hanna, M.G.; Johnson, N.E.; Kissel, J.T.; Sansone, V.A.; Shieh, P.B.; Tawil, R.N.; Trivedi, J.; Cannon, S.C. Review of the diagnosis and treatment of periodic paralysis. Muscle Nerve 2018, 57, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Dufendach, K.A.; Timothy, K.; Ackerman, M.J.; Blevins, B.; Pflaumer, A.; Etheridge, S.; Perry, J.; Blom, N.A.; Temple, J.; Chowdhury, D. Clinical outcomes and modes of death in Timothy syndrome: A multicenter international study of a rare disorder. JACC Clin. Electrophysiol. 2018, 4, 459–466. [Google Scholar] [CrossRef]
- Reichenbach, H.; Meister, E.; Theile, H. The heart-hand syndrome. A new variant of disorders of heart conduction and syndactylia including osseous changes in hands and feet. Kinderarztl. Prax. 1992, 60, 54–56. [Google Scholar]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K. CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef]
- Krause, U.; Gravenhorst, V.; Kriebel, T.; Ruschewski, W.; Paul, T. A rare association of long QT syndrome and syndactyly: Timothy syndrome (LQT 8). Clin. Res. Cardiol. 2011, 100, 1123–1127. [Google Scholar] [CrossRef]
- Borbás, J.; Vámos, M.; Hategan, L.; Hanák, L.; Farkas, N.; Szakács, Z.; Csupor, D.; Tél, B.; Kupó, P.; Csányi, B. Geno-and phenotypic characteristics and clinical outcomes of CACNA1C gene mutation associated Timothy syndrome,“cardiac only” Timothy syndrome and isolated long QT syndrome 8: A systematic review. Front. Cardiovasc. Med. 2022, 9, 1021009. [Google Scholar] [CrossRef]
- Diep, V.; Seaver, L.H. Long QT syndrome with craniofacial, digital, and neurologic features: Is it useful to distinguish between Timothy syndrome types 1 and 2? Am. J. Med. Genet. A 2015, 167, 2780–2785. [Google Scholar] [CrossRef] [PubMed]
- Bauer, R.; Timothy, K.W.; Golden, A. Update on the molecular genetics of Timothy syndrome. Front. Pediatr. 2021, 9, 668546. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.A.; Turner, C.; Timothy, K.W.; Seller, N.; Hares, D.L.; James, A.F.; Hancox, J.C.; Uzun, O.; Boyce, D.; Stuart, A.G. A multicentre study of patients with Timothy syndrome. EP Eur. 2018, 20, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Hiippala, A.; Tallila, J.; Myllykangas, S.; Koskenvuo, J.W.; Alastalo, T.P. Expanding the phenotype of Timothy syndrome type 2: An adolescent with ventricular fibrillation but normal development. Am. J. Med. Genet. A 2015, 167, 629–634. [Google Scholar] [CrossRef]
- Napolitano, C.; Bloise, R.; Priori, S.G. Timothy syndrome. In Cardiac Electrophysiology: From Cell to Bedside; Elsevier: Amsterdam, The Netherlands, 2014; pp. 953–957. [Google Scholar]
- Algra, A.; Tijssen, J.; Roelandt, J.; Pool, J.; Lubsen, J. QT interval variables from 24 hour electrocardiography and the two year risk of sudden death. Heart 1993, 70, 43–48. [Google Scholar] [CrossRef]
- Gussak, I.; Brugada, P.; Brugada, J.; Wright, R.S.; Kopecky, S.L.; Chaitman, B.R.; Bjerregaard, P. Idiopathic short QT interval: A new clinical syndrome? Cardiology 2001, 94, 99–102. [Google Scholar] [CrossRef]
- Hancox, J.C.; Du, C.; Butler, A.; Zhang, Y.; Dempsey, C.E.; Harmer, S.C.; Zhang, H. Pro-arrhythmic effects of gain-of-function potassium channel mutations in the short QT syndrome. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2023, 378, 20220165. [Google Scholar] [CrossRef] [PubMed]
- Thorsen, K.; Dam, V.S.; Kjaer-Sorensen, K.; Pedersen, L.N.; Skeberdis, V.A.; Jurevičius, J.; Treinys, R.; Petersen, I.M.; Nielsen, M.S.; Oxvig, C. Loss-of-activity-mutation in the cardiac chloride-bicarbonate exchanger AE3 causes short QT syndrome. Nat. Commun. 2017, 8, 1696. [Google Scholar] [CrossRef]
