
Unraveling the Genetic Architecture of Obesity: A Path to Personalized Medicine

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Genetic Architecture
4. Genomic and Epigenomic Landscapes of Obesity
5. Genetic and Polygenic Risk Scores for Obesity
6. Nutrigenetics and Obesity: Impact on Underlying Pathologies
7. The Gut Microbiome and Metabolome in Obesity: Implications for Metabolic Dysfunction and Personalized Approaches
8. Pharmacotherapy for Monogenic Obesity Disorders: A Model for Personalized Targeting
9. Challenges in Personalized Medicine for Obesity
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- World Health Organization. Obesity: Preventing and Managing the Global Epidemic, Report of a WHO Consultation; Technical Report Series 894; WHO: Geneva, Switzerland, 2000. [Google Scholar]
- World Health Organization. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 3 April 2024).
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Hubert, H.B.; Feinleib, M.; McNamara, P.M.; Castelli, W.P. Obesity as an independent risk factor for cardiovascular disease: A 26-year follow-up of participants in the Framingham Heart Study. Circulation 1983, 67, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Iseki, K.; Ikemiya, Y.; Kinjo, K.; Inoue, T.; Iseki, C.; Takishita, S. Body mass index and the risk of development of end-stage renal disease in a screened cohort. Kidney Int. 2004, 65, 1870–1876. [Google Scholar] [CrossRef] [PubMed]
- Anandacoomarasamy, A.; Caterson, I.; Sambrook, P.; Fransen, M.; March, L. The impact of obesity on the musculoskeletal system. Int. J. Obes. 2008, 32, 211–222. [Google Scholar] [CrossRef]
- Scott, K.M.; McGee, M.A.; Wells, J.E.; Oakley Browne, M.A. Obesity and mental disorders in the adult general population. J. Psychosom. Res. 2008, 64, 97–105. [Google Scholar] [CrossRef]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef]
- Dalgaard, K.; Landgraf, K.; Heyne, S.; Lempradl, A.; Longinotto, J.; Gossens, K.; Ruf, M.; Orthofer, M.; Strogantsev, R.; Selvaraj, M.; et al. Trim28 Haploinsufficiency Triggers Bi-stable Epigenetic Obesity. Cell 2016, 164, 353–364. [Google Scholar] [CrossRef]
- Koza, R.A.; Nikonova, L.; Hogan, J.; Rim, J.S.; Mendoza, T.; Faulk, C.; Skaf, J.; Kozak, L.P. Changes in gene expression foreshadow diet-induced obesity in genetically identical mice. PLoS Genet. 2006, 2, e81. [Google Scholar] [CrossRef]
- Lajunen, H.R.; Kaprio, J.; Keski-Rahkonen, A.; Rose, R.J.; Pulkkinen, L.; Rissanen, A.; Silventoinen, K. Genetic and environmental effects on body mass index during adolescence: A prospective study among Finnish twins. Int. J. Obes. 2009, 33, 559–567. [Google Scholar] [CrossRef]
- Piché, M.-E.; Poirier, P.; Lemieux, I.; Després, J.-P. Overview of epidemiology and contribution of obesity and body fat distribution to cardiovascular disease: An update. Prog. Cardiovasc. Dis. 2018, 61, 103–113. [Google Scholar] [CrossRef]
- Loos, R.J.F.; Yeo, G.S.H. The genetics of obesity: From discovery to biology. Nat. Rev. Genet. 2022, 23, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Cass, S. The age of the zettabyte cisco: The future of internet traffic is video [dataflow]. IEEE Spectr. 2014, 51, 68. [Google Scholar] [CrossRef]
- Hood, L.; Flores, M. A personal view on systems medicine and the emergence of proactive P4 medicine: Predictive, preventive, personalized and participatory. New Biotechnol. 2012, 29, 613–624. [Google Scholar] [CrossRef]
- Toledo, R.A.; Sekiya, T.; Longuini, V.C.; Coutinho, F.L.; Lourenço, D.M., Jr.; Toledo, S.P. Narrowing the gap of personalized medicine in emerging countries: The case of multiple endocrine neoplasias in Brazil. Clinics 2012, 67 (Suppl. S1), 3–6. [Google Scholar] [CrossRef]
- Fenech, M.; El-Sohemy, A.; Cahill, L.; Ferguson, L.R.; French, T.A.; Tai, E.S.; Milner, J.; Koh, W.P.; Xie, L.; Zucker, M.; et al. Nutrigenetics and nutrigenomics: Viewpoints on the current status and applications in nutrition research and practice. J. Nutr. Nutr. 2011, 4, 69–89. [Google Scholar] [CrossRef]
- Boender, A.J.; van Rozen, A.J.; Adan, R.A. Nutritional state affects the expression of the obesity-associated genes Etv5, Faim2, Fto, and Negr1. Obesity 2012, 20, 2420–2425. [Google Scholar] [CrossRef]
- Ho, E.V.; Klenotich, S.J.; McMurray, M.S.; Dulawa, S.C. Activity-Based Anorexia Alters the Expression of BDNF Transcripts in the Mesocorticolimbic Reward Circuit. PLoS ONE 2016, 11, e0166756. [Google Scholar] [CrossRef]
- Horstmann, A.; Kovacs, P.; Kabisch, S.; Boettcher, Y.; Schloegl, H.; Tönjes, A.; Stumvoll, M.; Pleger, B.; Villringer, A. Common genetic variation near MC4R has a sex-specific impact on human brain structure and eating behavior. PLoS ONE 2013, 8, e74362. [Google Scholar] [CrossRef]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef]
- Frayling, T.M.; Timpson, N.J.; Weedon, M.N.; Zeggini, E.; Freathy, R.M.; Lindgren, C.M.; Perry, J.R.; Elliott, K.S.; Lango, H.; Rayner, N.W.; et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 2007, 316, 889–894. [Google Scholar] [CrossRef]
- Akiyama, M.; Okada, Y.; Kanai, M.; Takahashi, A.; Momozawa, Y.; Ikeda, M.; Iwata, N.; Ikegawa, S.; Hirata, M.; Matsuda, K.; et al. Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nat. Genet. 2017, 49, 1458–1467. [Google Scholar] [CrossRef] [PubMed]
