
Atypical Manifestations of Cowden Syndrome in Pediatric Patients

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Histological Examination
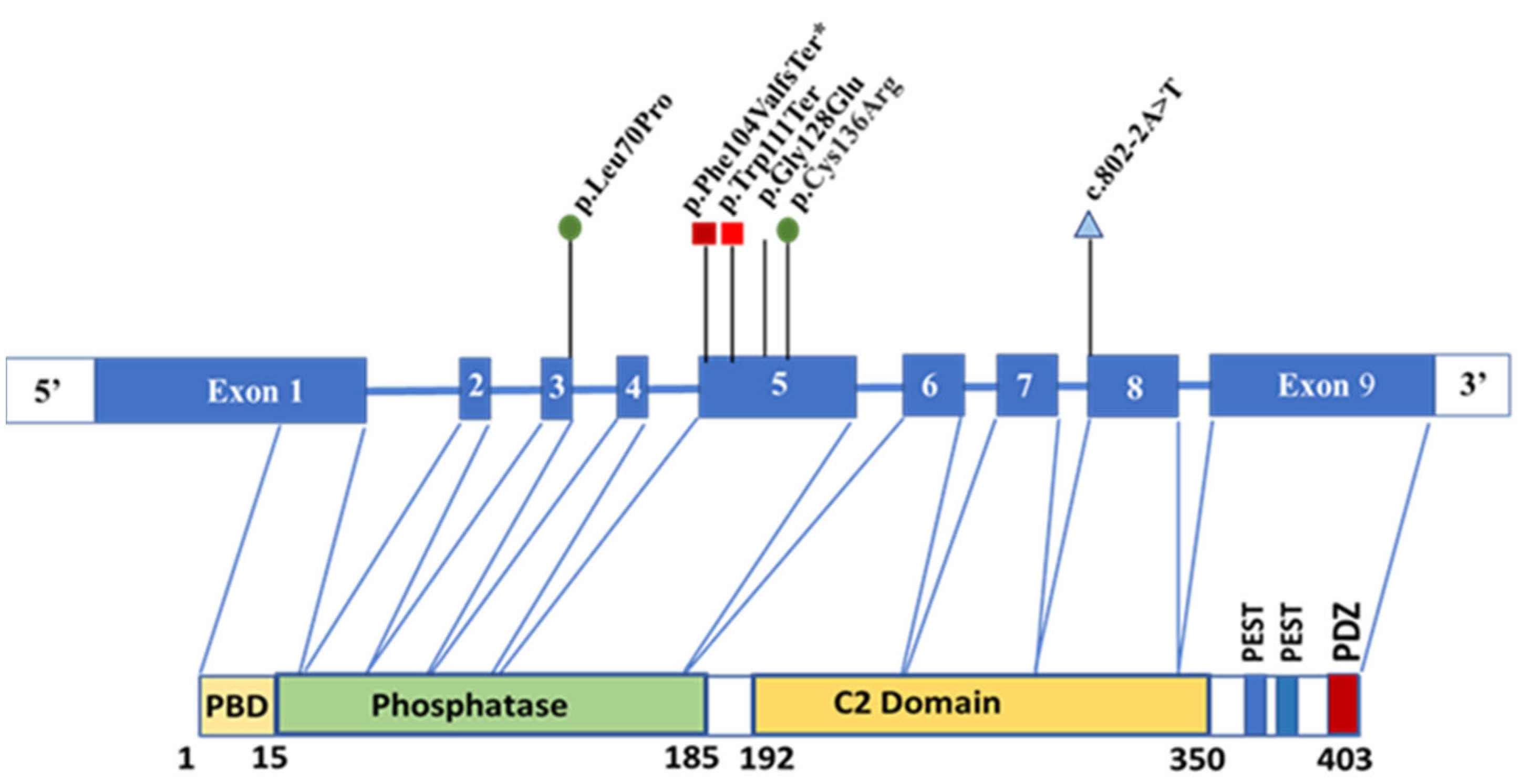
2.3. Genetic Testing
3. Results
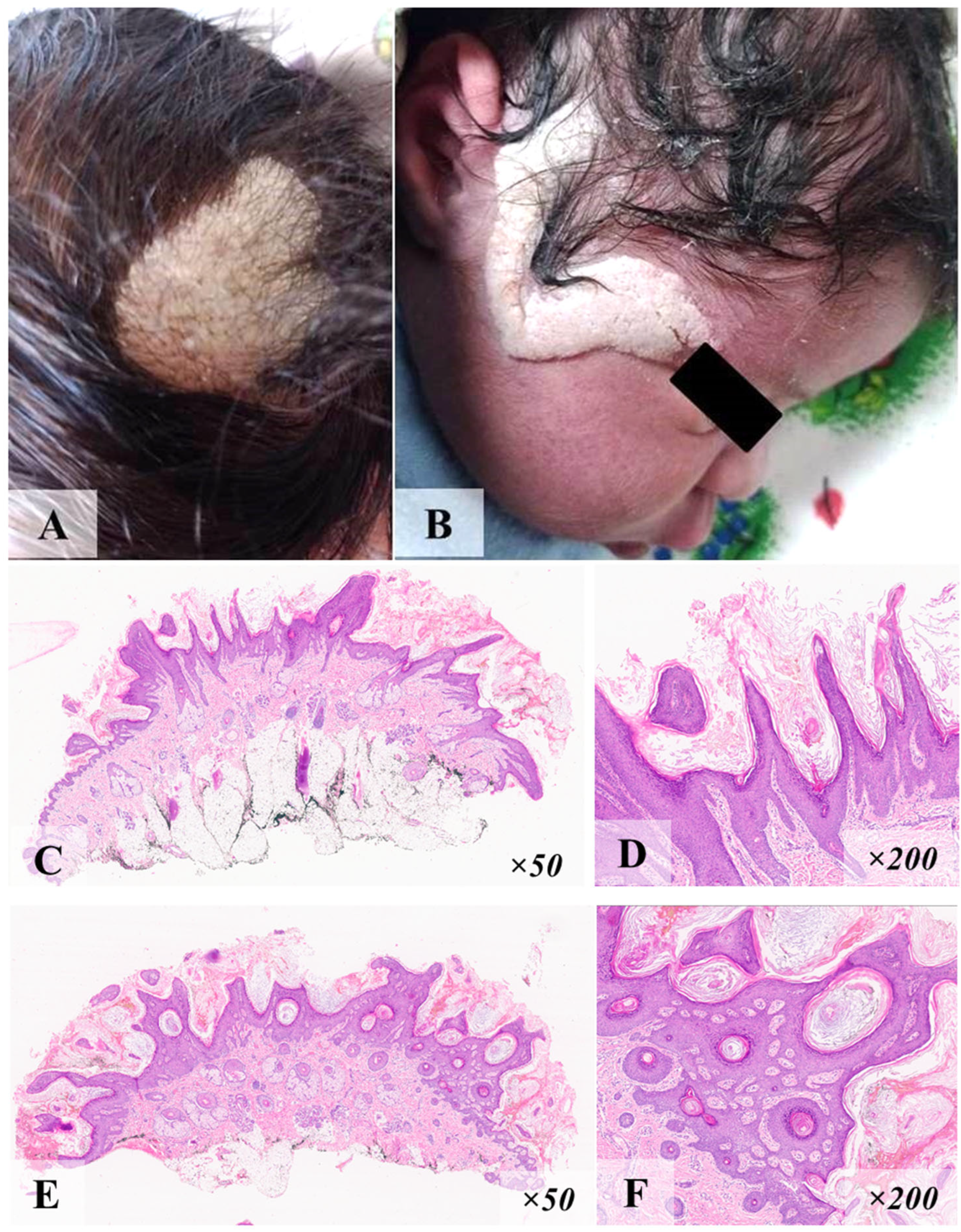
3.1. Case N°1
3.2. Case N°2
3.3. Case N°3
3.4. Case N°4
3.5. Case N°5
3.6. Case N°6
4. Discussion
- -
- ASD/expressed developmental delay (ID4, ID5, ID6);
- -
- dermatological features, namely, cyst (ID3), nevus of Jadassohn (ID1), lipoma (ID5, ID6), papillomas on the skin (ID8), fibrolipoma (ID12), and “café-au-lait” spots (ID13);
- -
- anomalies of vascular development (ID13);
- -
- gastrointestinal polyps (ID1, ID5);
- -
- thyroid pathology including multinodular goiter (ID11), thyroid nodule (ID5) follicular adenoma (ID13), and papillary thyroid cancer (ID8);
- -
- germ cell tumor (ID8, ID11, ID12).
