
A Five-Gene Signature for the Prediction of Event-Free Survival of Both Pediatric and Adult Acute Myeloid Leukemia




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Sources
- TARGET childhood AML data: We retrieved the TARGET AML RNA-seq data from the Genomic Data Commons (GDC) at portal.gdc.cancer.gov/, accessed on 20 November 2020. This dataset comprises 187 subjects and 21,047 genes with nonzero read counts. Additionally, clinical metadata, including EFS and censored status, are accessible. EFS represents the time patients survive without disease recurrence, progression, or further treatment [15]. This dataset was already processed and normalized to account for variations in the sequencing depth and library size, and it was log2 transformed, allowing for accurate comparison.
- TCGA adult AML data: The TCGA RNA-seq gene expression data were acquired from the Cancer Genomics Portal at https://www.cbioportal.org/, accessed on 16 February 2019. This dataset consists of 173 samples, encompassing 20,531 raw gene counts. Normalization was conducted through log2 transformation and quantile normalization. Similarly to the childhood AML data, EFS, censored status, and other pertinent clinical information were also included.
- Other validation data: For further validation, we incorporated two independent microarray datasets—GSE37642 [13] and GSE12417 [14], accessed on 8 December 2020. Both datasets represent adult AML, providing only OS information. GSE37642 encompasses 136 patients with 21,225 probes, while GSE12417 includes 163 patients with 21,225 probes. The microarray data were preprocessed with log2 transformation and quantile normalization.
2.2. Methods
3. Results
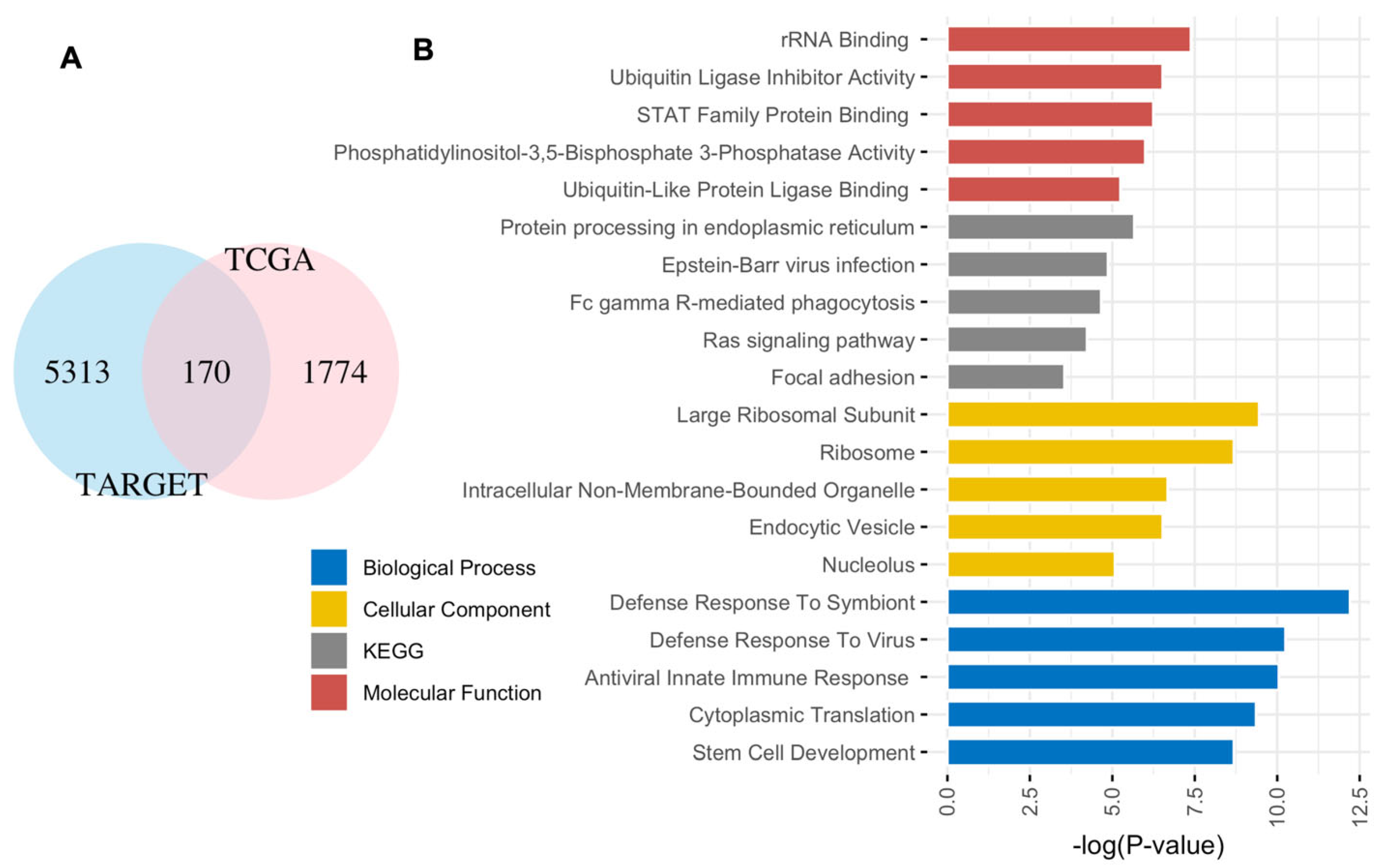
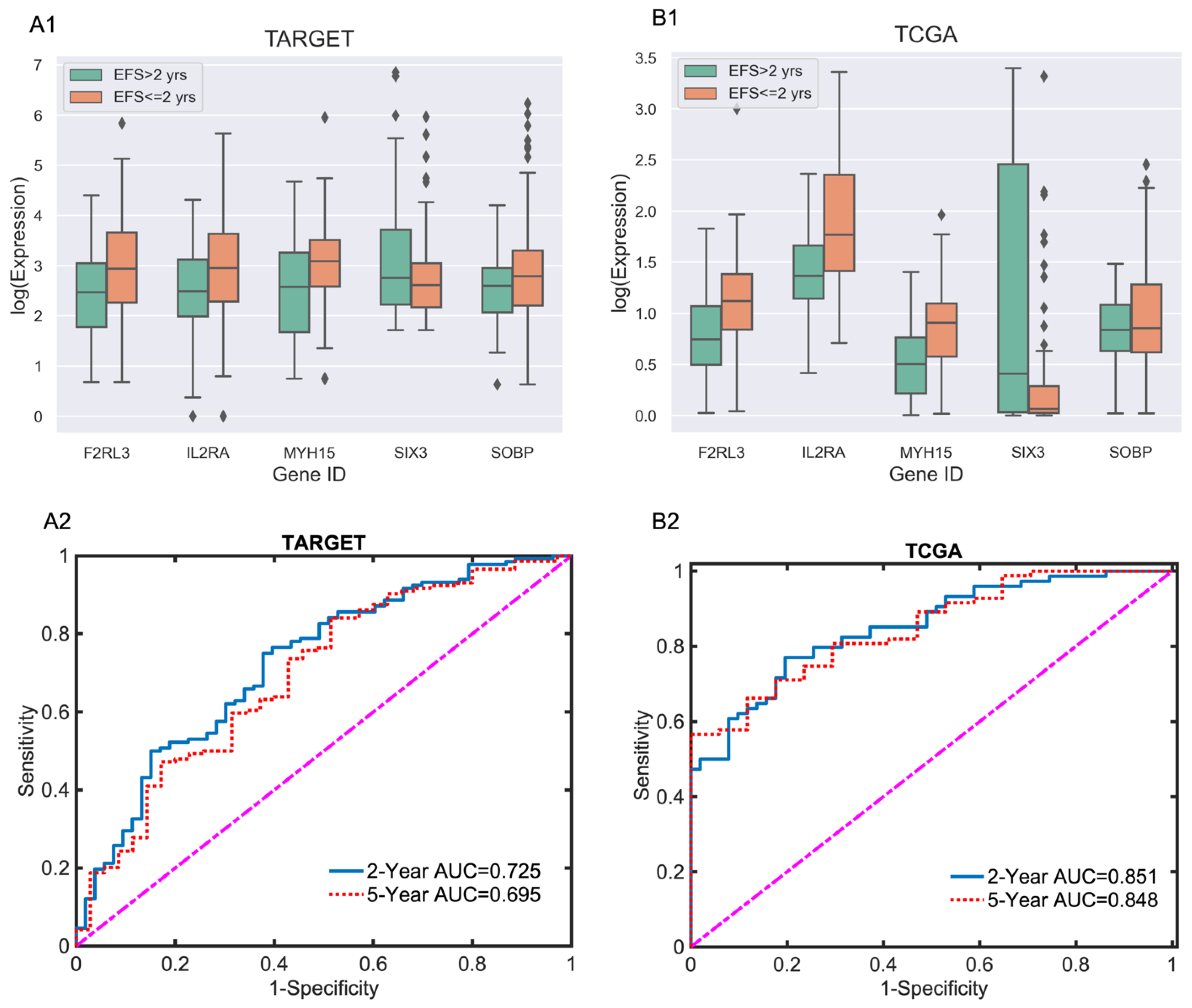
3.1. Five-Gene EFS Risk Score Models for Adult and Pediatric AML
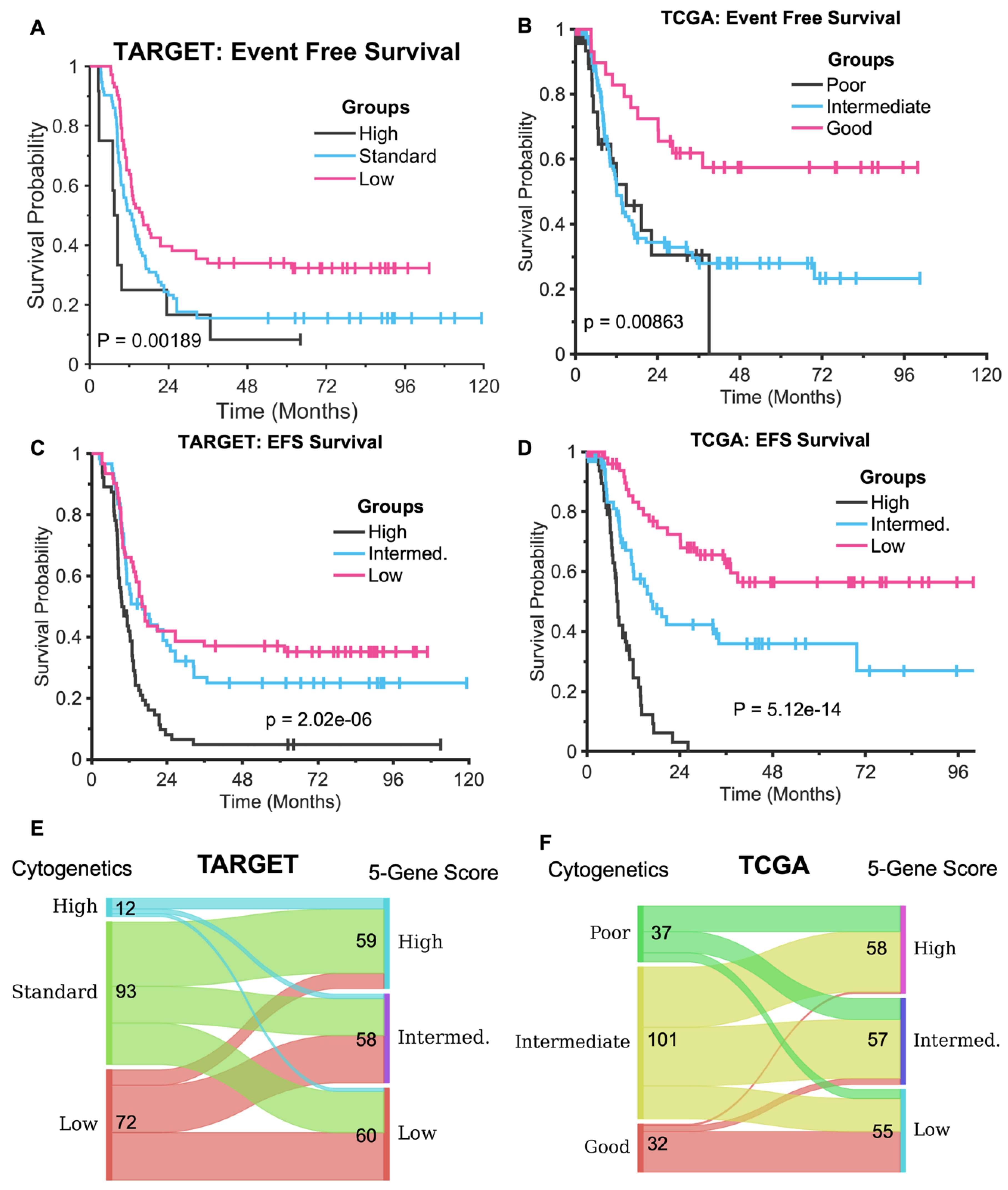
3.2. The Five-Gene Signature Demonstrates Superior EFS Stratification Compared to Cytogenetics Method