- Rohde, K.; Keller, M.; la Cour Poulsen, L.; Blüher, M.; Kovacs, P.; Böttcher, Y. Genetics and epigenetics in obesity. Metabolism 2019, 92, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Graff, M.; Scott, R.A.; Justice, A.E.; Young, K.L.; Feitosa, M.F.; Barata, L.; Winkler, T.W.; Chu, A.Y.; Mahajan, A.; Hadley, D.; et al. Genome-wide physical activity interactions in adiposity—A meta-analysis of 200,452 adults. PLoS Genet. 2017, 13, e1006528. [Google Scholar] [CrossRef] [PubMed]
- Maes, H.H.; Neale, M.C.; Eaves, L.J. Genetic and environmental factors in relative body weight and human adiposity. Behav. Genet. 1997, 27, 325–351. [Google Scholar] [CrossRef]
- Wardle, J.; Carnell, S.; Haworth, C.M.; Plomin, R. Evidence for a strong genetic influence on childhood adiposity despite the force of the obesogenic environment. Am. J. Clin. Nutr. 2008, 87, 398–404. [Google Scholar] [CrossRef]
- Riveros-McKay, F.; Mistry, V.; Bounds, R.; Hendricks, A.; Keogh, J.M.; Thomas, H.; Henning, E.; Corbin, L.J.; O’Rahilly, S.; Zeggini, E.; et al. Genetic architecture of human thinness compared to severe obesity. PLoS Genet. 2019, 15, e1007603. [Google Scholar] [CrossRef]
- Walley, A.J.; Asher, J.E.; Froguel, P. The genetic contribution to non-syndromic human obesity. Nat. Rev. Genet. 2009, 10, 431–442. [Google Scholar] [CrossRef]
- MacArthur, J.; Bowler, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E.; Junkins, H.; McMahon, A.; Milano, A.; Morales, J.; et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017, 45, D896–D901. [Google Scholar] [CrossRef]
- Su, L.N.; Wang, Y.B.; Wnag, C.G.; Wei, H.P. Network analysis identifies common genes associated with obesity in six obesity-related diseases. J. Zhejiang Univ. Sci. B 2017, 18, 727–732. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef]
- Chiang, K.M.; Chang, H.C.; Yang, H.C.; Chen, C.H.; Chen, H.H.; Lee, W.J.; Pan, W.H. Genome-wide association study of morbid obesity in Han Chinese. BMC Genet. 2019, 20, 97. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, I.; Ohseto, H.; Ueno, F.; Oonuma, T.; Narita, A.; Obara, T.; Ishikuro, M.; Murakami, K.; Noda, A.; Hozawa, A.; et al. Genome-wide association study based on clustering by obesity-related variables uncovers a genetic architecture of obesity in the Japanese and the UK populations. Heliyon 2024, 10, e36023. [Google Scholar] [CrossRef] [PubMed]
- El Hajj Chehadeh, S.; Osman, W.; Nazar, S.; Jerman, L.; Alghafri, A.; Sajwani, A.; Alawlaqi, M.; AlObeidli, M.; Jelinek, H.F.; AlAnouti, F.; et al. Implication of genetic variants in overweight and obesity susceptibility among the young Arab population of the United Arab Emirates. Gene 2020, 739, 144509. [Google Scholar] [CrossRef] [PubMed]
- Grarup, N.; Moltke, I.; Andersen, M.K.; Dalby, M.; Vitting-Seerup, K.; Kern, T.; Mahendran, Y.; Jørsboe, E.; Larsen, C.V.L.; Dahl-Petersen, I.K.; et al. Loss-of-function variants in ADCY3 increase risk of obesity and type 2 diabetes. Nat. Genet. 2018, 50, 172–174. [Google Scholar] [CrossRef]
- Saeed, S.; Bonnefond, A.; Tamanini, F.; Mirza, M.U.; Manzoor, J.; Janjua, Q.M.; Din, S.M.; Gaitan, J.; Milochau, A.; Durand, E.; et al. Loss-of-function mutations in ADCY3 cause monogenic severe obesity. Nat. Genet. 2018, 50, 175–179. [Google Scholar] [CrossRef]
- Siljee, J.E.; Wang, Y.; Bernard, A.A.; Ersoy, B.A.; Zhang, S.; Marley, A.; Von Zastrow, M.; Reiter, J.F.; Vaisse, C. Subcellular localization of MC4R with ADCY3 at neuronal primary cilia underlies a common pathway for genetic predisposition to obesity. Nat. Genet. 2018, 50, 180–185. [Google Scholar] [CrossRef]
- Yengo, L.; Sidorenko, J.; Kemper, K.E.; Zheng, Z.; Wood, A.R.; Weedon, M.N.; Frayling, T.M.; Hirschhorn, J.; Yang, J.; Visscher, P.M. Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum. Mol. Genet. 2018, 27, 3641–3649. [Google Scholar] [CrossRef]
- Cole, J.B.; Florez, J.C.; Hirschhorn, J.N. Comprehensive genomic analysis of dietary habits in UK Biobank identifies hundreds of genetic associations. Nat. Commun. 2020, 11, 1467. [Google Scholar] [CrossRef]
- Acosta, A. Precision medicine and obesity. Gastroenterol. Clin. 2021, 50, 127–139. [Google Scholar]
- Cordero, P.; Li, J.; Oben, J.A. Epigenetics of obesity: Beyond the genome sequence. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 361–366. [Google Scholar] [CrossRef]
- Barres, R.; Kirchner, H.; Rasmussen, M.; Yan, J.; Kantor, F.R.; Krook, A.; Näslund, E.; Zierath, J.R. Weight loss after gastric bypass surgery in human obesity remodels promoter methylation. Cell Rep. 2013, 3, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Milagro, F.I.; Campión, J.; Cordero, P.; Goyenechea, E.; Gómez-Uriz, A.M.; Abete, I.; Zulet, M.A.; Martínez, J.A. A dual epigenomic approach for the search of obesity biomarkers: DNA methylation in relation to diet-induced weight loss. Faseb J. 2011, 25, 1378–1389. [Google Scholar] [CrossRef] [PubMed]
- Rönn, T.; Volkov, P.; Davegårdh, C.; Dayeh, T.; Hall, E.; Olsson, A.H.; Nilsson, E.; Tornberg, A.; Dekker Nitert, M.; Eriksson, K.F.; et al. A six months exercise intervention influences the genome-wide DNA methylation pattern in human adipose tissue. PLoS Genet. 2013, 9, e1003572. [Google Scholar] [CrossRef]