- Raschwitz, L.S.; El-Battrawy, I.; Schlentrich, K.; Besler, J.; Veith, M.; Roterberg, G.; Liebe, V.; Schimpf, R.; Lang, S.; Wolpert, C. Differences in short QT syndrome subtypes: A systematic literature review and pooled analysis. Front. Genet. 2020, 10, 1312. [Google Scholar] [CrossRef]
- Giustetto, C.; Di Monte, F.; Wolpert, C.; Borggrefe, M.; Schimpf, R.; Sbragia, P.; Leone, G.; Maury, P.; Anttonen, O.; Haissaguerre, M. Short QT syndrome: Clinical findings and diagnostic–therapeutic implications. Eur. Heart J. 2006, 27, 2440–2447. [Google Scholar] [CrossRef]
- Campuzano, O.; Sarquella-Brugada, G.; Cesar, S.; Arbelo, E.; Brugada, J.; Brugada, R. Recent advances in short QT syndrome. Front. Cardiovasc. Med. 2018, 5, 149. [Google Scholar] [CrossRef] [PubMed]
- Dewi, I.P.; Dharmadjati, B.B. Short QT syndrome: The current evidences of diagnosis and management. J. Arrhythmia 2020, 36, 962–966. [Google Scholar] [CrossRef] [PubMed]
- El-Battrawy, I.; Schlentrich, K.; Besler, J.; Liebe, V.; Schimpf, R.; Lang, S.; Odening, K.E.; Wolpert, C.; Zhou, X.; Borggrefe, M. Sex-differences in short QT syndrome: A systematic literature review and pooled analysis. Eur. J. Prev. Cardiol. 2020, 27, 1335–1338. [Google Scholar] [CrossRef]
- Patel, U.; Pavri, B.B. Short QT syndrome: A review. Cardiol. Rev. 2009, 17, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Bjerregaard, P. Diagnosis and management of short QT syndrome. Heart Rhythm 2018, 15, 1261–1267. [Google Scholar] [CrossRef]
- Veltmann, C.; Borggrefe, M. A’Schwartz score’for short QT syndrome. Nat. Rev. Cardiol. 2011, 8, 251–252. [Google Scholar] [CrossRef]
- Gaita, F.; Giustetto, C.; Bianchi, F.; Schimpf, R.; Haissaguerre, M.; Calò, L.; Brugada, R.; Antzelevitch, C.; Borggrefe, M.; Wolpert, C. Short QT Syndrome: Pharmacological Treatment. J. Am. Coll. Cardiol. 2004, 43, 1494–14999. [Google Scholar] [CrossRef]
- Wolpert, C.; Schimpf, R.; Giustetto, C.; Antzelevitch, C.; Cordeiro, J.; Dumaine, R.; Brugada, R.; Hong, K.; Bauersfeld, U.; Gaita, F. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J. Cardiovasc. Electrophysiol. 2005, 16, 54–58. [Google Scholar] [CrossRef]
- Milberg, P.; Tegelkamp, R.; Osada, N.; Schimpf, R.; Wolpert, C.; Breithardt, G.; Borggrefe, M.; Eckardt, L. Reduction of dispersion of repolarization and prolongation of postrepolarization refractoriness explain the antiarrhythmic effects of quinidine in a model of short QT syndrome. J. Cardiovasc. Electrophysiol. 2007, 18, 658–664. [Google Scholar] [CrossRef]
- Schimpf, R.; Veltmann, C.; Giustetto, C.; Gaita, F.; Borggrefe, M.; Wolpert, C. In vivo effects of mutant HERG K+ channel inhibition by disopyramide in patients with a short QT-1 syndrome: A pilot study. J. Cardiovasc. Electrophysiol. 2007, 18, 1157–1160. [Google Scholar] [CrossRef]
- Bjerregaard, P.; Gussak, I. Atrial fibrillation in the setting of familial short QT interval. Heart Rhythm 2004, 1, S165. [Google Scholar]
- Bayliss, C.E.; Beanlands, D.S.; Baird, R. The pacemaker-twiddler’s syndrome: A new complication of implantable transvenous pacemakers. Can. Med. Assoc. J. 1968, 99, 371. [Google Scholar] [PubMed]
- Gomez, J.O.; Doukky, R.; Pietrasik, G.; Wigant, R.R.; Mungee, S.; Baman, T.S. Prevalence and predictors of Twiddler’s syndrome. Pacing Clin. Electrophysiol. 2023, 46, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Montisci, R.; Soro, C.; Demelas, R.; Agus, E.; Follesa, A.; Siragusa, G.; Nissardi, V. A case series of the twiddler syndrome. Eur. Heart J. Case Rep. 2024, 8, ytae004. [Google Scholar] [CrossRef]
