
Thymidine Kinase 1 Expression Correlates with Tumor Aggressiveness and Metastatic Potential in OSCC
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Sources
2.2. Identification of Differentially Expressed Genes
2.3. Gene Ontology Enrichment Analysis
2.4. Protein–Protein Interaction Network and Hub Gene Identification
2.5. Expression and Survival Analysis of Hub Genes
2.6. Analysis of Gene Expression Across Cancer Types and Disease Stages
2.7. Reagents and Antibodies
2.8. Cell Culture and Migratory Subpopulation Enrichment
2.9. Transwell Migration Assay
2.10. Wound Healing Assay
2.11. siRNA Transfection
2.12. Western Blotting
2.13. Statistical Analysis
3. Results
3.1. Identification of Consistently Dysregulated Genes in OSCC from Public GEO Datasets
3.2. Functional Enrichment and Identification of Hub Genes Among Consistently Dysregulated DEGs
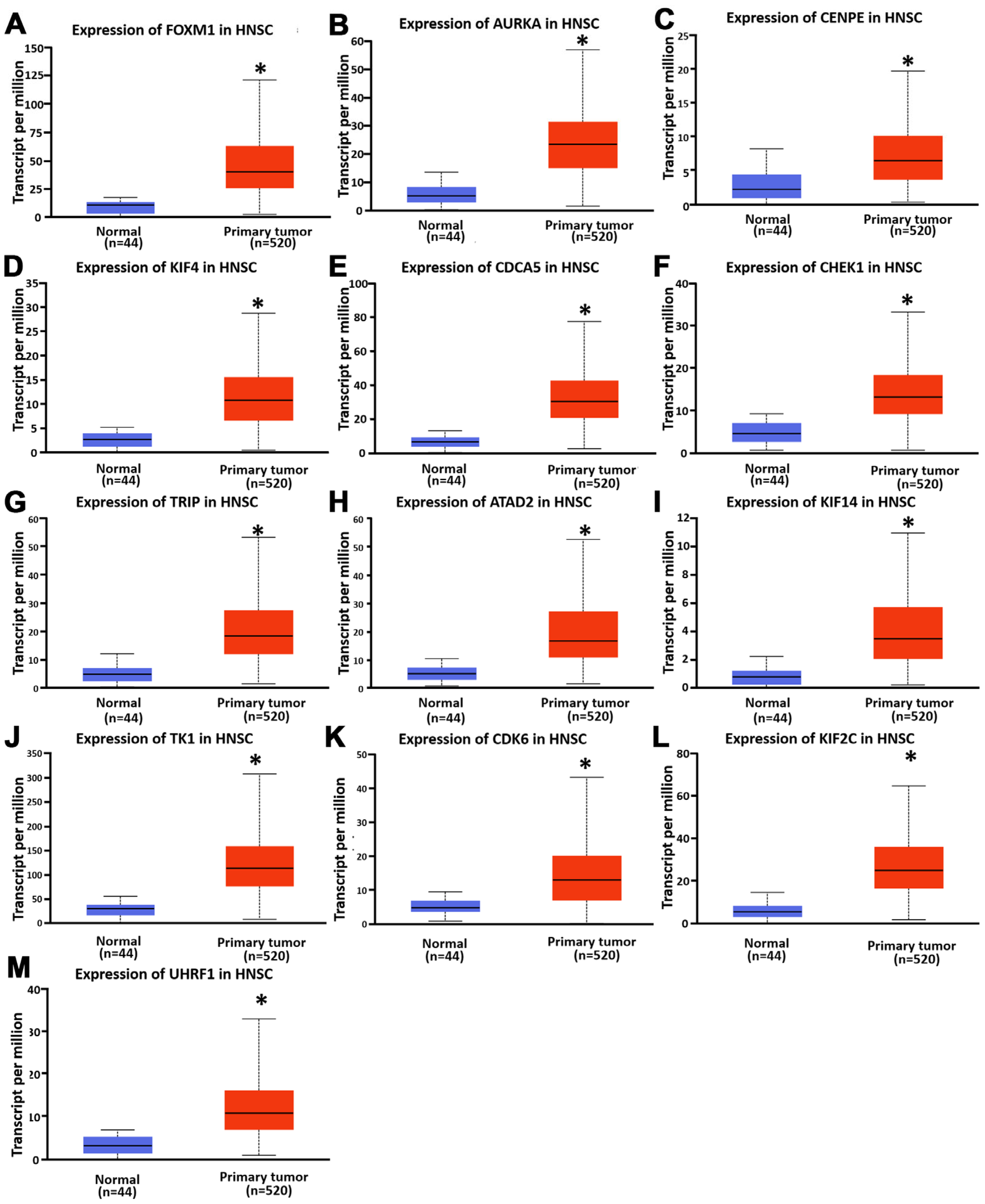
3.3. Expression and Prognostic Significance of Hub Genes in HNSC
3.4. TK1 Is Overexpressed in Multiple Cancers and Correlates with OSCC Progression
3.5. TK1 Expression Promotes OSCC Cell Motility and Invasiveness
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of Variance |
| ATAD2 | ATPase Family AAA Domain Containing 2 |
| AURKA | Aurora Kinase A |
| CC | cellular component |
| CDCA5 | Cell Division Cycle Associated 5 |
| CDK6 | Cyclin-Dependent Kinase 6 |
| CHEK1 | Checkpoint Kinase 1 |
| CPTAC | Clinical Proteomic Tumor Analysis Consortium |
| CENPE | Centromere Protein E |
| DEGs | differentially expressed genes |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMNC | Density of Maximum Neighborhood Component |
| EPC | Edge Percolated Component |
| EMT | epithelial–mesenchymal transition |
| FBS | Fetal Bovine Serum |
| FLT | 3′-Deoxy-3′-[18F] fluorothymidine |
| FOXM1 | Forkhead Box M1 |
| GEO | Gene Expression Omnibus |
| GSE | Gene Expression Series |
| GO | Gene Ontology |
| HGF-1 | Human Gingival Fibroblast-1 |
| HNSC | head and neck squamous cell carcinoma |
| HR | hazard ratio |
| IHC | immunohistochemistry |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KIF2C | Kinesin Family Member 2C |
| KIF4A | Kinesin Family Member 4A |
| KIF14 | Kinesin Family Member 14 |
| KM | Kaplan–Meier |
| MCC | Maximal Clique Centrality |
| MNC | Maximum Neighborhood Component |
| MCODE | Molecular Complex Detection |
| mRNA | Messenger Ribonucleic Acid |
| OS | overall survival |
| OSCC | oral squamous cell carcinoma |
| PCR | Polymerase Chain Reaction |
| PPI | protein–protein interaction |
| qPCR | Quantitative Polymerase Chain Reaction |
| siRNA | Small Interfering RNA |
| STRING | Search Tool for the Retrieval of Interacting Genes/Proteins |