- Voisin, S.; Eynon, N.; Yan, X.; Bishop, D.J. Exercise training and DNA methylation in humans. Acta Physiol. 2015, 213, 39–59. [Google Scholar] [CrossRef]
- Lindholm, M.E.; Marabita, F.; Gomez-Cabrero, D.; Rundqvist, H.; Ekström, T.J.; Tegnér, J.; Sundberg, C.J. An integrative analysis reveals coordinated reprogramming of the epigenome and the transcriptome in human skeletal muscle after training. Epigenetics 2014, 9, 1557–1569. [Google Scholar] [CrossRef]
- Demerath, E.W.; Guan, W.; Grove, M.L.; Aslibekyan, S.; Mendelson, M.; Zhou, Y.H.; Hedman, Å.K.; Sandling, J.K.; Li, L.A.; Irvin, M.R.; et al. Epigenome-wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum. Mol. Genet. 2015, 24, 4464–4479. [Google Scholar] [CrossRef]
- Dick, K.J.; Nelson, C.P.; Tsaprouni, L.; Sandling, J.K.; Aïssi, D.; Wahl, S.; Meduri, E.; Morange, P.E.; Gagnon, F.; Grallert, H.; et al. DNA methylation and body-mass index: A genome-wide analysis. Lancet 2014, 383, 1990–1998. [Google Scholar] [CrossRef]
- Irvin, M.R.; Zhi, D.; Joehanes, R.; Mendelson, M.; Aslibekyan, S.; Claas, S.A.; Thibeault, K.S.; Patel, N.; Day, K.; Jones, L.W.; et al. Epigenome-wide association study of fasting blood lipids in the Genetics of Lipid-lowering Drugs and Diet Network study. Circulation 2014, 130, 565–572. [Google Scholar] [CrossRef]
- Aslibekyan, S.; Demerath, E.W.; Mendelson, M.; Zhi, D.; Guan, W.; Liang, L.; Sha, J.; Pankow, J.S.; Liu, C.; Irvin, M.R.; et al. Epigenome-wide study identifies novel methylation loci associated with body mass index and waist circumference. Obesity 2015, 23, 1493–1501. [Google Scholar] [CrossRef]
- Campanella, G.; Gunter, M.J.; Polidoro, S.; Krogh, V.; Palli, D.; Panico, S.; Sacerdote, C.; Tumino, R.; Fiorito, G.; Guarrera, S.; et al. Epigenome-wide association study of adiposity and future risk of obesity-related diseases. Int. J. Obes. 2018, 42, 2022–2035. [Google Scholar] [CrossRef]
- Lee, Y.C.; Christensen, J.J.; Parnell, L.D.; Smith, C.E.; Shao, J.; McKeown, N.M.; Ordovás, J.M.; Lai, C.Q. Using Machine Learning to Predict Obesity Based on Genome-Wide and Epigenome-Wide Gene-Gene and Gene-Diet Interactions. Front. Genet. 2021, 12, 783845. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.; Drong, A.; Lehne, B.; Loh, M.; Scott, W.R.; Kunze, S.; Tsai, P.C.; Ried, J.S.; Zhang, W.; Yang, Y.; et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 2017, 541, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Meeks, K.A.C.; Henneman, P.; Venema, A.; Burr, T.; Galbete, C.; Danquah, I.; Schulze, M.B.; Mockenhaupt, F.P.; Owusu-Dabo, E.; Rotimi, C.N.; et al. An epigenome-wide association study in whole blood of measures of adiposity among Ghanaians: The RODAM study. Clin. Epigenetics 2017, 9, 103. [Google Scholar] [CrossRef]
- Villicaña, S.; Castillo-Fernandez, J.; Hannon, E.; Christiansen, C.; Tsai, P.C.; Maddock, J.; Kuh, D.; Suderman, M.; Power, C.; Relton, C.; et al. Genetic impacts on DNA methylation help elucidate regulatory genomic processes. Genome Biol. 2023, 24, 176. [Google Scholar] [CrossRef]
- Claussnitzer, M.; Dankel, S.N.; Kim, K.H.; Quon, G.; Meuleman, W.; Haugen, C.; Glunk, V.; Sousa, I.S.; Beaudry, J.L.; Puviindran, V.; et al. FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N. Engl. J. Med. 2015, 373, 895–907. [Google Scholar] [CrossRef]
- Kilpeläinen, T.O.; Qi, L.; Brage, S.; Sharp, S.J.; Sonestedt, E.; Demerath, E.; Ahmad, T.; Mora, S.; Kaakinen, M.; Sandholt, C.H.; et al. Physical activity attenuates the influence of FTO variants on obesity risk: A meta-analysis of 218,166 adults and 19,268 children. PLoS Med. 2011, 8, e1001116. [Google Scholar] [CrossRef]
- Loos, R.J.; Yeo, G.S. The bigger picture of FTO: The first GWAS-identified obesity gene. Nat. Rev. Endocrinol. 2014, 10, 51–61. [Google Scholar] [CrossRef]
- Ang, M.Y.; Takeuchi, F.; Kato, N. Deciphering the genetic landscape of obesity: A data-driven approach to identifying plausible causal genes and therapeutic targets. J. Hum. Genet. 2023, 68, 823–833. [Google Scholar] [CrossRef]
- Chung, J.Y.; Jung, H.U.; Kim, D.J.; Baek, E.J.; Kim, H.K.; Kang, J.O.; Lim, J.E.; Oh, B. Identification of five genetic variants with differential effects on obesity-related traits based on age. Front. Genet. 2022, 13, 970657. [Google Scholar] [CrossRef]
- Felix, J.F.; Bradfield, J.P.; Monnereau, C.; van der Valk, R.J.; Stergiakouli, E.; Chesi, A.; Gaillard, R.; Feenstra, B.; Thiering, E.; Kreiner-Møller, E.; et al. Genome-wide association analysis identifies three new susceptibility loci for childhood body mass index. Hum. Mol. Genet. 2016, 25, 389–403. [Google Scholar] [CrossRef]
- Hägg, S.; Ganna, A.; Van Der Laan, S.W.; Esko, T.; Pers, T.H.; Locke, A.E.; Berndt, S.I.; Justice, A.E.; Kahali, B.; Siemelink, M.A.; et al. Gene-based meta-analysis of genome-wide association studies implicates new loci involved in obesity. Hum. Mol. Genet. 2015, 24, 6849–6860. [Google Scholar] [CrossRef] [PubMed]