- Dattilo, G.; Scarano, M.; Casale, M.; Sergi, M.; Quattrocchi, S.; Parato, M.; Imbalzano, E. An atypical manifestation of Twiddler syndrome. International J. Cardiol. 2015, 186, 1–2. [Google Scholar] [CrossRef]
- Salahuddin, M.; Cader, F.A.; Nasrin, S.; Chowdhury, M.Z. The pacemaker-twiddler’s syndrome: An infrequent cause of pacemaker failure. BMC Res. Notes 2016, 9, 32. [Google Scholar] [CrossRef]
- Tahirovic, E.; Haxhibeqiri-Karabdic, I. Twiddler’s syndrome: Case report and literature review. Heart Views 2018, 19, 27–31. [Google Scholar] [CrossRef]
- Roberts, A.E.; Allanson, J.E.; Tartaglia, M.; Gelb, B.D. Noonan syndrome. Lancet 2013, 381, 333–342. [Google Scholar] [CrossRef]
- Vos, E.; Leenders, E.; Werkman, S.R.; Ten Cate, F.E.U.; Draaisma, J.M. The added value of the electrocardiogram in Noonan syndrome. Cardiol. Young 2022, 32, 936–943. [Google Scholar] [CrossRef]
- Noonan, J.A. Associated noncardiac malformations in children with congenital heart disease. Midwest. Soc. Pediat Res. 1963, 63, 468–470. [Google Scholar]
- Allanson, J.E.; Roberts, A.E. Noonan syndrome. In Cassidy and Allanson’s Management of Genetic Syndromes; Wiley: Hoboken, NJ, USA, 2021; pp. 651–669. [Google Scholar]
- Tartaglia, M.; Zampino, G.; Gelb, B. Noonan syndrome: Clinical aspects and molecular pathogenesis. Mol. Syndromol. 2010, 1, 2–26. [Google Scholar] [CrossRef] [PubMed]
- Tajan, M.; Paccoud, R.; Branka, S.; Edouard, T.; Yart, A. The RASopathy family: Consequences of germline activation of the RAS/MAPK pathway. Endocrine Rev. 2018, 39, 676–700. [Google Scholar] [CrossRef] [PubMed]
- Grant, A.R.; Cushman, B.J.; Cavé, H.; Dillon, M.W.; Gelb, B.D.; Gripp, K.W.; Lee, J.A.; Mason-Suares, H.; Rauen, K.A.; Tartaglia, M. Assessing the gene–disease association of 19 genes with the RASopathies using the ClinGen gene curation framework. Hum. Mutat. 2018, 39, 1485–1493. [Google Scholar] [CrossRef]
- Capri, Y.; Flex, E.; Krumbach, O.H.; Carpentieri, G.; Cecchetti, S.; Lißewski, C.; Adariani, S.R.; Schanze, D.; Brinkmann, J.; Piard, J. Activating mutations of RRAS2 are a rare cause of Noonan syndrome. Am. J. Hum. Genet. 2019, 104, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Linglart, L.; Gelb, B.D. Congenital heart defects in Noonan syndrome: Diagnosis, management, and treatment. Am. J. Med. Genet. Part C Semin. Med. Genet. 2020, 184, 73–80. [Google Scholar] [CrossRef]
- Sun, L.; Xie, Y.-M.; Wang, S.-S.; Zhang, Z.-W. Cardiovascular abnormalities and gene mutations in children with Noonan syndrome. Front. Genet. 2022, 13, 915129. [Google Scholar] [CrossRef]
- Raaijmakers, R.; Noordam, C.; Noonan, J.; Croonen, E.; Van Der Burgt, C.; Draaisma, J. Are ECG abnormalities in Noonan syndrome characteristic for the syndrome? Eur. J. Pediatr. 2008, 167, 1363–1367. [Google Scholar] [CrossRef]
- Armengol, A.; Brohet, C.; Lintermans, J.; Vliers, A. Left ventricle in Noonan’s syndrome. Electro-vecto-echo and angiocardiographic aspects. Arch. Mal. Coeur Vaiss. 1987, 80, 445–453. [Google Scholar]
- Bertola, D.R.; Chong, A.K.; Sugayama, S.M.; Albano, L.M.J.; Wagenführ, J.; Moysés, R.L.; Gonzalez, C.H. Cardiac findings in 31 patients with Noonan’s syndrome. Arq. Bras. Cardiol. 2000, 75, 409–412. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Kuroda, H.; Ikegawa, T.; Kawai, S.; Ono, S.; Kim, K.-S.; Yanagi, S.; Kurosawa, K.; Aoki, Y.; Iwamoto, M. Electrocardiographic Changes with Age in Japanese Patients with Noonan Syndrome. J. Cardiovasc. Dev. Dis. 2023, 11, 10. [Google Scholar] [CrossRef]
- Zenker, M.; Edouard, T.; Blair, J.C.; Cappa, M. Noonan syndrome: Improving recognition and diagnosis. Arch. Dis. Child. 2022, 107, 1073–1078. [Google Scholar] [CrossRef]
- Van der Burgt, I. Noonan syndrome. Orphanet J. Rare Dis. 2007, 2, 4. [Google Scholar] [CrossRef]