| TCGA | The Cancer Genome Atlas |
| TIMER | Tumor Immune Estimation Resource |
| TK1 | Thymidine Kinase 1 |
| TRIP13 | Thyroid Hormone Receptor Interactor 13 |
| UALCAN | University of Alabama at Birmingham Cancer Data Analysis Portal |
| UHRF1 | Ubiquitin-like with PHD and RING Finger Domains 1 |
| WB | Western blot |
References
- Abdulla, R.; Devasia Puthenpurackal, J.; Pinto, S.M.; Rekha, P.D.; Subbannayya, Y. Serum autoantibody profiling of oral squamous cell carcinoma patients reveals NUBP2 as a potential diagnostic marker. Front. Oncol. 2023, 13, 1167691. [Google Scholar] [CrossRef] [PubMed]
- Kisoda, S.; Mouri, Y.; Kitamura, N.; Yamamoto, T.; Miyoshi, K.; Kudo, Y. The role of partial-EMT in the progression of head and neck squamous cell carcinoma. J. Oral Biosci. 2022, 64, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, J.; Zhu, H. Risk factors for cervical lymph node metastasis of head and neck squamous cell carcinoma: A retrospective population-based study. J. Stomatol. Oral Maxillofac. Surg. 2022, 123, e484–e488. [Google Scholar] [CrossRef] [PubMed]
- Lehyanti, J.; Even, C.; Fessart, E.; Wagner-Ballon, C.; Moreira, A.; Houessinon, A. Management of oligometastatic head and neck squamous cell carcinoma: A systematic review. Oral Oncol. 2024, 159, 107085. [Google Scholar] [CrossRef]
- Chen, F.; Tang, L.; Xia, T.; He, E.; Hu, G.; Li, Y.; Zhang, M.; Zhou, J.; Eriksson, S.; Skog, S. Serum thymidine kinase 1 levels predict cancer-free survival following neoadjuvant, surgical and adjuvant treatment of patients with locally advanced breast cancer. Mol. Clin. Oncol. 2013, 1, 894–902. [Google Scholar] [CrossRef]
- Ji, Y.; Wu, X.B.; Chen, J.Y.; Hu, B.; Zhu, Q.K.; Zhu, X.F.; Zheng, M.F. Serum thymidine kinase 1 levels correlate with clinical characteristics of esophageal squamous cell carcinoma. Int. J. Clin. Exp. Med. 2015, 8, 12850–12857. [Google Scholar]
- Wei, Y.T.; Luo, Y.Z.; Feng, Z.Q.; Huang, Q.X.; Mo, A.S.; Mo, S.X. TK1 overexpression is associated with the poor outcomes of lung cancer patients: A systematic review and meta-analysis. Biomark. Med. 2018, 12, 403–413. [Google Scholar] [CrossRef]
- Singh, S.; Kumar, R.; Kumar, U.; Kumari, R. Clinical Significance and Role of TK1, CEA, CA 19-9 and CA 72-4 levels in Diagnosis of Colorectal Cancers. Asian Pac. J. Cancer Prev. 2020, 21, 3133–3136. [Google Scholar] [CrossRef]
- Meirovitz, A.; Gross, M.; Leibovici, V.; Sheva, K.; Popovzer, A.; Barak, V. Clinical Applicability of the Proliferation Marker Thymidine Kinase 1 in Head and Neck Cancer Patients. Anticancer Res. 2021, 41, 1083–1087. [Google Scholar] [CrossRef]