- Heid, I.M.; Jackson, A.U.; Randall, J.C.; Winkler, T.W.; Qi, L.; Steinthorsdottir, V.; Thorleifsson, G.; Zillikens, M.C.; Speliotes, E.K.; Mägi, R.; et al. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat. Genet. 2010, 42, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Hinney, A.; Nguyen, T.T.; Scherag, A.; Friedel, S.; Brönner, G.; Müller, T.D.; Grallert, H.; Illig, T.; Wichmann, H.E.; Rief, W.; et al. Genome wide association (GWA) study for early onset extreme obesity supports the role of fat mass and obesity associated gene (FTO) variants. PLoS ONE 2007, 2, e1361. [Google Scholar] [CrossRef]
- Kilpeläinen, T.O.; Carli, J.F.; Skowronski, A.A.; Sun, Q.; Kriebel, J.; Feitosa, M.F.; Hedman, Å.K.; Drong, A.W.; Hayes, J.E.; Zhao, J.; et al. Genome-wide meta-analysis uncovers novel loci influencing circulating leptin levels. Nat. Commun. 2016, 7, 10494. [Google Scholar] [CrossRef]
- Lee, W.J.; Lim, J.E.; Kang, J.O.; Ha, T.W.; Jung, H.U.; Kim, D.J.; Baek, E.J.; Kim, H.K.; Chung, J.Y.; Oh, B. Smoking-Interaction Loci Affect Obesity Traits: A Gene-Smoking Stratified Meta-Analysis of 545,131 Europeans. Lifestyle Genom. 2022, 15, 87–97. [Google Scholar] [CrossRef]
- Li, S.; Zhao, J.H.; Luan, J.; Luben, R.N.; Rodwell, S.A.; Khaw, K.T.; Ong, K.K.; Wareham, N.J.; Loos, R.J. Cumulative effects and predictive value of common obesity-susceptibility variants identified by genome-wide association studies. Am. J. Clin. Nutr. 2010, 91, 184–190. [Google Scholar] [CrossRef]
- Liu, Y.J.; Liu, X.G.; Wang, L.; Dina, C.; Yan, H.; Liu, J.F.; Levy, S.; Papasian, C.J.; Drees, B.M.; Hamilton, J.J.; et al. Genome-wide association scans identified CTNNBL1 as a novel gene for obesity. Hum. Mol. Genet. 2008, 17, 1803–1813. [Google Scholar] [CrossRef]
- Liu, S.; Wilson, J.G.; Jiang, F.; Griswold, M.; Correa, A.; Mei, H. Multi-variant study of obesity risk genes in African Americans: The Jackson Heart Study. Gene 2016, 593, 315–321. [Google Scholar] [CrossRef]
- Liu, L.; Fan, Q.; Zhang, F.; Guo, X.; Liang, X.; Du, Y.; Li, P.; Wen, Y.; Hao, J.; Wang, W.; et al. A Genomewide Integrative Analysis of GWAS and eQTLs Data Identifies Multiple Genes and Gene Sets Associated with Obesity. Biomed Res. Int. 2018, 2018, 3848560. [Google Scholar] [CrossRef]
- Lv, D.; Zhang, D.D.; Wang, H.; Zhang, Y.; Liang, L.; Fu, J.F.; Xiong, F.; Liu, G.L.; Gong, C.X.; Luo, F.H.; et al. Genetic variations in SEC16B, MC4R, MAP2K5 and KCTD15 were associated with childhood obesity and interacted with dietary behaviors in Chinese school-age population. Gene 2015, 560, 149–155. [Google Scholar] [CrossRef]
- Mägi, R.; Manning, S.; Yousseif, A.; Pucci, A.; Santini, F.; Karra, E.; Querci, G.; Pelosini, C.; McCarthy, M.I.; Lindgren, C.M.; et al. Contribution of 32 GWAS-identified common variants to severe obesity in European adults referred for bariatric surgery. PLoS ONE 2013, 8, e70735. [Google Scholar] [CrossRef] [PubMed]
- Mejía-Benítez, A.; Klünder-Klünder, M.; Yengo, L.; Meyre, D.; Aradillas, C.; Cruz, E.; Pérez-Luque, E.; Malacara, J.M.; Garay, M.E.; Peralta-Romero, J.; et al. Analysis of the contribution of FTO, NPC1, ENPP1, NEGR1, GNPDA2 and MC4R genes to obesity in Mexican children. BMC Med. Genet. 2013, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.F.; Zhang, L.; Liu, Y.; Li, J.; Shen, H.; Liu, Y.Z.; Tian, Q.; He, H.; Wu, S.; Ran, S.; et al. Meta-analysis of genome-wide association data identifies novel susceptibility loci for obesity. Hum. Mol. Genet. 2014, 23, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Pulit, S.L.; Stoneman, C.; Morris, A.P.; Wood, A.R.; Glastonbury, C.A.; Tyrrell, J.; Yengo, L.; Ferreira, T.; Marouli, E.; Ji, Y.; et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum. Mol. Genet. 2019, 28, 166–174. [Google Scholar] [CrossRef]
- Rask-Andersen, M.; Jacobsson, J.A.; Moschonis, G.; Ek, A.E.; Chrousos, G.P.; Marcus, C.; Manios, Y.; Fredriksson, R.; Schiöth, H.B. The MAP2K5-linked SNP rs2241423 is associated with BMI and obesity in two cohorts of Swedish and Greek children. BMC Med. Genet. 2012, 13, 36. [Google Scholar] [CrossRef]
- Shungin, D.; Winkler, T.W.; Croteau-Chonka, D.C.; Ferreira, T.; Locke, A.E.; Mägi, R.; Strawbridge, R.J.; Pers, T.H.; Fischer, K.; Justice, A.E.; et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature 2015, 518, 187–196. [Google Scholar] [CrossRef]
- Tang, J.; Xu, H.; Xin, Z.; Mei, Q.; Gao, M.; Yang, T.; Zhang, X.; Levy, D.; Liu, C.T. Identifying BMI-associated genes via a genome-wide multi-omics integrative approach using summary data. Hum. Mol. Genet. 2024, 33, 733–738. [Google Scholar] [CrossRef]
- Wang, K.; Li, W.D.; Zhang, C.K.; Wang, Z.; Glessner, J.T.; Grant, S.F.; Zhao, H.; Hakonarson, H.; Price, R.A. A genome-wide association study on obesity and obesity-related traits. PLoS ONE 2011, 6, e18939. [Google Scholar] [CrossRef]
- Taylor, J.Y.; Huang, Y.; Zhao, W.; Wright, M.L.; Wang, Z.; Hui, Q.; Potts-Thompson, S.; Barcelona, V.; Prescott, L.; Yao, Y.; et al. Epigenome-wide association study of BMI in Black populations from InterGEN and GENOA. Obesity 2023, 31, 243–255. [Google Scholar] [CrossRef]
- Do, W.L.; Sun, D.; Meeks, K.; Dugué, P.A.; Demerath, E.; Guan, W.; Li, S.; Chen, W.; Milne, R.; Adeyemo, A.; et al. Epigenome-wide meta-analysis of BMI in nine cohorts: Examining the utility of epigenetically predicted BMI. Am. J. Hum. Genet. 2023, 110, 273–283. [Google Scholar] [CrossRef]