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CS/PHTS | Cowden syndrome (or PTEN hamartoma tumor syndrome) |
| CS | Cowden syndrome |
| ASD | Autism spectrum disorder |
| BRRS | Bannayan–Riley–Ruvalcaba syndrome |
| ACMG | American College of Medical Genetics and Genomics |
| PM | Pathogenic moderate |
| PS | Pathogenic strong |
| M | Macrocephaly |
| DD | Developmental delay |
| DSD | Delayed speech development |
| WT | Wild type |
| PCT | Polychemotherapy |
| R-CHOP | Rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, prednisone |
| TIRADS | Thyroid Imaging Reporting and Data System |
| MRI | Magnetic resonance imaging |
| NGS | Next-generation sequencing |
| MALT | Mucosa-associated lymphoid tissue |
| RCC | Renal-cell carcinoma |
| TC | Thyroid cancer |
References
- Garofola, C.; Jamal, Z.; Gross, G.P. Cowden Disease; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Nelen, M.R.; Kremer, H.; Konings, I.B.; Schoute, F.; van Essen, A.J.; Koch, R.; Woods, C.G.; Fryns, J.-P.; Hamel, B.; Hoefsloot, L.H.; et al. Novel PTEN mutations in patients with Cowden disease: Absence of clear genotype–phenotype correlations. Eur. J. Hum. Genet. 1999, 7, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Molvi, M.; Sharma, Y.K.; Dash, K. Cowden Syndrome: Case Report, Update and Proposed Diagnostic and Surveillance Routines. Indian J. Dermatol. 2015, 60, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Cavaillé, M.; Crampon, D.; Achim, V.; Bubien, V.; Uhrhammer, N.; Privat, M.; Ponelle-Chachuat, F.; Gay-Bellile, M.; Lepage, M.; Ouedraogo, Z.G.; et al. Diagnosis of PTEN mosaicism: The relevance of additional tumor DNA sequencing. A case report and review of the literature. BMC Med. Genom. 2023, 16, 166. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, L.A.; Schuurs-Hoeijmakers, J.; Spier, I.; Haadsma, M.L.; Eijkelenboom, A.; Cremer, K.; Mensenkamp, A.R.; Aretz, S.; Vos, J.R.; Hoogerbrugge, N. Catch them if you are aware: PTEN postzygotic mosaicism in clinically suspicious patients with PTEN Hamartoma Tumour Syndrome and literature review. Eur. J. Med. Genet. 2022, 65, 104533. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Smith, C.; Marushchak, T.; Koehler, K.; Holmes, H.; Raskind, W.; Walsh, T.; Bennett, R.L. A mosaic PTEN mutation causing Cowden syndrome identified by deep sequencing. Anesthesia Analg. 2013, 15, 1004–1007. [Google Scholar] [CrossRef]
- Mester, J.; Charis, E. PTEN hamartoma tumor syndrome. Handb. Clin. Neurol. 2015, 132, 129–137. [Google Scholar]
- Tan, M.-H.; Mester, J.; Peterson, C.; Yang, Y.; Chen, J.-L.; Rybicki, L.A.; Milas, K.; Pederson, H.; Remzi, B.; Orloff, M.S.; et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am. J. Hum. Genet. 2011, 88, 42–56. [Google Scholar] [CrossRef]
- Takayama, T.; Muguruma, N.; Igarashi, M.; Ohsumi, S.; Oka, S.; Kakuta, F.; Kubo, Y.; Kumagai, H.; Sasaki, M.; Sugai, T.; et al. Clinical Guidelines for Diagnosis and Management of Cowden Syndrome/PTEN Hamartoma Tumor Syndrome in Children and Adults-Secondary Publication. J. Anus Rectum Colon. 2023, 7, 284–300. [Google Scholar] [CrossRef]
- Tischkowitz, M.; Colas, C.; Pouwels, S.; Hoogerbrugge, N. PHTS Guideline Development Group; European Reference Network GENTURIS. Cancer Surveillance Guideline for individuals with PTEN hamartoma tumor syndrome. Eur. J. Hum. Genet. 2020, 28, 1387–1393. [Google Scholar] [CrossRef]