- Vergara, P.; Altizio, S.; Falasconi, G.; Pannone, L.; Gulletta, S.; Della Bella, P. Electrophysiological substrate in patients with Barlow’s disease. Arrhythm. Electrophysiol. Rev. 2021, 10, 33. [Google Scholar] [CrossRef]
- Cheng, T.O. John B. Barlow: The man and his syndrome. Int. J. Cardiol. 2014, 177, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J. Conjoint clinic on the clinical significance of late systolic murmurs and non-ejection systolic clicks. J. Chronic Dis. 1965, 18, 665–673. [Google Scholar] [CrossRef]
- Tamura, K.; Fukuda, Y.; Ishizaki, M.; Masuda, Y.; Yamanaka, N.; Ferrans, V.J. Abnormalities in elastic fibers and other connective-tissue components of floppy mitral valve. Am. Heart J. 1995, 129, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Rabkin, E.; Aikawa, M.; Stone, J.R.; Fukumoto, Y.; Libby, P.; Schoen, F.J. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation 2001, 104, 2525–2532. [Google Scholar] [CrossRef]
- Basso, C.; Perazzolo Marra, M.; Rizzo, S.; De Lazzari, M.; Giorgi, B.; Cipriani, A.; Frigo, A.C.; Rigato, I.; Migliore, F.; Pilichou, K. Arrhythmic mitral valve prolapse and sudden cardiac death. Circulation 2015, 132, 556–566. [Google Scholar] [CrossRef]
- Kubala, M.; Essayagh, B.; Michelena, H.I.; Enriquez-Sarano, M.; Tribouilloy, C. Arrhythmic mitral valve prolapse in 2023: Evidence-based update. Front. Cardiovasc. Med. 2023, 10, 1130174. [Google Scholar] [CrossRef]
- Lab, M. Mechanically dependent changes in action potentials recorded from the intact frog ventricle. Circ. Res 1978, 42, 519–528. [Google Scholar] [CrossRef]
- Korovesis, T.G.; Koutrolou-Sotiropoulou, P.; Katritsis, D.G. Arrhythmogenic mitral valve prolapse. Arrhythm. Electrophysiol. Rev. 2022, 11, e16. [Google Scholar] [CrossRef] [PubMed]
- Guy, T.S.; Hill, A.C. Mitral valve prolapse. Annu. Rev. Med. 2012, 63, 277–292. [Google Scholar] [CrossRef]
- Han, H.C.; Ha, F.J.; Teh, A.W.; Calafiore, P.; Jones, E.F.; Johns, J.; Koshy, A.N.; O’Donnell, D.; Hare, D.L.; Farouque, O. Mitral valve prolapse and sudden cardiac death: A systematic review. J. Am. Heart Assoc. 2018, 7, e010584. [Google Scholar] [CrossRef]
- Savage, D.D.; Levy, D.; Garrison, R.J.; Castelli, W.P.; Kligfield, P.; Devereux, R.B.; Anderson, S.J.; Kannel, W.B.; Feinleib, M. Mitral valve prolapse in the general population. 3. Dysrhythmias: The Framingham Study. Am. Heart J. 1983, 106, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Brunec-Keller, M.; Scharf, C.; Radulovic, J.; Berdat, P.A.; CH, A.J.; Vogt, P.; Duru, F.; Caselli, S. Barlow disease: Effect of mitral valve repair on ventricular arrhythmias in 82 patients in a retrospective long-term study. J. Cardiovasc. Surg. 2022, 63, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Essayagh, B.; Sabbag, A.; Antoine, C.; Benfari, G.; Yang, L.-T.; Maalouf, J.; Asirvatham, S.; Michelena, H.; Enriquez-Sarano, M. Presentation and outcome of arrhythmic mitral valve prolapse. J. Am. Coll. Cardiol. 2020, 76, 637–649. [Google Scholar] [CrossRef]
- Lotan, D.; Salazar-Mendiguchía, J.; Mogensen, J.; Rathore, F.; Anastasakis, A.; Kaski, J.; Garcia-Pavia, P.; Olivotto, I.; Charron, P.; Biagini, E. Clinical profile of cardiac involvement in Danon disease: A multicenter European registry. Circ. Genom. Precis. Med. 2020, 13, e003117. [Google Scholar] [CrossRef]
- Danon, M.J.; Oh, S.J.; DiMauro, S.; Manaligod, J.R.; Eastwood, A.; Naidu, S.; Schliselfeld, L.H. Lysosomal glycogen storage disease with normal acid maltase. Neurology 1981, 31, 51. [Google Scholar] [CrossRef]
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.; Oh, S.J.; Koga, Y. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef]