- Alfano, R.; Zugna, D.; Barros, H.; Bustamante, M.; Chatzi, L.; Ghantous, A.; Herceg, Z.; Keski-Rahkonen, P.; de Kok, T.M.; Nawrot, T.S.; et al. Cord blood epigenome-wide meta-analysis in six European-based child cohorts identifies signatures linked to rapid weight growth. BMC Med. 2023, 21, 17. [Google Scholar] [CrossRef] [PubMed]
- Ali, O.; Cerjak, D.; Kent, J.W., Jr.; James, R.; Blangero, J.; Carless, M.A.; Zhang, Y. Methylation of SOCS3 is inversely associated with metabolic syndrome in an epigenome-wide association study of obesity. Epigenetics 2016, 11, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kassam, I.; Lau, S.H.; Kooner, J.S.; Wilson, R.; Peters, A.; Winkelmann, J.; Chambers, J.C.; Chow, V.T.; Khor, C.C.; et al. Impact of BMI and waist circumference on epigenome-wide DNA methylation and identification of epigenetic biomarkers in blood: An EWAS in multi-ethnic Asian individuals. Clin. Epigenetics 2021, 13, 195. [Google Scholar] [CrossRef]
- Dhana, K.; Braun, K.V.E.; Nano, J.; Voortman, T.; Demerath, E.W.; Guan, W.; Fornage, M.; van Meurs, J.B.J.; Uitterlinden, A.G.; Hofman, A.; et al. An Epigenome-Wide Association Study of Obesity-Related Traits. Am. J. Epidemiol. 2018, 187, 1662–1669. [Google Scholar] [CrossRef]
- Kvaløy, K.; Page, C.M.; Holmen, T.L. Epigenome-wide methylation differences in a group of lean and obese women—A HUNT Study. Sci. Rep. 2018, 8, 16330. [Google Scholar] [CrossRef]
- Li, W.; Xia, M.; Zeng, H.; Lin, H.; Teschendorff, A.E.; Gao, X.; Wang, S. Longitudinal analysis of epigenome-wide DNA methylation reveals novel loci associated with BMI change in East Asians. Clin. Epigenetics 2024, 16, 70. [Google Scholar] [CrossRef]
- Nikpay, M.; Ravati, S.; Dent, R.; McPherson, R. Epigenome-Wide Study Identified Methylation Sites Associated with the Risk of Obesity. Nutrients 2021, 13, 1984. [Google Scholar] [CrossRef]
- Sayols-Baixeras, S.; Subirana, I.; Fernández-Sanlés, A.; Sentí, M.; Lluís-Ganella, C.; Marrugat, J.; Elosua, R. DNA methylation and obesity traits: An epigenome-wide association study. The REGICOR study. Epigenetics 2017, 12, 909–916. [Google Scholar] [CrossRef]
- Vehmeijer, F.O.L.; Küpers, L.K.; Sharp, G.C.; Salas, L.A.; Lent, S.; Jima, D.D.; Tindula, G.; Reese, S.; Qi, C.; Gruzieva, O.; et al. DNA methylation and body mass index from birth to adolescence: Meta-analyses of epigenome-wide association studies. Genome Med. 2020, 12, 105. [Google Scholar] [CrossRef]
- Wang, X.; Pan, Y.; Zhu, H.; Hao, G.; Huang, Y.; Barnes, V.; Shi, H.; Snieder, H.; Pankow, J.; North, K.; et al. An epigenome-wide study of obesity in African American youth and young adults: Novel findings, replication in neutrophils, and relationship with gene expression. Clin. Epigenetics 2018, 10, 3. [Google Scholar] [CrossRef]
- Xie, T.; Gorenjak, V.; Stathopoulou, M.G.; Dadé, S.; Marouli, E.; Masson, C.; Murray, H.; Lamont, J.; Fitzgerald, P.; Deloukas, P.; et al. Epigenome-wide association study detects a novel loci associated with central obesity in healthy subjects. BMC Med. Genom. 2021, 14, 233. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Fan, B.; Huang, J.; Cowling, B.J.; Au Yeung, S.L.R.; Baccarelli, A.; Leung, G.M.; Schooling, C.M. Environment- and epigenome-wide association study of obesity in ‘Children of 1997’ birth cohort. eLife 2023, 12, e82377. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Lopez, O.; Cuervo, M.; Goni, L.; Milagro, F.I.; Riezu-Boj, J.I.; Martinez, J.A. Modeling of an integrative prototype based on genetic, phenotypic, and environmental information for personalized prescription of energy-restricted diets in overweight/obese subjects. Am. J. Clin. Nutr. 2020, 111, 459–470. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Wade, K.H.; Zahid, S.; Brancale, J.; Xia, R.; Distefano, M.; Senol-Cosar, O.; Haas, M.E.; Bick, A.; et al. Polygenic Prediction of Weight and Obesity Trajectories from Birth to Adulthood. Cell 2019, 177, 587–596.e589. [Google Scholar] [CrossRef]
- Goodarzi, M.O. Genetics of obesity: What genetic association studies have taught us about the biology of obesity and its complications. Lancet Diabetes Endocrinol. 2018, 6, 223–236. [Google Scholar] [CrossRef]
- Barrea, L.; Annunziata, G.; Bordoni, L.; Muscogiuri, G.; Colao, A.; Savastano, S. Nutrigenetics-personalized nutrition in obesity and cardiovascular diseases. Int. J. Obes. Suppl. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- He, Z.; Zhang, R.; Jiang, F.; Zhang, H.; Zhao, A.; Xu, B.; Jin, L.; Wang, T.; Jia, W.; Jia, W.; et al. FADS1-FADS2 genetic polymorphisms are associated with fatty acid metabolism through changes in DNA methylation and gene expression. Clin. Epigenetics 2018, 10, 113. [Google Scholar] [CrossRef]
- Bäck, M.; Xhaard, C.; Rouget, R.; Thuillier, Q.; Plunde, O.; Larsson, S.C.; Girerd, N.; Ferreira, J.P.; Boivin, J.M.; Bozec, E.; et al. Fatty acid desaturase genetic variations and dietary omega-3 fatty acid intake associate with arterial stiffness. Eur. Heart J. Open 2022, 2, oeac016. [Google Scholar] [CrossRef]
- Hosseinpour-Niazi, S.; Mirmiran, P.; Hosseini, S.; Hadaegh, F.; Ainy, E.; Daneshpour, M.S.; Azizi, F. Effect of TCF7L2 on the relationship between lifestyle factors and glycemic parameters: A systematic review. Nutr. J. 2022, 21, 59. [Google Scholar] [CrossRef]