- Hendricks, L.A.J.; Hoogerbrugge, N.; Schuurs-Hoeijmakers, J.H.M.; Vos, J.R. A review on age-related cancer risks in PTEN hamartoma tumor syndrome. Clin. Genet. 2021, 99, 219–225. [Google Scholar] [CrossRef]
- Tuli, G.; Munarin, J.; Mussa, A.; Carli, D.; Gastaldi, R.; Borgia, P.; Vigone, M.C.; Abbate, M.; Ferrero, G.B.; De Sanctis, L. Thyroid nodular disease and PTEN mutation in a multicentre series of children with PTEN hamartoma tumor syndrome (PHTS). Endocrine 2021, 74, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Ciaccio, C.; Saletti, V.; D’Arrigo, S.; Esposito, S.; Alfei, E.; Moroni, I.; Tonduti, D.; Chiapparini, L.; Pantaleoni, C.; Milani, D. Clinical spectrum of PTEN mutation in pediatric patients. A bicenter experience. Eur. J. Med. Genet. 2019, 62, 103596. [Google Scholar] [CrossRef] [PubMed]
- Jonker, L.; Lebbink, C.; Jongmans, M.; Nievelstein, R.; Merks, J.; van Dijkum, E.N.; Links, T.; Hoogerbrugge, N.; van Trotsenburg, A.; van Santen, H. Recommendations on surveillance for differentiated thyroid carcinoma in children with PTEN hamartoma tumor syndrome. Eur. Thyroid. J. 2020, 9, 234–242. [Google Scholar] [CrossRef]
- Zelenova, E.; Belysheva, T.; Sofronov, D.; Semenova, V.; Radjabova, G.; Vishnevskaya, Y.; Kletskaya, I.; Sharapova, E.; Karasev, I.; Romanov, D.; et al. Cutaneous Metastasis of Rectal Cancer as a Diagnostic Challenge: A Clinical Case and Literature Review. Diagnostics 2024, 14, 2420. [Google Scholar] [CrossRef]
- Bricheva, E.B.; Nagaeva, E.V.; Brovin, D.N.; Bondarenko, E.V.; Sheremeta, M.S.; Bezlepkina, O.B.; Olina, T.S.; Kovalenko, T.V. Thyroid cancer in a child with Cowden syndrome. Probl. Endocrinol. 2024, 70, 84–90. (In Russian) [Google Scholar] [CrossRef]
- Plamper, M.; Gohlke, B.; Woelfle, J. PTEN hamartoma tumor syndrome in childhood and adolescence—A comprehensive review and presentation of the German pediatric guideline. Mol. Cell. Pediatr. 2022, 9, 3. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Galli, E.; D’alò, F.; Cuccaro, A.; Alma, E.; Maiolo, E.; Brugnoletti, F.; Larocca, L.M.; Zollino, M.; Bacigalupo, A.P.; Hohaus, S. Burkitt Lymphoma as Fourth Neoplasia in a Patient Affected by Cowden Syndrome with a Novel PTEN Germline Pathogenic Variant. Mediterr. J. Hematol. Infect. Dis. 2020, 12, e2020034. [Google Scholar] [CrossRef]
- Cavaillé, M.; Ponelle-Chachuat, F.; Uhrhammer, N.; Viala, S.; Gay-Bellile, M.; Privat, M.; Bidet, Y.; Bignon, Y.-J. Early Onset Multiple Primary Tumors in Atypical Presentation of Cowden Syndrome Identified by Whole-Exome-Sequencing. Front. Genet. 2018, 9, 353. [Google Scholar] [CrossRef]
- Hagelstrom, R.T.; Ford, J.; Reiser, G.M.; Nelson, M.; Pickering, D.L.; Althof, P.A.; Sanger, W.G.; Coccia, P.F. Breast Cancer and Non-Hodgkin Lymphoma in a Young Male with Cowden Syndrome. Pediatr. Blood Cancer 2016, 63, 544–546. [Google Scholar] [CrossRef]
- Bubien, V.; Bonnet, F.; Brouste, V.; Hoppe, S.; Barouk-Simonet, E.; David, A.; Edery, P.; Bottani, A.; Layet, V.; Caron, O.; et al. High cumulative risks of cancer in patients with PTEN hamartoma tumor syndrome. J. Med. Genet. 2013, 50, 255–263. [Google Scholar] [CrossRef]