3.3. The Five-Gene Signature Predicts the Overall Survival of TARGET and TCGA and Two Independent Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bolouri, H.; Farrar, J.E.; Triche, T., Jr.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Burd, A.; Levine, R.L.; Ruppert, A.S.; Mims, A.S.; Borate, U.; Stein, E.M.; Patel, P.; Baer, M.R.; Stock, W.; Deininger, M.; et al. Precision medicine treatment in acute myeloid leukemia using prospective genomic profiling: Feasibility and preliminary efficacy of the Beat AML Master Trial. Nat. Med. 2020, 26, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, S.; O’Connor, C.; Cañete, A.; Bittencourt-Silvestre, J.; Sarrou, E.; Prendergast, Á.; Choi, J.; Johnston, P.; Wells, C.A.; Gibson, B.; et al. Age-specific biological and molecular profiling distinguishes paediatric from adult acute myeloid leukamias. Nat. Commun. 2018, 9, 5280. [Google Scholar] [CrossRef]
- Tazi, Y.; Arango-Ossa, J.E.; Zhou, Y.; Bernard, E.; Thomas, I.; Gilkes, A.; Freeman, S.; Pradat, Y.; Johnson, S.J.; Hills, R.; et al. Unified classification and risk-stratification in Acute Myeloid Leukemia. Nat. Commun. 2022, 13, 4622. [Google Scholar] [CrossRef]
- Yu, S.; Yang, S.; Hu, L.; Duan, W.; Zhao, T.; Qin, Y.; Wang, Y.; Lai, Y.; Shi, H.; Tang, F.; et al. Genetic abnormalities predict outcomes in patients with core binding factor acute myeloid leukemia. Ann. Hematol. 2025; Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Elcheva, I. A six-gene prognostic signature for both adult and pediatric acute myeloid leukemia identified with machine learning. Am. J. Transl. Res. 2022, 14, 6210–6221. [Google Scholar]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Aung, M.M.K.; Mills, M.L.; Bittencourt-Silvestre, J.; Keeshan, K. Insights into the molecular profiles of adult and paediatric acute myeloid leukaemia. Mol. Oncol. 2021, 15, 2253–2272. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, Z.; Spiegelman, V.S.; Wang, H.G. Distinct noncoding RNAs and RNA binding proteins associated with high-risk pediatric and adult acute myeloid leukemias detected by regulatory network analysis. Cancer Rep. 2022, 5, e1592. [Google Scholar] [CrossRef]