- Choi, J.H. Variation in the TAS2R38 Bitterness Receptor Gene Was Associated with Food Consumption and Obesity Risk in Koreans. Nutrients 2019, 11, 1973. [Google Scholar] [CrossRef]
- Bloss, C.S.; Wineinger, N.E.; Darst, B.F.; Schork, N.J.; Topol, E.J. Impact of direct-to-consumer genomic testing at long term follow-up. J. Med. Genet. 2013, 50, 393–400. [Google Scholar] [CrossRef] [PubMed]
- San-Cristobal, R.; Milagro, F.I.; Martínez, J.A. Future challenges and present ethical considerations in the use of personalized nutrition based on genetic advice. J. Acad. Nutr. Diet. 2013, 113, 1447–1454. [Google Scholar] [CrossRef] [PubMed]
- Arkadianos, I.; Valdes, A.M.; Marinos, E.; Florou, A.; Gill, R.D.; Grimaldi, K.A. Improved weight management using genetic information to personalize a calorie controlled diet. Nutr. J. 2007, 6, 29. [Google Scholar] [CrossRef]
- Gardner, C.D.; Trepanowski, J.F.; Del Gobbo, L.C.; Hauser, M.E.; Rigdon, J.; Ioannidis, J.P.A.; Desai, M.; King, A.C. Effect of Low-Fat vs Low-Carbohydrate Diet on 12-Month Weight Loss in Overweight Adults and the Association With Genotype Pattern or Insulin Secretion: The DIETFITS Randomized Clinical Trial. Jama 2018, 319, 667–679. [Google Scholar] [CrossRef]
- Ley, R.E.; Peterson, D.A.; Gordon, J.I. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006, 124, 837–848. [Google Scholar] [CrossRef]
- Davis, C.D. The Gut Microbiome and Its Role in Obesity. Nutr. Today 2016, 51, 167–174. [Google Scholar] [CrossRef]
- Duncan, S.H.; Lobley, G.E.; Holtrop, G.; Ince, J.; Johnstone, A.M.; Louis, P.; Flint, H.J. Human colonic microbiota associated with diet, obesity and weight loss. Int. J. Obes. 2008, 32, 1720–1724. [Google Scholar] [CrossRef]
- O’Toole, P.W.; Marchesi, J.R.; Hill, C. Next-generation probiotics: The spectrum from probiotics to live biotherapeutics. Nat. Microbiol. 2017, 2, 17057. [Google Scholar] [CrossRef]
- Boicean, A.; Ichim, C.; Sasu, S.M.; Todor, S.B. Key Insights into Gut Alterations in Metabolic Syndrome. J. Clin. Med. 2025, 14, 2678. [Google Scholar] [CrossRef]
- Abdalkareem Jasim, S.; Jade Catalan Opulencia, M.; Alexis Ramírez-Coronel, A.; Kamal Abdelbasset, W.; Hasan Abed, M.; Markov, A.; Raheem Lateef Al-Awsi, G.; Azamatovich Shamsiev, J.; Thaeer Hammid, A.; Nader Shalaby, M.; et al. The emerging role of microbiota-derived short-chain fatty acids in immunometabolism. Int. Immunopharmacol. 2022, 110, 108983. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Guo, L.; Leon Swisher, C.; Shah, N.; Huang, L.; Napier, L.A.; Kirkness, E.F.; Spector, T.D.; Caskey, C.T.; Thorens, B.; et al. Profound Perturbation of the Metabolome in Obesity Is Associated with Health Risk. Cell Metab. 2019, 29, 488–500.e482. [Google Scholar] [CrossRef] [PubMed]
- Stroeve, J.H.; Saccenti, E.; Bouwman, J.; Dane, A.; Strassburg, K.; Vervoort, J.; Hankemeier, T.; Astrup, A.; Smilde, A.K.; van Ommen, B.; et al. Weight loss predictability by plasma metabolic signatures in adults with obesity and morbid obesity of the DiOGenes study. Obesity 2016, 24, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Geidenstam, N.; Magnusson, M.; Danielsson, A.P.H.; Gerszten, R.E.; Wang, T.J.; Reinius, L.E.; Mulder, H.; Melander, O.; Ridderstråle, M. Amino Acid Signatures to Evaluate the Beneficial Effects of Weight Loss. Int. J. Endocrinol. 2017, 2017, 6490473. [Google Scholar] [CrossRef]
- Landrier, J.F.; Derghal, A.; Mounien, L. MicroRNAs in Obesity and Related Metabolic Disorders. Cells 2019, 8, 859. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J.; Wu, C. Modulation of Gut Microbiota and Immune System by Probiotics, Pre-biotics, and Post-biotics. Front. Nutr. 2021, 8, 634897. [Google Scholar] [CrossRef]
- Anvarbatcha, R.; Kunnathodi, F.; Arafat, A.A.; Azmi, S.; Mustafa, M.; Ahmad, I.; Alotaibi, H.F. Harnessing Probiotics: Exploring the Role of the Gut Microbiome in Combating Obesity. Probiotics Antimicrob. Proteins 2025. [Google Scholar] [CrossRef]
- Roager, H.M.; Christensen, L.H. Personal diet-microbiota interactions and weight loss. Proc. Nutr. Soc. 2022, 81, 243–254. [Google Scholar] [CrossRef]
- Saeed, S.; Arslan, M.; Froguel, P. Genetics of Obesity in Consanguineous Populations: Toward Precision Medicine and the Discovery of Novel Obesity Genes. Obesity 2018, 26, 474–484. [Google Scholar] [CrossRef]
- Clément, K.; van den Akker, E.; Argente, J.; Bahm, A.; Chung, W.K.; Connors, H.; De Waele, K.; Farooqi, I.S.; Gonneau-Lejeune, J.; Gordon, G.; et al. Efficacy and safety of setmelanotide, an MC4R agonist, in individuals with severe obesity due to LEPR or POMC deficiency: Single-arm, open-label, multicentre, phase 3 trials. Lancet Diabetes Endocrinol. 2020, 8, 960–970. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Matarese, G.; Lord, G.M.; Keogh, J.M.; Lawrence, E.; Agwu, C.; Sanna, V.; Jebb, S.A.; Perna, F.; Fontana, S.; et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J. Clin. Investig. 2002, 110, 1093–1103. [Google Scholar] [CrossRef]
- Collet, T.H.; Dubern, B.; Mokrosinski, J.; Connors, H.; Keogh, J.M.; Mendes de Oliveira, E.; Henning, E.; Poitou-Bernert, C.; Oppert, J.M.; Tounian, P.; et al. Evaluation of a melanocortin-4 receptor (MC4R) agonist (Setmelanotide) in MC4R deficiency. Mol. Metab. 2017, 6, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Ayers, K.L.; Glicksberg, B.S.; Garfield, A.S.; Longerich, S.; White, J.A.; Yang, P.; Du, L.; Chittenden, T.W.; Gulcher, J.R.; Roy, S.; et al. Melanocortin 4 Receptor Pathway Dysfunction in Obesity: Patient Stratification Aimed at MC4R Agonist Treatment. J. Clin. Endocrinol. Metab. 2018, 103, 2601–2612. [Google Scholar] [CrossRef] [PubMed]