- Xu, J.; Li, Z.; Liu, Y.; Zhang, X.; Niu, F.; Zheng, H.; Wang, L.; Kang, L.; Wang, K.; Xu, B. Danon disease: A case report and literature review. Diagn. Pathol. 2021, 16, 39. [Google Scholar] [CrossRef]
- Zhai, Y.; Miao, J.; Peng, Y.; Wang, Y.; Dong, J.; Zhao, X. Clinical features of Danon disease and insights gained from LAMP-2 deficiency models. Trends Cardiovasc. Med. 2023, 33, 81–89. [Google Scholar] [CrossRef]
- D’souza, R.S.; Levandowski, C.; Slavov, D.; Graw, S.L.; Allen, L.A.; Adler, E.; Mestroni, L.; Taylor, M.R. Danon disease: Clinical features, evaluation, and management. Circ. Heart Fail. 2014, 7, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Boucek, D.; Jirikowic, J.; Taylor, M. Natural history of Danon disease. Genet. Med. 2011, 13, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Prall, F.R.; Drack, A.; Taylor, M.; Ku, L.; Olson, J.L.; Gregory, D.; Mestroni, L.; Mandava, N. Ophthalmic manifestations of Danon disease. Ophthalmology 2006, 113, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Cenacchi, G.; Papa, V.; Pegoraro, V.; Marozzo, R.; Fanin, M.; Angelini, C. Danon disease: Review of natural history and recent advances. Neuropathol. Appl. Neurobiol. 2020, 46, 303–322. [Google Scholar] [CrossRef]
- Cheng, Z.; Fang, Q. Danon disease: Focusing on heart. J. Hum. Genet. 2012, 57, 407–410. [Google Scholar] [CrossRef]
- Li, X.; Li, J.; Lu, M. Danon disease manifesting as dilated cardiomyopathy in a 37-year-old woman. Eur. Heart J. Cardiovasc. Imaging 2024, 25, e163. [Google Scholar] [CrossRef]
- Hong, K.N.; Eshraghian, E.A.; Arad, M.; Argirò, A.; Brambatti, M.; Bui, Q.; Caspi, O.; de Frutos, F.; Greenberg, B.; Ho, C.Y. International consensus on differential diagnosis and management of patients with Danon disease: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2023, 82, 1628–1647. [Google Scholar] [CrossRef]
- Lee, T.H.; Salomon, D.R.; Rayment, C.M.; Antman, E.M. Hypotension and sinus arrest with exercise-induced hyperkalemia and combined verapamil/propranolol therapy. Am. J. Med. 1986, 80, 1203–1204. [Google Scholar] [CrossRef]
- Lizyness, K.; Dewald, O. BRASH syndrome. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Shah, P.; Gozun, M.; Keitoku, K.; Kimura, N.; Yeo, J.; Czech, T.; Nishimura, Y. Clinical characteristics of BRASH syndrome: Systematic scoping review. Eur. J. Int. Med. 2022, 103, 57–61. [Google Scholar] [CrossRef]
- Majeed, H.; Khan, U.; Khan, A.M.; Khalid, S.N.; Farook, S.; Gangu, K.; Sagheer, S.; Sheikh, A.B. BRASH syndrome: A systematic review of reported cases. Curr. Probl. Cardiol. 2023, 48, 101663. [Google Scholar] [CrossRef] [PubMed]
- Farkas, J.D.; Long, B.; Koyfman, A.; Menson, K. BRASH syndrome: Bradycardia, renal failure, AV blockade, shock, and hyperkalemia. J. Emerg. Med. 2020, 59, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Kemnic, T.; Hildebrandt, K.R. BRASH syndrome. BMJ Case Rep. CP 2020, 13, e233825. [Google Scholar] [CrossRef] [PubMed]
- Ceasovschih, A.; Șorodoc, V.; Covantsev, S.; Balta, A.; Uzokov, J.; Kaiser, S.E.; Almaghraby, A.; Lionte, C.; Stătescu, C.; Sascău, R.A. Electrocardiogram features in non-cardiac diseases: From mechanisms to practical aspects. J. Multidiscip. Healthc. 2024, 17, 1695–1719. [Google Scholar] [CrossRef]
- Bundgaard, H.; Jøns, C.; Lodder, E.M.; Izarzugaza, J.M.; Romero Herrera, J.A.; Pehrson, S.; Tfelt-Hansen, J.; Ahlberg, G.; Olesen, M.S.; Holst, A.G. A novel familial cardiac arrhythmia syndrome with widespread ST-segment depression. N. Engl. J. Med. 2018, 379, 1780–1781. [Google Scholar] [CrossRef]