- Ng, S.W.K.; Murphy, T.; King, I.; Zhang, T.; Mah, M.; Lu, Z.; Stickle, N.; Ibrahimova, N.; Arruda, A.; Mitchell, A.; et al. A clinical laboratory-developed LSC17 stemness score assay for rapid risk assessment of patients with acute myeloid leukemia. Blood Adv. 2022, 6, 1064–1073. [Google Scholar] [CrossRef]
- Wagner, S.; Vadakekolathu, J.; Tasian, S.K.; Altmann, H.; Bornhäuser, M.; Pockley, A.G.; Ball, G.R.; Rutella, S. A parsimonious 3-gene signature predicts clinical outcomes in an acute myeloid leukemia multicohort study. Blood Adv. 2019, 3, 1330–1346. [Google Scholar] [CrossRef] [PubMed]
- Duployez, N.; Marceau-Renaut, A.; Villenet, C.; Petit, A.; Rousseau, A.; Ng, S.W.K.; Paquet, A.; Gonzales, F.; Barthélémy, A.; Leprêtre, F.; et al. The stem cell-associated gene expression signature allows risk stratification in pediatric acute myeloid leukemia. Leukemia 2019, 33, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Herold, T.; Jurinovic, V.; Batcha, A.M.N.; Bamopoulos, S.A.; Rothenberg-Thurley, M.; Ksienzyk, B.; Hartmann, L.; Greif, P.A.; Phillippou-Massier, J.; Krebs, S.; et al. A 29-gene and cytogenetic score for the prediction of resistance to induction treatment in acute myeloid leukemia. Haematologica 2018, 103, 456–465. [Google Scholar] [CrossRef]
- Metzeler, K.H.; Hummel, M.; Bloomfield, C.D.; Spiekermann, K.; Braess, J.; Sauerland, M.C.; Heinecke, A.; Radmacher, M.; Marcucci, G.; Whitman, S.P.; et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood 2008, 112, 4193–4201. [Google Scholar] [CrossRef]
- Delgado, A.; Guddati, A.K. Clinical endpoints in oncology—A primer. Am. J. Cancer Res. 2021, 11, 1121–1131. [Google Scholar] [PubMed] [PubMed Central]
- Liu, H.; Wang, Y.; Qi, C.; Xie, T.; Peng, Z.; Li, J.; Shen, L.; Zhang, X. Evaluation of Event-Free Survival Surrogating Overall Survival as the Endpoint in Neoadjuvant Clinical Trials of Gastroesophageal Adenocarcinoma. Front. Oncol. 2022, 12, 835389. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Simon, N.; Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Cox’s Proportional Hazards Model via Coordinate Descent. J. Stat. Softw. 