- Clément, K.; Biebermann, H.; Farooqi, I.S.; Van der Ploeg, L.; Wolters, B.; Poitou, C.; Puder, L.; Fiedorek, F.; Gottesdiener, K.; Kleinau, G.; et al. MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency. Nat. Med. 2018, 24, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.D.; Roland, B.L.; Cole, R.L.; Trevaskis, J.L.; Weyer, C.; Koda, J.E.; Anderson, C.M.; Parkes, D.G.; Baron, A.D. Leptin responsiveness restored by amylin agonism in diet-induced obesity: Evidence from nonclinical and clinical studies. Proc. Natl. Acad. Sci. USA 2008, 105, 7257–7262. [Google Scholar] [CrossRef]
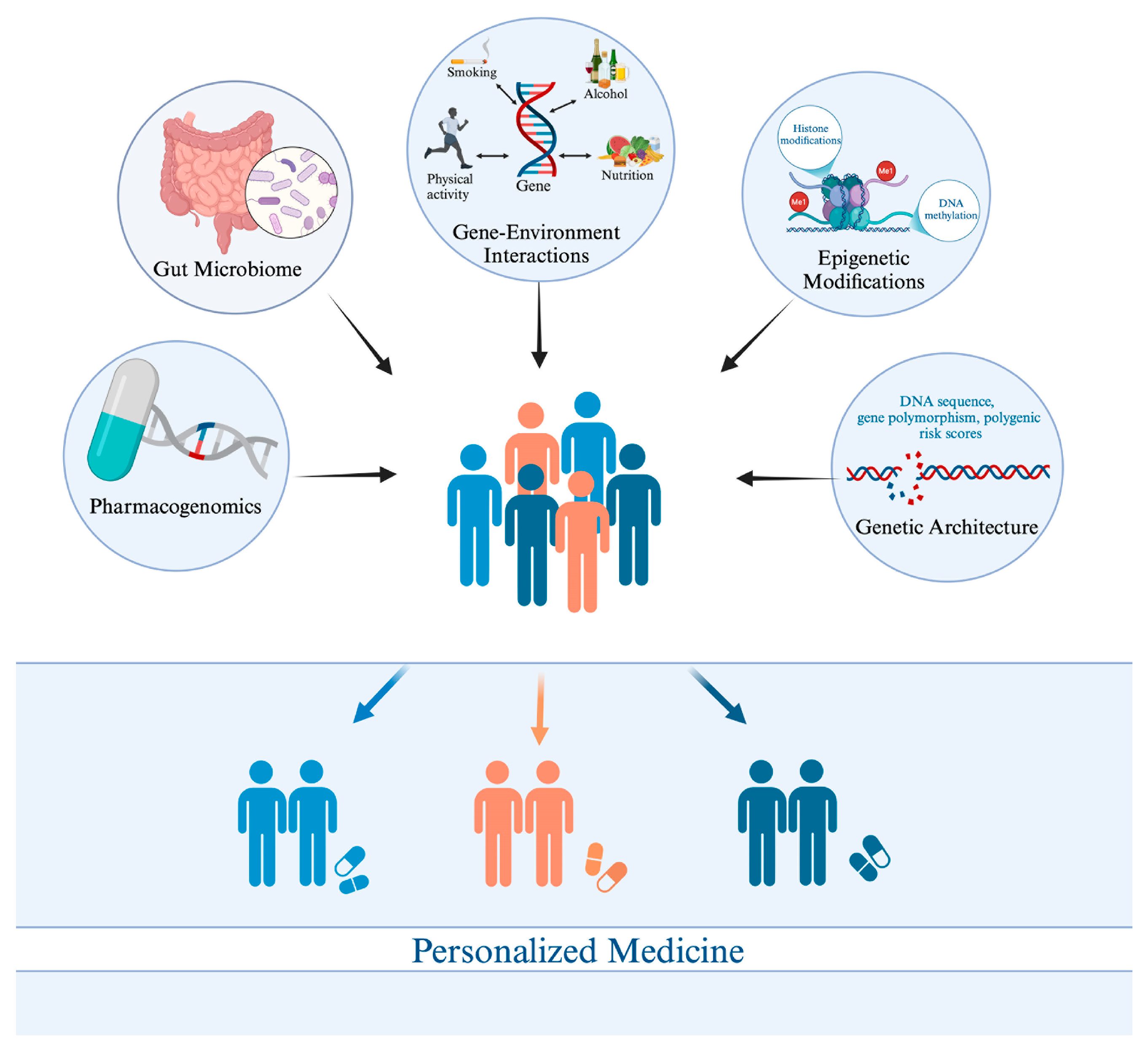
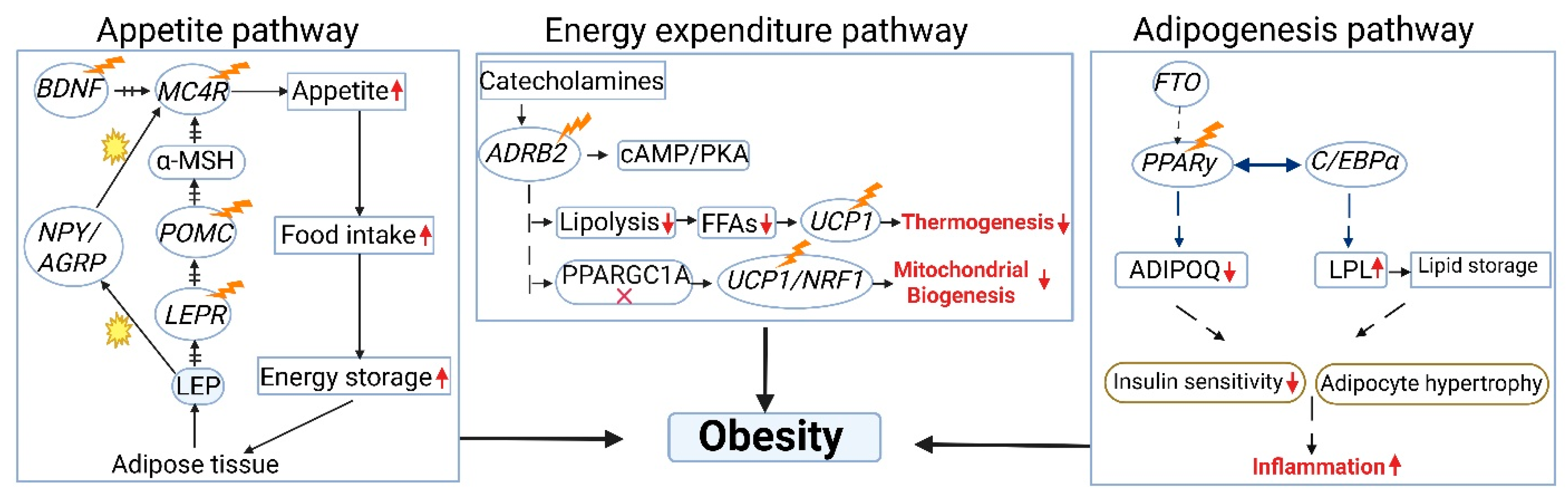

{kind=link}
{kind=link}
| Feature | Monogenic Obesity | Polygenic Obesity |
|---|---|---|
| Genetic Cause | Single, highly penetrant mutation in a key gene | Cumulative effect of multiple common variants with small individual effects |
| Example Genes | LEP, LEPR, POMC, MC4R | FTO, TCF7L2, IRS1 |
| Typical Onset | Severe, early-onset (often in infancy or early childhood) | Variable, can present at any age, but often develops in adulthood |
| Prevalence | Rare (<1% of obesity cases) | Common (>95% of obesity cases) |
| Effect on Specific Pathways | Disrupts a key regulatory pathway (e.g., leptin signaling) | Influences multiple pathways (appetite, metabolism, adipogenesis) |
| Environmental Influence | Less susceptible to environmental factors | More susceptible to environmental fators |
| Potential for Targeted Therapy | High (e.g., leptin replacement, MC4R agonists) | Emerging (e.g., personalized dietary recommendations based on GRS, PRS) |
| Study ID | Study Population | Key Gene(s)/Locus (Selected) | Key Findings |
|---|---|---|---|
| Akiyama et al., 2017 [23] | Japanese | GPR101, GPR75, KCNQ1 | Associated with increased BMI |
| Ang et al., 2023 [60] | Primarily European ancestry | MTOR, MAP2K5, RBFOX1, EP300, DNM1 | Associated with increased obesity risk |
| Chehadeh et al., 2020 [35] | Young Emirati adults | FTO, MC4R, TMEM18 | FTO variant rs3751812 linked to obesity in males, while MC4R and TMEM18 associated with females |
| Chiang et al., 2019 [33] | Han Chinese | FTO | Associated with morbid obesity |
| Chung et al., 2022 [61] | European ancestry | APOE | C allele of rs429358 increasingly associated with lower body fat percentage with age |
| Felix et al., 2016 [62] | European ancestry | ELP3, RAB27B, ADAM23 | Associated with increased adiposity in childhood |
| Hägg et al., 2015 [63] | European ancestry | TXNDC12, PEX2, SSFA2 | Associated with increased adiposity |
| Heid et al., 2010 [64] | European ancestry | RSPO3, VEGFA, GRB14, LYPLAL1, ITPR2, HOXC13, ADAMTS | Associated with increased WHR |
| Hinney et al., 2007 [65] | German ancestry | FTO | rs1121980 associated with increased obesity risk |
| Kilpeläinen et al., 2011 [66] | European ancestry | LEP, SLC32A1, GCKR, CCNL1, FTO | Associated with increased adiposity and increased leptin |
| Lee, 2022 [67] | European population | INPP4B, CHRNB4 | Associated with decreased BMI in smokers |
| Li et al., 2010 [68] | European descent | FTO, TMEM18, MC4R, FAIM2 | Variants in these genes showed associations with increased BMI and obesity |
| Liu et al., 2008 [69] | U.S. and French Caucasian populations | CTNNBL1 | rs6013029 particularly associated with increased BMI and fat mass |
| Liu et al., 2016 [70] | African American adults | NEGR1, NRXN3, BDNF, ADCY3, FTO | Multi-variant interactions involving these genes were associated with obesity risk |
| Liu et al., 2018 [71] | European and non-European ancestry | TUFM, SPI, APOBR, CPEB4 | TUFM, SPI, APOBR, and CPEB4 associated with BMI and WHR, respectively |
| Locke et al., 2015 [21] | European ancestry | FTO | Associated with increased BMI |
| Lv et al., 2015 [72] | Chinese children and adolescents | MC4R, SEC16B, MAP2K5, KCTD15 | Associated with increased obesity risk |
| Mägi et al., 2013 [73] | European adults | FTO, NEGR1 | FTO (rs9939609) and NEGR1 (rs2815752) loci associated with obesity |
| Mejía-Benítez et al., 2013 [74] | Mexican children | GNPDA2 | GNPDA2 (rs10938397) locus associated with BMI |
| Pei et al., 2014 [75] | Diverse ethnic backgrounds | CTSS, NLK, FTO, MC4R, TMEM18 | CTSS and NLK were linked to lower fat mass; FTO and MC4R to higher BMI; TMEM18 to lower BMI |
| Pulit et al., 2019 [76] | European ancestry | FTO | Associated with increased BMI and WHR |
| Rask-Andersen et al., 2012 [77] | Swedish and Greek children | AP2K5 | AP2K5-linked SNP rs2241423 associated with lower BMI |
| Shungin et al., 2015 [78] | Mainly European ancestry | LEKR1, CCDC92, VEGFA, RSPO3 | Positively associated with body fat distribution |
| Takahashi et al., 2024 [34] | Japanese adults | FTO, MC4R, SEC16B, BDNF | FTO, MC4R, and SEC16B positively associated with higher BMI, while BDNF showed negative associations |
| Tang et al., 2024 [79] | European ancestry | US, STX4, CCNT2, FUBP1, NDUFS3, RAPSN | Associated with increased BMI |