- Christensen, A.H.; Vissing, C.R.; Pietersen, A.; Tfelt-Hansen, J.; Hartvig Lindkær Jensen, T.; Pehrson, S.; Henriksen, F.L.; Sandgaard, N.C.F.; Iversen, K.K.; Jensen, H.K. Electrocardiographic findings, arrhythmias, and left ventricular involvement in familial ST-depression syndrome. Circ. Arrhythm. Electrophysiol. 2022, 15, e010688. [Google Scholar] [CrossRef]
- Frosted, R.; Paludan-Müller, C.; Vad, O.B.; Olesen, M.S.; Bundgaard, H.; van Dam, P.; Christensen, A.H. CineECG analysis provides new insights into Familial ST-segment Depression Syndrome. Europace 2023, 25, euad116. [Google Scholar] [CrossRef]
- Christensen, A.H.; Nyholm, B.C.; Vissing, C.R.; Pietersen, A.; Tfelt-Hansen, J.; Olesen, M.S.; Pehrson, S.; Iversen, K.K.; Jensene, H.K.; Bundgaard, H. Natural history and clinical characteristics of the first 10 Danish families with familial ST-depression syndrome. J. Am. Coll. Cardiol. 2021, 77, 2617–2619. [Google Scholar] [CrossRef]
- Stătescu, C.; Anghel, L.; Benchea, L.C.; Tudurachi, B.S.; Leonte, A.; Zăvoi, A.; Zota, I.M.; Prisacariu, C.; Radu, R.; Șerban, I.L.; et al. A Systematic Review on the Risk Modulators of Myocardial Infarction in the "Young"-Implications of Lipoprotein (a). Int. J. Mol. Sci. 2023, 24, 5927. [Google Scholar] [CrossRef]
- Protonotarios, N.; Tsatsopoulou, A.; Patsourakos, P.; Alexopoulos, D.; Gezerlis, P.; Simitsis, S.; Scampardonis, G. Cardiac abnormalities in familial palmoplantar keratosis. Heart 1986, 56, 321–326. [Google Scholar] [CrossRef]
- Protonotarios, A.; Asimaki, A.; Basso, C.; Xylouri, Z.; Monda, E.; Protonotarios, I.; Crisci, G.; Abrahms, D.J.; Anastasakis, A.; Antoniades, L. Naxos Disease and Related Cardio-Cutaneous Syndromes. JACC Adv. 2025, 4, 101547. [Google Scholar] [CrossRef]
- Leopoulou, M.; Mattsson, G.; LeQuang, J.A.; Pergolizzi, J.V.; Varrassi, G.; Wallhagen, M.; Magnusson, P. Naxos disease–a narrative review. Expert. Rev. Cardiovasc. Ther. 2020, 18, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, S.M.; Kumar, P.; Basavaraja, G. Carvajal syndrome. Int. J. Trichol. 2016, 8, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Protonotarios, I.; Asimaki, A.; Xylouri, Z.; Protonotarios, A.; Tsatsopoulou, A. Clinical and molecular aspects of naxos disease. Heart Fail. Clin. 2022, 18, 89–99. [Google Scholar] [CrossRef]
- Laxmi, R.K.; Prithvish, C.; Sanjay, H. Cardiocutaneous Syndrome: Naxos Disease. Int. J. Preclin. Clin. 2022, 3, 83–86. [Google Scholar] [CrossRef]
- Corrado, D.; Anastasakis, A.; Basso, C.; Bauce, B.; Blomström-Lundqvist, C.; Bucciarelli-Ducci, C.; Cipriani, A.; De Asmundis, C.; Gandjbakhch, E.; Jiménez-Jáimez, J. Proposed diagnostic criteria for arrhythmogenic cardiomyopathy: European Task Force consensus report. Int. J. Cardiol. 2024, 395, 131447. [Google Scholar] [CrossRef]
- Finsterer, J.; Stöllberger, C.; Wollmann, E.; Dertinger, S.; Laccone, F. Autosomal dominant Carvajal plus syndrome due to the novel desmoplakin mutation c. 1678A> T (p. Ile560Phe). Mol. Genet. Metab. Rep. 2016, 8, 1–3. [Google Scholar]
- Brandão, M.; Bariani, R.; Rigato, I.; Bauce, B. Desmoplakin cardiomyopathy: Comprehensive review of an increasingly recognized entity. J. Clin. Med. 2023, 12, 2660. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Huerta, L. Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. J. Am. Acad. Dermatol. 1998, 39, 418–421. [Google Scholar] [CrossRef]
- Sun, Q.; Wine Lee, L.; Hall, E.K.; Choate, K.A.; Elder, R.W. Hair and skin predict cardiomyopathies: Carvajal and erythrokeratodermia cardiomyopathy syndromes. Pediatr. Derm. 2021, 38, 31–38. [Google Scholar] [CrossRef]