- Haibach, H.; Burns, T.W.; Carlson, H.E.; Burman, K.D.; Deftos, L.J. Multiple hamartoma syndrome (Cowden’s disease) associated with renal cell carcinoma and primary neuroendocrine carcinoma of the skin (Merkel cell carcinoma). Am. J. Clin. Pathol. 1992, 97, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Mester, J.L.; Zhou, M.; Prescott, N.; Eng, C. Papillary renal cell carcinoma is associated with PTEN hamartoma tumor syndrome. Urology 2012, 79, 1187.e1–1187.e7. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Wang, X.; Evans, A.J.; Campbell, S.C.; Nguyen, J.K.; Farncombe, K.M.; Eng, C. Early-onset renal cell carcinoma in PTEN harmatoma tumor syndrome. NPJ Genom. Med. 2020, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Smpokou, P.; Fox, V.L.; Tan, W.H. PTEN hamartoma tumor syndrome: Early tumor development in children. Arch. Dis. Child. 2015, 100, 34–37. [Google Scholar] [CrossRef]
- Kouzuki, K.; Umeda, K.; Kawasaki, H.; Isobe, K.; Akazawa, R.; Tasaka, K.; Tanaka, K.; Kubota, H.; Saida, S.; Kato, I.; et al. Immature teratoma of the ovary associated with Cowden syndrome. Pediatr. Blood Cancer 2022, 69, e29555. [Google Scholar] [CrossRef]
- Cho, M.Y.; Kim, H.S.; Eng, C.; Kim, D.S.; Kang, S.J.; Eom, M.; Yi, S.Y.; Bronner, M.P. First report of ovarian dysgerminoma in Cowden syndrome with germline PTEN mutation and PTEN-related 10q loss of tumor heterozygosity. Am. J. Surg. Pathol. 2008, 32, 1258–1264. [Google Scholar] [CrossRef]
- Tullius, B.P.; Shankar, S.P.; Cole, S.; Triano, V.; Aradhya, S.; Huang, E.C.; Sanchez, T.; Pawar, A. Novel heterozygous mutation in the PTEN gene associated with ovarian germ cell tumor complicated by growing teratoma syndrome and overgrowth in a two-year-old female. Pediatr. Blood Cancer 2019, 66, e27788. [Google Scholar] [CrossRef]
- Devi, M.; Leonard, N.; Silverman, S.; Al-Qahtani, M.; Girgis, R. Testicular mixed germ cell tumor in an adolescent with cowden disease. Oncology 2007, 72, 194–196. [Google Scholar] [CrossRef]
- Hildenbrand, C.; Burgdorf, W.H.; Lautenschlager, S. Cowden syndrome-diagnostic skin signs. Dermatology 2001, 202, 362–366. [Google Scholar] [CrossRef]
- Plana-Pla, A.; Condal, L.; Jaka, A.; Blanco, I.; Castellanos, E.; Bielsa, I. Verrucous epidermal nevus as a manifestation of a type 2 mosaic PTEN mutation in Cowden syndrome. Pediatr. Dermatol. 2023, 40, 179–181. [Google Scholar] [CrossRef]
- Isa, H.M.; Mohamed, Z.S.; Isa, Z.H.; Busehail, M.Y.; Alaradi, Z.A. Cowden Syndrome: A Rare Cause of Intestinal Polyposis. Cureus 2024, 16, e64838. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, L.A.; Hoogerbrugge, N.; Venselaar, H.; Aretz, S.; Spier, I.; Legius, E.; Brems, H.; de Putter, R.; Claes, K.B.; Evans, D.G.; et al. Genotype-phenotype associations in a large PTEN Hamartoma Tumor Syndrome (PHTS) patient cohort. Eur. J. Med. Genet. 2022, 65, 104632. [Google Scholar] [CrossRef] [PubMed]
- Jenny, B.; Radovanovic, I.; Haenggeli, C.A.; Delavelle, J.; Rüfenacht, D.; Kaelin, A.; Blouin, J.L.; Bottani, A.; Rilliet, B. Association of multiple vertebral hemangiomas and severe paraparesis in a patient with a PTEN hamartoma tumor syndrome: Case report. J. Neurosurg. 2007, 107 (Suppl. S4), 307–313. [Google Scholar] [CrossRef]
- Paparo, L.; Rossi, G.B.; Delrio, P.; Rega, D.; Duraturo, F.; Liccardo, R.; Debellis, M.; Izzo, P.; De Rosa, M. Differential expression of PTEN gene correlates with phenotypic heterogeneity in three cases of patients showing clinical manifestations of PTEN hamartoma tumour syndrome. Hered. Cancer Clin. Pract. 2013, 11, 8. [Google Scholar] [CrossRef]