| Wang et al., 2011 [80] | Non-Hispanic Caucasians | FTO, NRXN3 | FTO associated with increased BMI and NRXN3 with higher WHR |
| Yengo et al., 2018 [39] | European ancestry | HSD17B12, STAG3L1, CAMKV | Associated with decreased adiposity |
| Study ID | Study Population | Key Gene(s)/Locus (Selected) | Key Findings |
| Taylor et al., 2023 [81] | Black/African American | SOCS3, RALGDS, PSKH1, FGD2, BMP6, TSLP | Hypomethylation in these genes was linked to higher BMI |
| Do et al., 2023 [82] | Multi-ethnic | TOP1MT, TNFRSF13B, LGALS3BP | Hypermethylation at TOP1MT was positively associated and another two were negatively associated with BMI |
| Alfano et al., 2023 [83] | European population | ARID5B, KLF9, PCSK5 | Methylation in these genes positively associated with rapid weight growth |
| Ali et al., 2016 [84] | Northern European ancestry | SOCS3 | Hypomethylation of SOCS3 associated with higher risk of obesity |
| Aslibekyan et al., 2015 [51] | European American | CPT1A, PHGDH CD38, LINC00263 | Hypermethylation at CPT1A and PHGDH associated with lower adiposity and CD38 and LINC00263 associated with increased adiposity |
| Campanella et al., 2018 [52] | European ancestry | ABCG1 | Methylation at the CpG site in the gene ABCG1 showed association with BMI, WC, WHR, and WHtR |
| Chen et al., 2021 [85] | Multi-ethnic Asian individuals | THADA, TNIK, RSRC1, ETAA1 | Methylation near THADA and TNIK is linked to lower BMI, while RSRC1 and ETAA1 are linked to higher BMI and WC |
| Demerath et al., 2015 [48] | African Americans | ABCG1, SREBF1, KDM2B, CPT1A, LGALS3BP, PBX1, BBS2, DHCR24 | Methylation near ABCG1, SREBF1, KDM2B, LGALS3BP, PBX1, and BBS2 was positively associated with BMI and/or WC, while CPT1A and DHCR24 showed negative associations |
| Dhana et al., 2018 [86] | European and African American | MSI2, LARS2, ABCG1, SREBF1, LGALS3BP, BRDT CPT1A, TMEM49 | Methylation at MSI2, LARS2, ABCG1, SREBF1, and LGALS3BP (positive) and CPT1A, TMEM49, and BRDT (negative) associated with BMI and/or WC |
| Kvaløy et al., 2018 [87] | Norwegian women | RPS6KA2, DMAP1, SETBP1 | Methylation at RPS6KA2, DMAP1, and SETBP1 negatively associated with BMI |
| Lee et al., 2021 [53] | European ancestry | CPT1A, ABCG1 | Methylation in CPT1A and ABCG1 associated with BMI |
| Li et al., 2024 [88] | Han Chinese | TRIM15, SLC38A4 | Hypermethylation at SLC38A4 associated with a decrease in BMI and TRIM15 associated with increase in BMI |
| Meeks et al., 2017 [55] | Sub-Saharan African | CPT1A | Hypomethylation of CPT1A associated with increased BMI, obesity, WC, and abdominal obesity |
| Nikpay et al., 2021 [89] | Primarily European ancestry | CCNL1, SLC5A11, MAST3, POMC, ADCY3, DNAJC27 | Hypomethylation at CCNL1 and SLC5A11 and hypermethylation at MAST3, POMC, ADCY3, and DNAJC27 associated with increased BMI |
| Sayols-Baixeras et al., 2017 [90] | European ancestry | SREBF1, NOTCH4 SLC7A11, CPT1A, SYNGAP1, GRIK1, CACNA1C, CUX1 | Hypermethylation of CUX1, SREBF1, SLC7A11, SYNGAP1, and GRIK1 and hypomethylation of CPT1A, CACNA1C, and NOTCH4 are linked to higher BMI and/or WC |
| Vehmeijer et al., 2020 [91] | Primarily European ancestry | SFRP5, SLC43A2, SFXN5 | Hypermethylation at CpGs in SFRP5, SLC43A2, and SFXN5 was positively associated with increased BMI |
| Wahl et al., 2017 [54] | European and Indian Asian | ABCG1, SREBF1, SOCS3, CPT1A | Hypermethylation at ABCG1, SREBF1, and SOCS3 and hypomethylation at CPT1A associated with increased BMI |
| Wang et al., 2018 [92] | African American | SBNO2, SOCS3, CISH, PIM3, KLF4 | Hypermethylation at SBNO2 and hypomethylation at SOCS3, CISH, PIM3, and KLF4 associated with higher obesity |
| Xie et al., 2021 [93] | European ancestry | ST8SIA5 | Hypermethylation near ST8SIA5 associated with higher WC |
| Zhao et al., 2023 [94] | Chinese ancestry | RPS6KA2, RPTOR, ZNF827, KSR1, NFIC | Hypomethylation at RPS6KA2, RPTOR, and ZNF827 and hypermethylation at KSR1 and NFIC are linked to higher BMI and WHR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunnathodi, F.; Arafat, A.A.; Alhazzani, W.; Mustafa, M.; Azmi, S.; Ahmad, I.; Selan, J.S.; Anvarbatcha, R.; Alotaibi, H.F. Unraveling the Genetic Architecture of Obesity: A Path to Personalized Medicine. Diagnostics 2025, 15, 1482. https://doi.org/10.3390/diagnostics15121482
Kunnathodi F, Arafat AA, Alhazzani W, Mustafa M, Azmi S, Ahmad I, Selan JS, Anvarbatcha R, Alotaibi HF. Unraveling the Genetic Architecture of Obesity: A Path to Personalized Medicine. Diagnostics. 2025; 15(12):1482. https://doi.org/10.3390/diagnostics15121482
Chicago/Turabian StyleKunnathodi, Faisal, Amr A. Arafat, Waleed Alhazzani, Mohammad Mustafa, Sarfuddin Azmi, Ishtiaque Ahmad, Jamala Saleh Selan, Riyasdeen Anvarbatcha, and Haifa F. Alotaibi. 2025. "Unraveling the Genetic Architecture of Obesity: A Path to Personalized Medicine" Diagnostics 15, no. 12: 1482. https://doi.org/10.3390/diagnostics15121482
APA StyleKunnathodi, F., Arafat, A. A., Alhazzani, W., Mustafa, M., Azmi, S., Ahmad, I., Selan, J. S., Anvarbatcha, R., & Alotaibi, H. F. (2025). Unraveling the Genetic Architecture of Obesity: A Path to Personalized Medicine. Diagnostics, 15(12), 1482. https://doi.org/10.3390/diagnostics15121482