- Malvi, P.; Janostiak, R.; Nagarajan, A.; Cai, G.; Wajapeyee, N. Loss of thymidine kinase 1 inhibits lung cancer growth and metastatic attributes by reducing GDF15 expression. PLoS Genet. 2019, 15, e1008439. [Google Scholar] [CrossRef]
- Troost, E.G.; Bussink, J.; Slootweg, P.J.; Peeters, W.J.; Merkx, M.A.; van der Kogel, A.J.; Oyen, W.J.; Kaanders, J.H. Histopathologic validation of 3′-deoxy-3′-18F-fluorothymidine PET in squamous cell carcinoma of the oral cavity. J. Nucl. Med. 2010, 51, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Molthoff, C.F.; Klabbers, B.M.; Berkhof, J.; Felten, J.T.; van Gelder, M.; Windhorst, A.D.; Slotman, B.J.; Lammertsma, A.A. Monitoring response to radiotherapy in human squamous cell cancer bearing nude mice: Comparison of 2′-deoxy-2′-[18F]fluoro-D-glucose (FDG) and 3′-[18F]fluoro-3′-deoxythymidine (FLT). Mol. Imaging Biol. 2007, 9, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, D.M.; Clarke, M.J.; Mladek, A.C.; Carlson, B.L.; Trump, D.P.; Jacobson, M.S.; Kemp, B.J.; Lowe, V.J.; Sarkaria, J.N. Using fluorodeoxythymidine to monitor anti-EGFR inhibitor therapy in squamous cell carcinoma xenografts. Head Neck 2008, 30, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Landi, M.T.; Dracheva, T.; Rotunno, M.; Figueroa, J.D.; Liu, H.; Dasgupta, A.; Mann, F.E.; Fukuoka, J.; Hames, M.; Bergen, A.W.; et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS ONE 2008, 3, e1651. [Google Scholar] [CrossRef]
- Lu, T.P.; Hsiao, C.K.; Lai, L.C.; Tsai, M.H.; Hsu, C.P.; Lee, J.M.; Chuang, E.Y. Identification of regulatory SNPs associated with genetic modifications in lung adenocarcinoma. BMC Res. Notes 2015, 8, 92. [Google Scholar] [CrossRef]
- Sanchez-Palencia, A.; Gomez-Morales, M.; Gomez-Capilla, J.A.; Pedraza, V.; Boyero, L.; Rosell, R.; Farez-Vidal, M.E. Gene expression profiling reveals novel biomarkers in nonsmall cell lung cancer. Int. J. Cancer 2011, 129, 355–364. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef]
- Hou, G.X.; Liu, P.; Yang, J.; Wen, S. Mining expression and prognosis of topoisomerase isoforms in non-small-cell lung cancer by using Oncomine and Kaplan-Meier plotter. PLoS ONE 2017, 12, e0174515. [Google Scholar] [CrossRef]
- Gyorffy, B. Transcriptome-level discovery of survival-associated biomarkers and therapy targets in non-small-cell lung cancer. Br. J. Pharmacol. 2024, 181, 362–374. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Q.; Zhou, X.; He, Y.; Guo, Q.; Shi, Q.; Eriksson, S.; Zhou, J.; He, E.; Skog, S. Thymidine kinase 1 expression in ovarian serous adenocarcinoma is superior to Ki-67: A new prognostic biomarker. Tumour Biol. 2017, 39, 1010428317706479. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Wang, Z.; Xu, M.; Li, B.; Huang, Z.; Qin, S.; Nice, E.C.; Tang, J.; Huang, C. Oral squamous cell carcinomas: State of the field and emerging directions. Int. J. Oral Sci. 2023, 15, 44. [Google Scholar] [CrossRef]