- Wang, W.; Murray, B.; Tichnell, C.; Gilotra, N.A.; Zimmerman, S.L.; Gasperetti, A.; Scheel, P.; Tandri, H.; Calkins, H.; James, C.A. Clinical characteristics and risk stratification of desmoplakin cardiomyopathy. Ep Eur. 2022, 24, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Garrod, D.; Chidgey, M. Desmosome structure, composition and function. Biochim. Biophys. Acta 2008, 1778, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, S.R.; Gard, J.J.; Carvajal-Huerta, L.; Ruiz-Cabezas, J.C.; Thiene, G.; Saffitz, J.E. Structural and molecular pathology of the heart in Carvajal syndrome. Cardiovasc. Pathol. 2004, 13, 26–32. [Google Scholar] [CrossRef]
- Di Lorenzo, F.; Marchionni, E.; Ferradini, V.; Latini, A.; Pezzoli, L.; Martino, A.; Romeo, F.; Iorio, A.; Bianchi, S.; Iascone, M. DSP-related cardiomyopathy as a distinct clinical entity? Emerging evidence from an Italian cohort. Int. J. Mol. Sci. 2023, 24, 2490. [Google Scholar] [CrossRef]
- López-Ayala, J.M.; Gómez-Milanés, I.; Sánchez Muñoz, J.J.; Ruiz-Espejo, F.; Ortíz, M.; González-Carrillo, J.; López-Cuenca, D.; Oliva-Sandoval, M.; Monserrat, L.; Valdés, M. Desmoplakin truncations and arrhythmogenic left ventricular cardiomyopathy: Characterizing a phenotype. Europace 2014, 16, 1838–1846. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, A.; Bauce, B.; De Lazzari, M.; Rigato, I.; Bariani, R.; Meneghin, S.; Pilichou, K.; Motta, R.; Aliberti, C.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy: Characterization of left ventricular phenotype and differential diagnosis with dilated cardiomyopathy. J. Am. Heart Assoc. 2020, 9, e014628. [Google Scholar] [CrossRef]
- Anghel, L.; Sascău, R.; Zota, I.M.; Stătescu, C. Well-Known and Novel Serum Biomarkers for Risk Stratification of Patients with Non-ischemic Dilated Cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 5688. [Google Scholar] [CrossRef]
- Smith, E.D.; Lakdawala, N.K.; Papoutsidakis, N.; Aubert, G.; Mazzanti, A.; McCanta, A.C.; Agarwal, P.P.; Arscott, P.; Dellefave-Castillo, L.M.; Vorovich, E.E. Desmoplakin cardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. Circulation 2020, 141, 1872–1884. [Google Scholar] [CrossRef]
- Augusto, J.B.; Eiros, R.; Nakou, E.; Moura-Ferreira, S.; Treibel, T.A.; Captur, G.; Akhtar, M.M.; Protonotarios, A.; Gossios, T.D.; Savvatis, K. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: A comprehensive genotype-imaging phenotype study. Eur. Heart J. Cardiovasc. Imaging 2020, 21, 326–336. [Google Scholar] [CrossRef]
- Ghawanmeh, M.; Simon Frances, B.; Kerai, A.; Patel, P.; Du, J.; Kumar, P. Management of recurrent myocarditis due to desmoplakin cardiomyopathy: Diagnostic and therapeutic challenges. Case Rep. 2022, 4, 59–62. [Google Scholar]
- Reza, N.; de Feria, A.; Chowns, J.L.; Hoffman-Andrews, L.; Vann, L.; Kim, J.; Marzolf, A.; Owens, A.T. Cardiovascular characteristics of patients with genetic variation in Desmoplakin (DSP). Cardiogenetics 2022, 12, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, A.P.; Chiampas, K.; Muller, S.A.; Gasperetti, A.; Yanek, L.R.; Carrick, R.T.; Gordon, C.; Tichnell, C.; Murray, B.; Calkins, H. Endurance exercise promotes episodes of myocardial injury in individuals with a pathogenic desmoplakin (DSP) variant. Heart Rhythm 2024, in press. [Google Scholar] [CrossRef] [PubMed]
- Lemus Barrios, G.A.; Lopez-Lopez, J.P.; Barbosa-Balaguera, S.; Correa, A.M. Left-dominant arrhythmogenic cardiomyopathy due to desmoplakin mutation: A case report. ESC Heart Fail. 2023, 10, 3190–3194. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ECG Syndrome | ECG Characteristics | ECG Aspect |
|---|---|---|
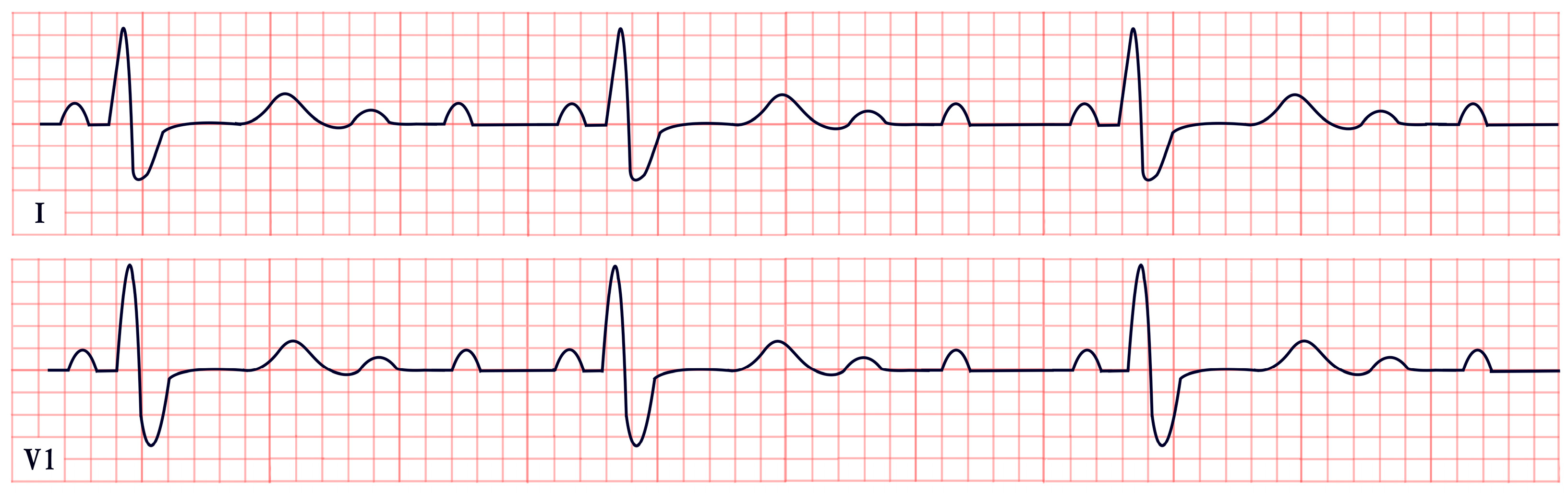