- Kubo, Y.; Urano, Y.; Hida, Y.; Ikeuchi, T.; Nomoto, M.; Kunitomo, K.; Arase, S. A novel PTEN mutation in a Japanese patient with Cowden disease. Br. J. Dermatol. 2000, 142, 1100–1105. [Google Scholar] [CrossRef]
- Ngeow, J.; Stanuch, K.; Mester, J.L.; Barnholtz-Sloan, J.S.; Eng, C. Second malignant neoplasms in patients with cowden syndrome with underlying germline PTEN mutations. J. Clin. Oncol. 2014, 32, 1818–1824. [Google Scholar] [CrossRef]
- Matsumoto, K.; Nosaka, K.; Shiomi, T.; Matsuoka, Y.; Umekita, Y. Tumor-to-tumor metastases in Cowden’s disease: An autopsy case report and review of the literature. Diagn. Pathol. 2015, 10, 172. [Google Scholar] [CrossRef]
- Pena-Couso, L.; Ercibengoa, M.; Mercadillo, F.; Gómez-Sánchez, D.; Inglada-Pérez, L.; Santos, M.; Lanillos, J.; Gutiérrez-Abad, D.; Hernández, A.; Carbonell, P.; et al. Considerations on diagnosis and surveillance measures of PTEN hamartoma tumor syndrome: Clinical and genetic study in a series of Spanish patients. Orphanet J. Rare Dis. 2022, 17, 85. [Google Scholar] [CrossRef]
- Gervas, P.; Molokov, A.; Schegoleva, A.; Kiselev, A.; Babyshkina, N.; Pisareva, L.; Tyukalov, Y.; Choynzonov, E.; Cherdyntseva, N. New germline mutations in non-BRCA genes among breast cancer women of Mongoloid origin. Mol. Biol. Rep. 2020, 47, 5315–5321. [Google Scholar] [CrossRef]
- Marsh, D.J.; Dahia, P.L.; Caron, S.; Kum, J.B.; Frayling, I.M.; Tomlinson, I.P.; Hughes, K.S.; Eeles, R.A.; Hodgson, S.V.; Murday, V.A.; et al. Germline PTEN mutations in Cowden syndrome-like families. J. Med. Genet. 1998, 35, 881–885. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Major Criteria | Minor Criteria |
|---|---|
| Macrocephaly (head circumference greater than 58 cm in women and greater than 60 cm in men) | Structural lesions of the thyroid gland (adenoma, adenomatous goiter, etc.) |
| Follicular carcinoma of the thyroid gland | Thyroid cancer (papillary carcinoma) |
| Breast cancer | Colorectal cancer |
| Endometrial cancer | Renal cell carcinoma |
| Gastrointestinal hamartomas (including ganglioneuromas but excluding hyperplastic polyps; ≥3) | Esophageal glycogen acanthosis (≥3) |
| Lhermitte–Duclos disease in adults (dysplastic gangliocytoma of the cerebellum) | Intellectual disability (IQ ≤ 75), autism spectrum disorder (ASD) |
| Macular pigmentation of the glans penis | Testicular lipomatosis |
| Multiple skin and mucous membrane lesions (≥3): -tricholemmomas; -acral keratosis; -cutaneous mucosal neuromas; -oral papillomas (especially on the gingiva and tongue). | Vascular anomalies |
| Lipoma (≥3) | |
| Family History (at Least One Relative Fulfils the Diagnostic Criteria) and/or the Presence of a Pathogenic Variant in the PTEN Gene in the Patient | No Family History, Genetic Status of the Patient Is Unknown/PTEN-wt |
|---|---|
|
|
| Patient, Sex, Age | Major Criteria | Minor Criteria | Additional Pediatric Criteria | Atypical Features (Age of Diagnosis, Years) |
|---|---|---|---|---|
| Case N°1, g.87933068, c.309_312del (p.Phe104ValfsTer8) | ||||
| ID1 Male 2y.o. | Macrocephaly | – | Macrosomia (0) | Soft epidermal nevus (0), one hyperplastic polyp of the colon (2) |
| ID2 Male 47y.o. | Macrocephaly | Lipomatosis | ||
| ID3 Male 16y.o. | Macrocephaly | – | Macrosomia (0), cyst on the skin in the left temporal region | |