- Jagadeesan, D.; Sathasivam, K.V.; Fuloria, N.K.; Balakrishnan, V.; Khor, G.H.; Ravichandran, M.; Solyappan, M.; Fuloria, S.; Gupta, G.; Ahlawat, A.; et al. Comprehensive insights into oral squamous cell carcinoma: Diagnosis, pathogenesis, and therapeutic advances. Pathol. Res. Pract. 2024, 261, 155489. [Google Scholar] [CrossRef]
- Zhu, C.; Zhang, N.; Zhong, A.; Xiao, K.; Lu, R.; Guo, L. A combined strategy of TK1, HE4 and CA125 shows better diagnostic performance than risk of ovarian malignancy algorithm (ROMA) in ovarian carcinoma. Clin. Chim. Acta 2022, 524, 43–50. [Google Scholar] [CrossRef]
- Song, X.; Skog, S.; Wei, L.; Qin, J.; Yang, R.; Li, J.; Zhou, J.; He, E.; Zhou, J. Nomogram model of serum thymidine kinase 1 combined with ultrasonography for prediction of central lymph node metastasis risk in patients with papillary thyroid carcinoma pre-surgery. Front. Endocrinol. 2024, 15, 1366219. [Google Scholar] [CrossRef]
- Sun, A.; Chen, H.; Shi, X.; Shang, Z.; Zhang, J. Diagnostic Value of Joint Detection of Serum TK1, TSGF, CA199, and CA724 for Gastric Cancer and Its Relationship with Clinicopathologic Features and Prognosis. Am. Surg. 2025, 91, 570–578. [Google Scholar] [CrossRef]
- Wu, M.; Ye, M. Transcription factor Dp-1 knockdown downregulates thymidine kinase 1 expression to protect against proliferation and epithelial-mesenchymal transition in cervical cancer. Funct. Integr. Genom. 2023, 23, 301. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-J.; Peng, P.-W.; Wu, C.-Y.; Chang, T.-M.; Liu, J.-F.; Lin, K.-C. Thymidine Kinase 1 Expression Correlates with Tumor Aggressiveness and Metastatic Potential in OSCC. Diagnostics 2025, 15, 1567. https://doi.org/10.3390/diagnostics15121567
Lee C-J, Peng P-W, Wu C-Y, Chang T-M, Liu J-F, Lin K-C. Thymidine Kinase 1 Expression Correlates with Tumor Aggressiveness and Metastatic Potential in OSCC. Diagnostics. 2025; 15(12):1567. https://doi.org/10.3390/diagnostics15121567
Chicago/Turabian StyleLee, Chia-Jung, Pei-Wen Peng, Chia-Yu Wu, Tsung-Ming Chang, Ju-Fang Liu, and Kuan-Chou Lin. 2025. "Thymidine Kinase 1 Expression Correlates with Tumor Aggressiveness and Metastatic Potential in OSCC" Diagnostics 15, no. 12: 1567. https://doi.org/10.3390/diagnostics15121567
APA StyleLee, C.-J., Peng, P.-W., Wu, C.-Y., Chang, T.-M., Liu, J.-F., & Lin, K.-C. (2025). Thymidine Kinase 1 Expression Correlates with Tumor Aggressiveness and Metastatic Potential in OSCC. Diagnostics, 15(12), 1567. https://doi.org/10.3390/diagnostics15121567