2011, 39, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Scrucca, L.; Santucci, A.; Aversa, F. Competing risk analysis using R: An easy guide for clinicians. Bone Marrow Transpl. 2007, 40, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Uno, H.; Cai, T.; Pencina, M.J.; D’Agostino, R.B.; Wei, L.J. On the C-statistics for evaluating overall adequacy of risk prediction procedures with censored survival data. Stat. Med. 2011, 30, 1105–1117. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mrózek, K.; Kohlschmidt, J.; Blachly, J.S.; Nicolet, D.; Carroll, A.J.; Archer, K.J.; Mims, A.S.; Larkin, K.T.; Orwick, S.; Oakes, C.C.; et al. Outcome prediction by the 2022 European LeukemiaNet genetic-risk classification for adults with acute myeloid leukemia: An Alliance study. Leukemia 2023, 37, 788–798. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, E.; Lu, S.X.; Pastore, A.; Chen, X.; Imig, J.; Chun-Wei Lee, S.; Hockemeyer, K.; Ghebrechristos, Y.E.; Yoshimi, A.; Inoue, D.; et al. Targeting an RNA-Binding Protein Network in Acute Myeloid Leukemia. Cancer Cell. 2019, 35, 369–384.e7. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhou, X.; Sun, S.C. Targeting ubiquitin signaling for cancer immunotherapy. Sig. Transduct. Target Ther. 2021, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Goyama, S.; Shrestha, M.; Schibler, J.; Rosenfeldt, L.; Miller, W.; O’Brien, E.; Mizukawa, B.; Kitamura, T.; Palumbo, J.S.; Mulloy, J.C. Protease-activated receptor-1 inhibits proliferation but enhances leukemia stem cell activity in acute myeloid leukemia. Oncogene 2017, 36, 2589–2598. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, F.; Yang, L.; Xiao, M.; Zhang, Z.; Shen, J.; Anuchapreeda, S.; Tima, S.; Chiampanichayakul, S.; Xiao, Z. PD-L1 regulates cell proliferation and apoptosis in acute myeloid leukemia by activating PI3K-AKT signaling pathway. Sci. Rep. 2022, 12, 11444. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nguyen, C.H.; Schlerka, A.; Grandits, A.M.; Koller, E.; van der Kouwe, E.; Vassiliou, G.S.; Staber, P.B.; Heller, G.; Wieser, R. IL2RA Promotes Aggressiveness and Stem Cell-Related Properties of Acute Myeloid Leukemia. Cancer Res. 2020, 80, 4527–4539. [Google Scholar] [CrossRef] [PubMed]