| ID4 Male 15y.o. | Macrocephaly | ASD | DSD | |
| Case N°2, g.87933091, c.332G>A (p.Trp111Ter) | ||||
| ID5 Male 14y.o. | Macrocephaly, one papilloma of the palatine tonsil (5) | Lipomatosis (11) thyroid nodule (14) | DSD, epicanthus, hypertelorism, extra left ventricular chord, resting bradycardia, kyphoscoliosis, hallux valgus | Single hyperplastic polyps of the colon (11) |
| ID6 Male 3y.o. | Macrocephaly | Lipoma up to 4 cm in size (2y10m) | Macrosomia (0), DSD, paroxysmal ventricular tachycardia | |
| ID7 Male 47y.o. | Macrocephaly | Multinodular goiter, lipoma of the ileum, vascular malformation in the cerebellum | Lymphofollicular hyperplasia of the colon (18), papillomas in axillary and inguinal areas | Diffuse large B-cell lymphoma GCB type (44) |
| Case N°3, g.87933139, c.380G>A (p.Gly127Glu), rs398123322 | ||||
| ID8 Female 15y.o. | Macrocephaly | Papillary thyroid cancer (11) | Macrosomia (0), ventricular septal defect, papillomas in the right axilla, and on the left hand | Germ cell tumor of the right ovary (7) |
| ID9 Male 10y.o. | Macrocephaly, penile lentiginosis, papillomatosis of the palatine tonsils (10) | – | Macrosomia (0), aplasia of the right testis | |
| ID10 Male 46y.o. | Macrocephaly, penile lentiginosis | – | ||
| Case N°4, g.87960892, c.802-2A>T, rs587782455 | ||||
| ID11 Female, 14y.o. | Macrocephaly | Multinodular goiter (10) | Macrosomia (0) | Germ cell tumor of the right ovary (4) |
| Case N°5, g.87925557, c.209T>C (p.Leu70Pro), rs121909226 | ||||
| ID12 Female 10y.o. | Macrocephaly | Fibrolipoma of suprascapular region (4 months) | Macrosomia (0), scaphocephaly | Germ cell tumor of the left ovary (8) |
| Case N°6, g.87933165, c.406T>C (p.Cys136Arg), rs786201044 | ||||
| ID13 Male 17y.o. | Macrocephaly | Lymphangioma of the right axillary region, follicular adenomas of the thyroid (14) | Macrosomia (0), MR, “café-au-lait” spots, gingival hypertrophy, chest deformation, scoliosis, pulmonary sclerosing pneumocytoma (13) | Papillary renal cell carcinoma (13) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zelenova, E.; Belysheva, T.; Sharapova, E.; Barinova, I.; Fedorova, A.; Semenova, V.; Vishnevskaya, Y.; Kletskaya, I.; Mitrofanova, A.; Sofronov, D.; et al. Atypical Manifestations of Cowden Syndrome in Pediatric Patients. Diagnostics 2025, 15, 1456. https://doi.org/10.3390/diagnostics15121456
Zelenova E, Belysheva T, Sharapova E, Barinova I, Fedorova A, Semenova V, Vishnevskaya Y, Kletskaya I, Mitrofanova A, Sofronov D, et al. Atypical Manifestations of Cowden Syndrome in Pediatric Patients. Diagnostics. 2025; 15(12):1456. https://doi.org/10.3390/diagnostics15121456
Chicago/Turabian StyleZelenova, Ekaterina, Tatiana Belysheva, Elena Sharapova, Irina Barinova, Alexandra Fedorova, Vera Semenova, Yana Vishnevskaya, Irina Kletskaya, Anna Mitrofanova, Denis Sofronov, and et al. 2025. "Atypical Manifestations of Cowden Syndrome in Pediatric Patients" Diagnostics 15, no. 12: 1456. https://doi.org/10.3390/diagnostics15121456
APA StyleZelenova, E., Belysheva, T., Sharapova, E., Barinova, I., Fedorova, A., Semenova, V., Vishnevskaya, Y., Kletskaya, I., Mitrofanova, A., Sofronov, D., Karasev, I., Romanov, D., Valiev, T., & Nasedkina, T. (2025). Atypical Manifestations of Cowden Syndrome in Pediatric Patients. Diagnostics, 15(12), 1456. https://doi.org/10.3390/diagnostics15121456