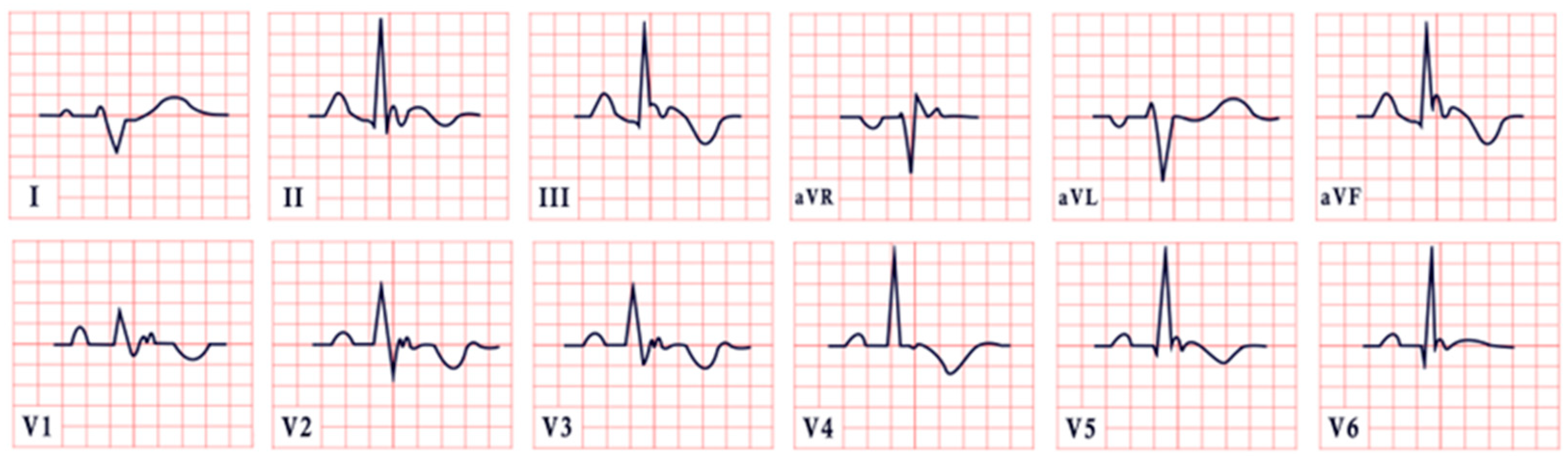
| Long QT Syndrome | -Prolonged QTc interval (>460 ms in females, >450 ms in males) -T-wave notching, inversion, and T-wave alternans | 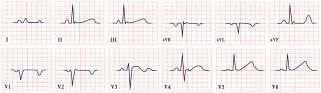 |
| Jervell and Lange-Nielsen Syndrome | -QTc prolongation >500 ms -Broad, notched, or biphasic T waves | 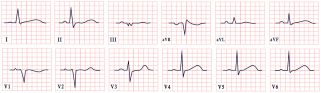 |
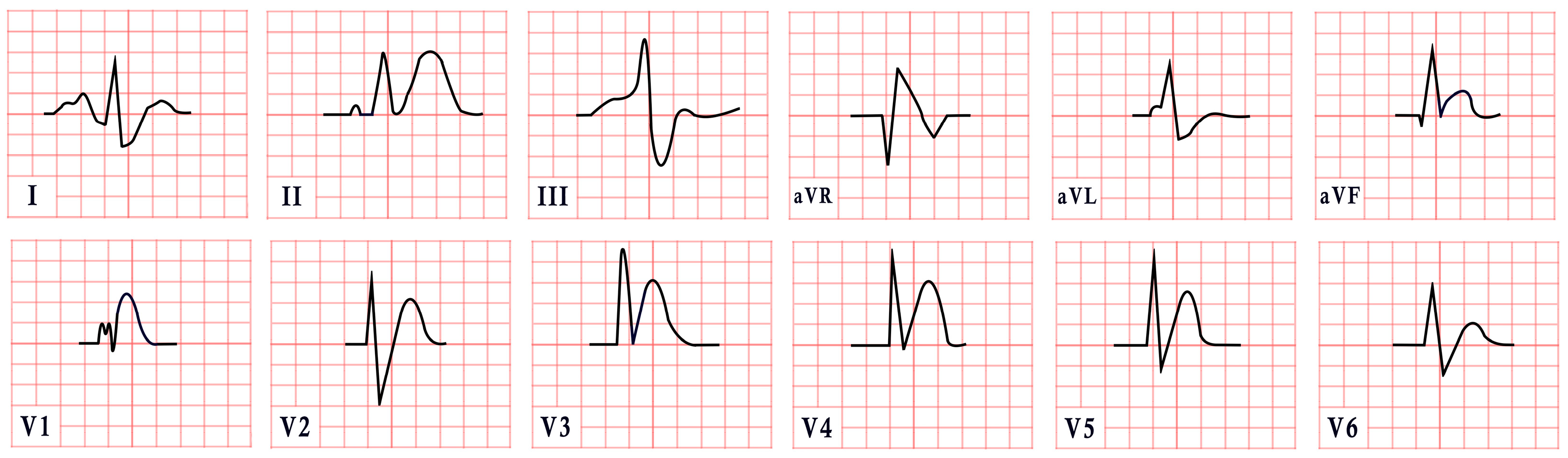
| Romano–Ward Syndrome | -LQT1: QTc = 466 ± 44 ms, broad-based, asymmetric, or peaked ‘infantile’ T waves -LQT2: QTc = 490 ± 49 ms, low-amplitude T waves -LQT3: QTc = 496 ± 49 ms, remote or asymmetric T waves | 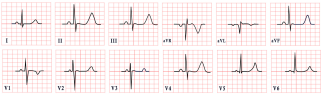 |
| Andersen–Tawil Syndrome | -Prominent U waves in V2–V4 -Q-U prolongation >655 ms -QTc may be normal | 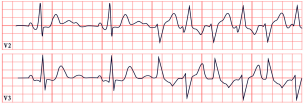 |
| Timothy Syndrome | -QTc > 480 ms (TS1), >600 ms (TS2) -2:1 AV block -T-wave alternans |  |
| Short QT Syndrome | -Short QTc < 370 ms with poor HR adaptation -Short or absent ST segment -Tall, narrow, peaked, or biphasic T waves -Prominent U waves | 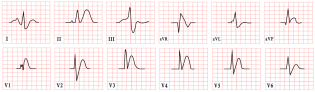 |
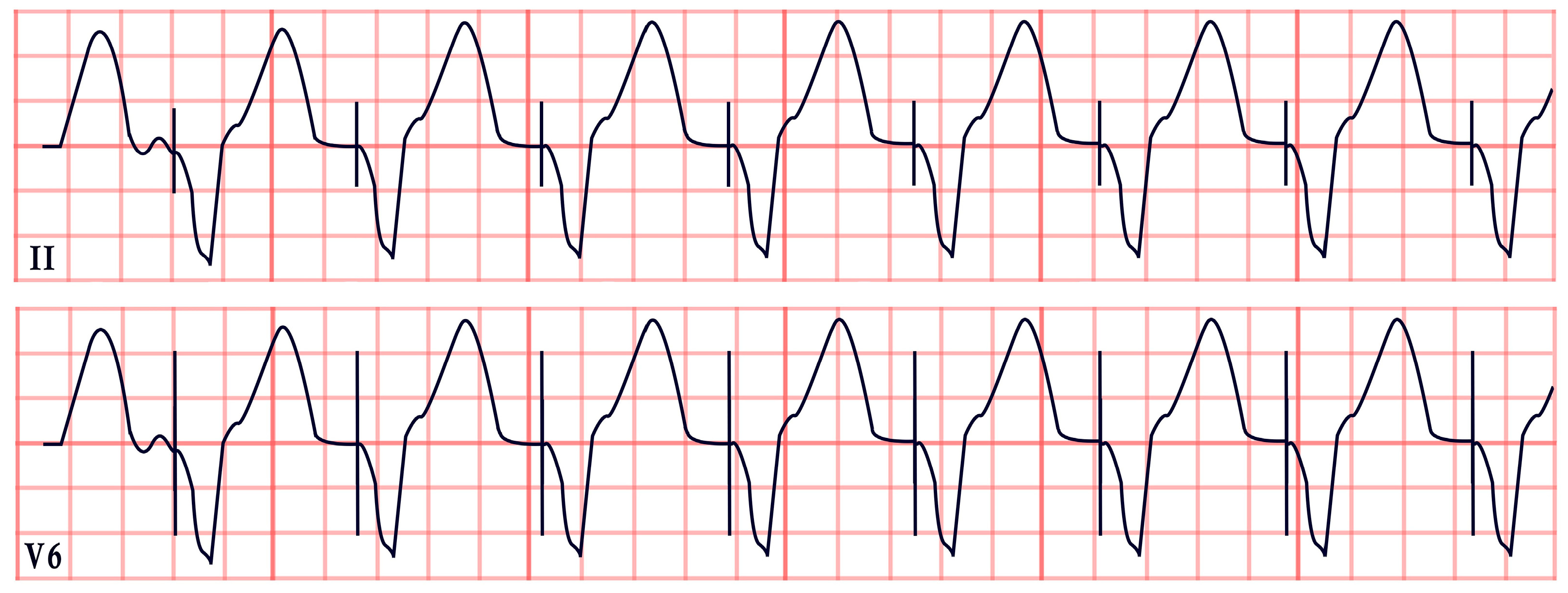
| Twiddler’s Syndrome | -Loss of capture, abnormal pacing morphology -Irregular/asynchronous pacing -Asystole | 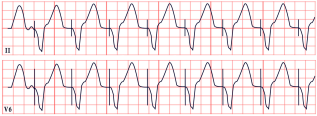 |
| Noonan Syndrome | -Left axis deviation -Abnormal R/S ratio in V4–V6 -Abnormal Q waves |  |
| Barlow’s Syndrome | -Inverted T waves -PVCs | 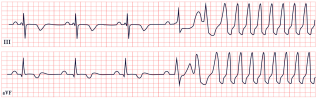 |
| Danon Disease | -Short PR interval, Delta waves -Abnormal Q waves -AV block -Inverted T waves -Left ventricular hypertrophy | 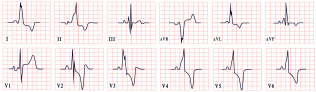 |
| BRASH Syndrome | -Peaked T waves -Bradycardia, junctional rhythm | 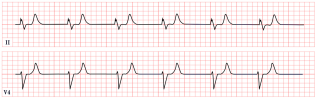 |
| Bundgaard Syndrome | -Persistent ST depression in II, V5, V6 -Deepens with exertion, non-ischemic |  |
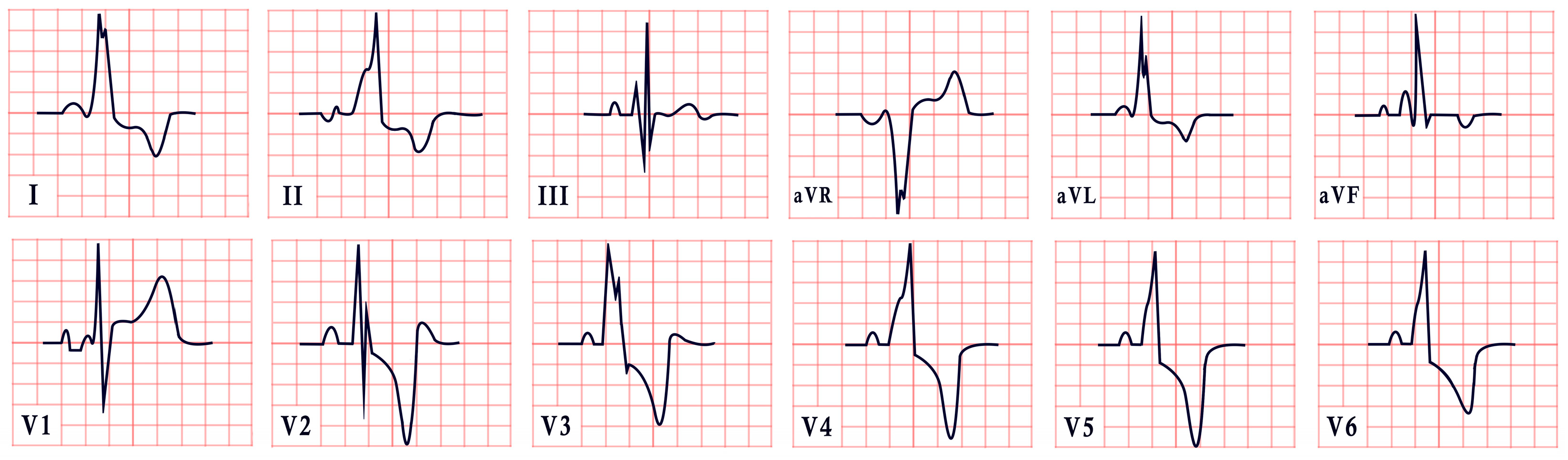
| Naxos Disease | -Inverted T waves in V1–V3/V1–V6 -Epsilon waves | 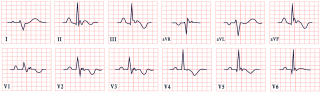 |
| Carvajal Syndrome | -Low QRS voltage -Inverted T waves in V2–V6 -Epsilon waves | 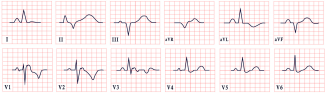 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceasovschih, A.; Balta, A.; Șorodoc, V.; Rathod, K.; El Gohary, A.; Covantsev, S.; Masszi, R.; Şener, Y.Z.; Corlăteanu, A.; Naqvi, S.H.R.; et al. Broad Electrocardiogram Syndromes Spectrum: From Common Emergencies to Particular Electrical Heart Disorders—Part II. Diagnostics 2025, 15, 1568. https://doi.org/10.3390/diagnostics15121568
Ceasovschih A, Balta A, Șorodoc V, Rathod K, El Gohary A, Covantsev S, Masszi R, Şener YZ, Corlăteanu A, Naqvi SHR, et al. Broad Electrocardiogram Syndromes Spectrum: From Common Emergencies to Particular Electrical Heart Disorders—Part II. Diagnostics. 2025; 15(12):1568. https://doi.org/10.3390/diagnostics15121568
Chicago/Turabian StyleCeasovschih, Alexandr, Anastasia Balta, Victorița Șorodoc, Krishnaraj Rathod, Ahmed El Gohary, Serghei Covantsev, Richárd Masszi, Yusuf Ziya Şener, Alexandru Corlăteanu, Syed Haseeb Raza Naqvi, and et al. 2025. "Broad Electrocardiogram Syndromes Spectrum: From Common Emergencies to Particular Electrical Heart Disorders—Part II" Diagnostics 15, no. 12: 1568. https://doi.org/10.3390/diagnostics15121568
APA StyleCeasovschih, A., Balta, A., Șorodoc, V., Rathod, K., El Gohary, A., Covantsev, S., Masszi, R., Şener, Y. Z., Corlăteanu, A., Naqvi, S. H. R., Grejdieru, A., Kounis, N. G., & Șorodoc, L. (2025). Broad Electrocardiogram Syndromes Spectrum: From Common Emergencies to Particular Electrical Heart Disorders—Part II. Diagnostics, 15(12), 1568. https://doi.org/10.3390/diagnostics15121568