- Ortiz Rojas, C.A.; Pereira-Martins, D.A.; Bellido More, C.C.; Sternadt, D.; Weinhäuser, I.; Hilberink, J.R.; Coelho-Silva, J.L.; Thomé, C.H.; Ferreira, G.A.; Ammatuna, E.; et al. A 4-gene prognostic index for enhancing acute myeloid leukaemia survival prediction. Br. J. Haematol. 2024, 204, 2287–2300. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Shiba, N.; Tsujimoto, S.; Yamato, G.; Hara, Y.; Kato, S.; Yoshida, K.; Ogawa, S.; Hayashi, Y.; Iwamoto, S.; et al. High IL2RA/CD25 expression is a prognostic stem cell biomarker for pediatric acute myeloid leukemia without a core-binding factor. Pediatr. Blood Cancer 2024, 71, e30803. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.C.; Mammucari, C.; Argentini, C.; Reggiani, C.; Schiaffino, S. Two novel/ancient myosins in mammalian skeletal muscles: MYH14/7b and MYH15 are expressed in extraocular muscles and muscle spindles. J. Physiol. 2010, 588 Pt 2, 353–364. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gladilin, E.; Ohse, S.; Boerries, M.; Busch, H.; Xu, C.; Schneider, M.; Meister, M.; Eils, R. TGFβ-induced cytoskeletal remodeling mediates elevation of cell stiffness and invasiveness in NSCLC. Sci. Rep. 2019, 9, 7667. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, G.; Gao, Y.; Li, K.; Lin, A.; Jiang, Z. Genomic analysis of biomarkers related to the prognosis of acute myeloid leukemia. Oncol. Lett. 2020, 20, 1824–1834. [Google Scholar] [CrossRef]
- Ma, T.L.; Zhu, P.; Chen, J.X.; Hu, Y.H.; Xie, J. SIX3 function in cancer: Progression and comprehensive analysis. Cancer Gene Ther. 2022, 29, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Wu, Y.; Guo, J.; Zhang, W.; Wang, R.; Yang, H.; Wang, Z. The prognostic signature based on glycolysis-immune related genes for acute myeloid leukemia patients. Immunobiology 2023, 228, 152355. [Google Scholar] [CrossRef] [PubMed]
- Tavares, A.L.P.; Jourdeuil, K.; Neilson, K.M.; Majumdar, H.D.; Moody, S.A. Sobp modulates the transcriptional activation of Six1 target genes and is required during craniofacial development. Development 2021, 148, dev199684. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Matsumoto, Y.; Chinen, Y.; Shimura, Y.; Nagoshi, H.; Sasaki, N.; Muramatsu, A.; Kuriyama, K.; Ohshiro, M.; Hirakawa, Y.; Iwai, T.; et al. Recurrent intragenic exon rearrangements of SOBP and AUTS2 in non-Hodgkin B-cell lymphoma. Int. J. Hematol. 2020, 111, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, J.; He, X.; Gao, Y.; Mahara, G.; Lin, Z.; Zhang, J. A novel approach to determine two optimal cut-points of a continuous predictor with a U-shaped relationship to hazard ratio in survival data: Simulation and application. BMC Med. Res. Methodol. 2019, 19, 96. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Eng, K.H.; Schiller, E.; Morrell, K. On representing the prognostic value of continuous gene expression biomarkers with the restricted mean survival curve. Oncotarget 2015, 6, 36308–36318. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Han, K.; Jung, I. Restricted Mean Survival Time for Survival Analysis: A Quick Guide for Clinical Researchers. Korean J. Radiol. 2022, 23, 495–499. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, D.; Liu, A.J.; Sheng, L.; Liu, Z.; Elcheva, I. A Five-Gene Signature for the Prediction of Event-Free Survival of Both Pediatric and Adult Acute Myeloid Leukemia. Diagnostics 2025, 15, 1421. https://doi.org/10.3390/diagnostics15111421
Chen D, Liu AJ, Sheng L, Liu Z, Elcheva I. A Five-Gene Signature for the Prediction of Event-Free Survival of Both Pediatric and Adult Acute Myeloid Leukemia. Diagnostics. 2025; 15(11):1421. https://doi.org/10.3390/diagnostics15111421
Chicago/Turabian StyleChen, Dechang, Alvin J. Liu, Li Sheng, Zhenqiu Liu, and Irina Elcheva. 2025. "A Five-Gene Signature for the Prediction of Event-Free Survival of Both Pediatric and Adult Acute Myeloid Leukemia" Diagnostics 15, no. 11: 1421. https://doi.org/10.3390/diagnostics15111421
APA StyleChen, D., Liu, A. J., Sheng, L., Liu, Z., & Elcheva, I. (2025). A Five-Gene Signature for the Prediction of Event-Free Survival of Both Pediatric and Adult Acute Myeloid Leukemia. Diagnostics, 15(11), 1421. https://doi.org/10.3390/diagnostics15111421