
Inflammation—A Possible Link between Myocarditis and Arrhythmogenic Cardiomyopathy

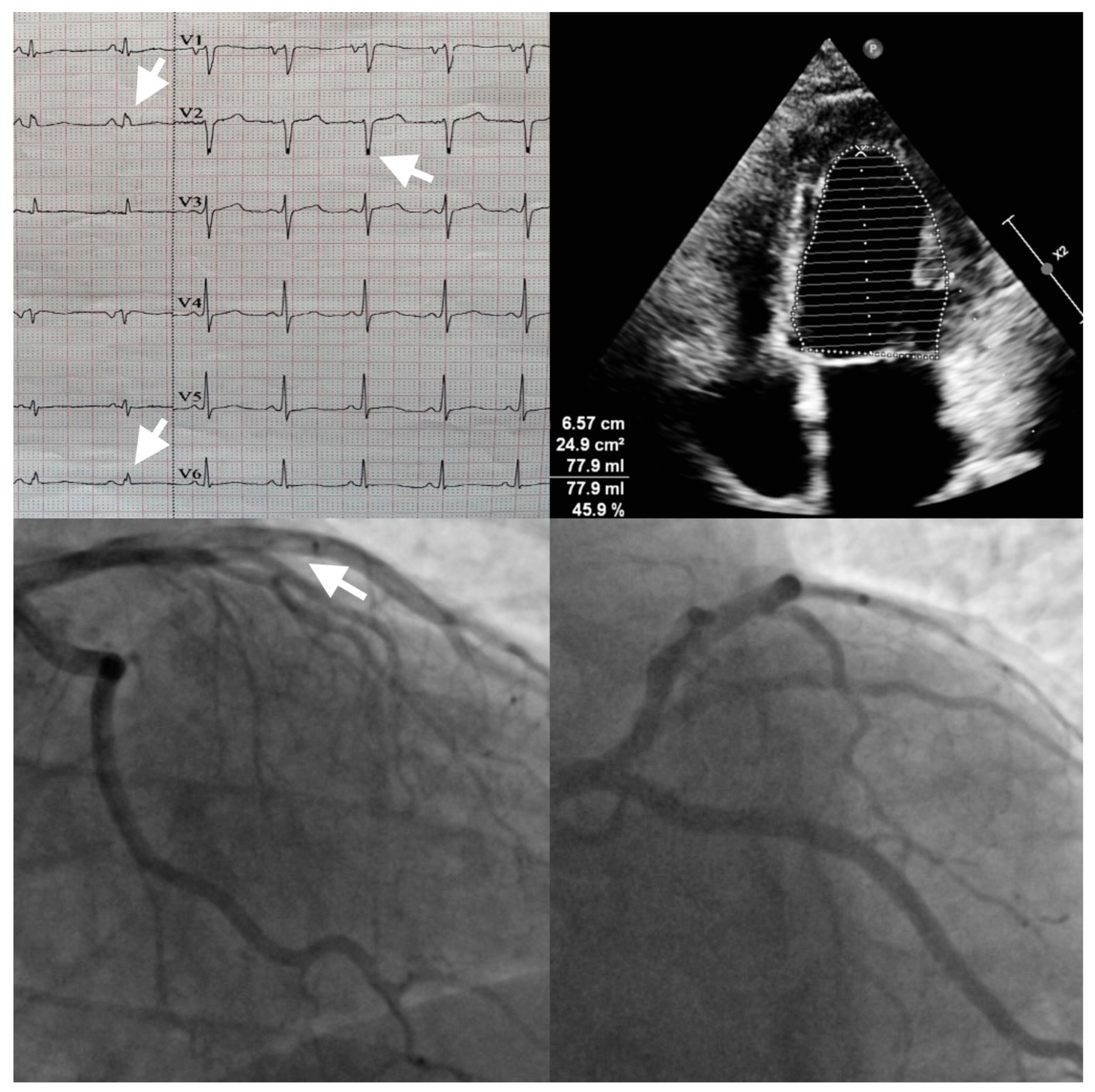{kind=link}
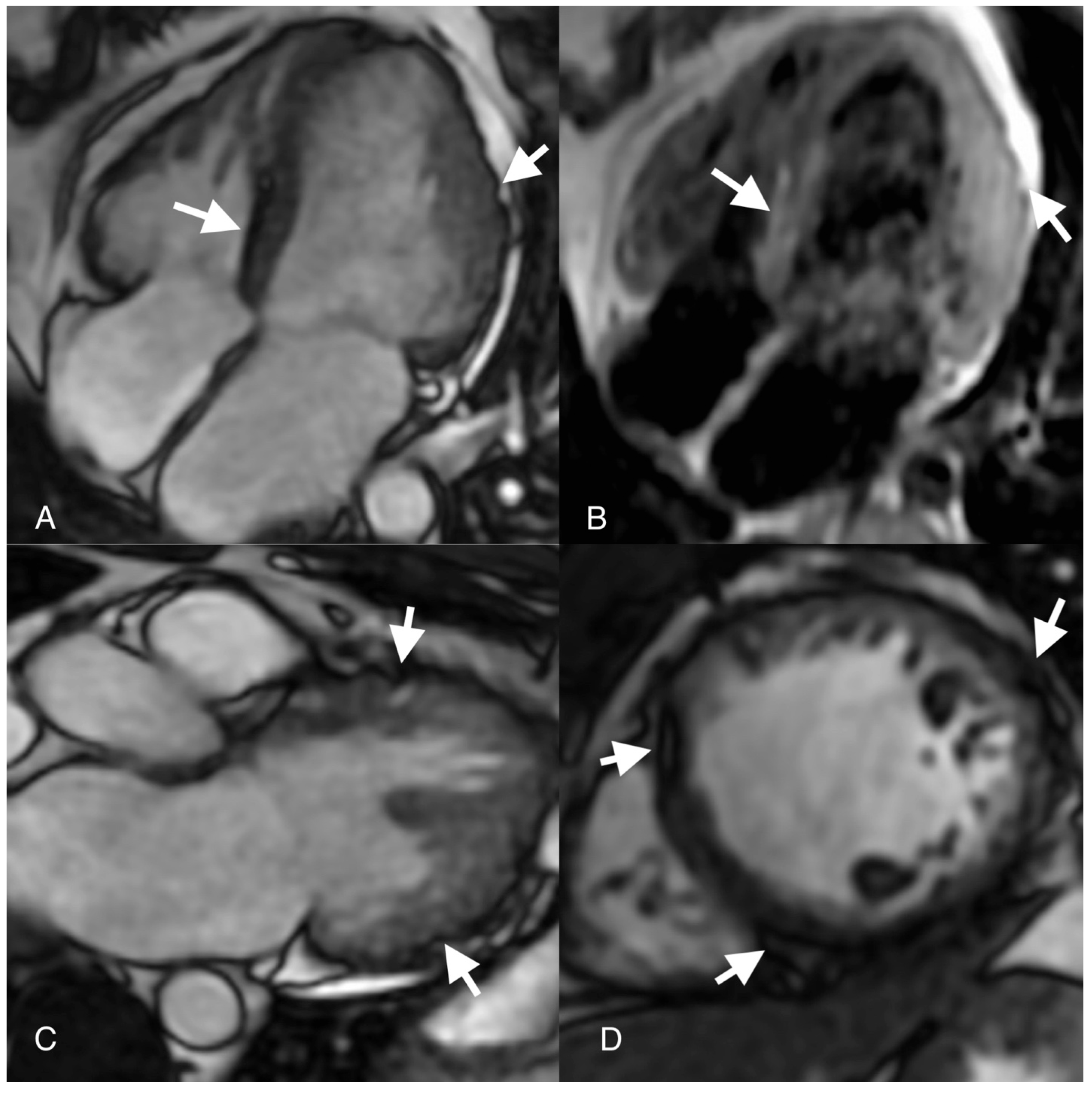{kind=link}
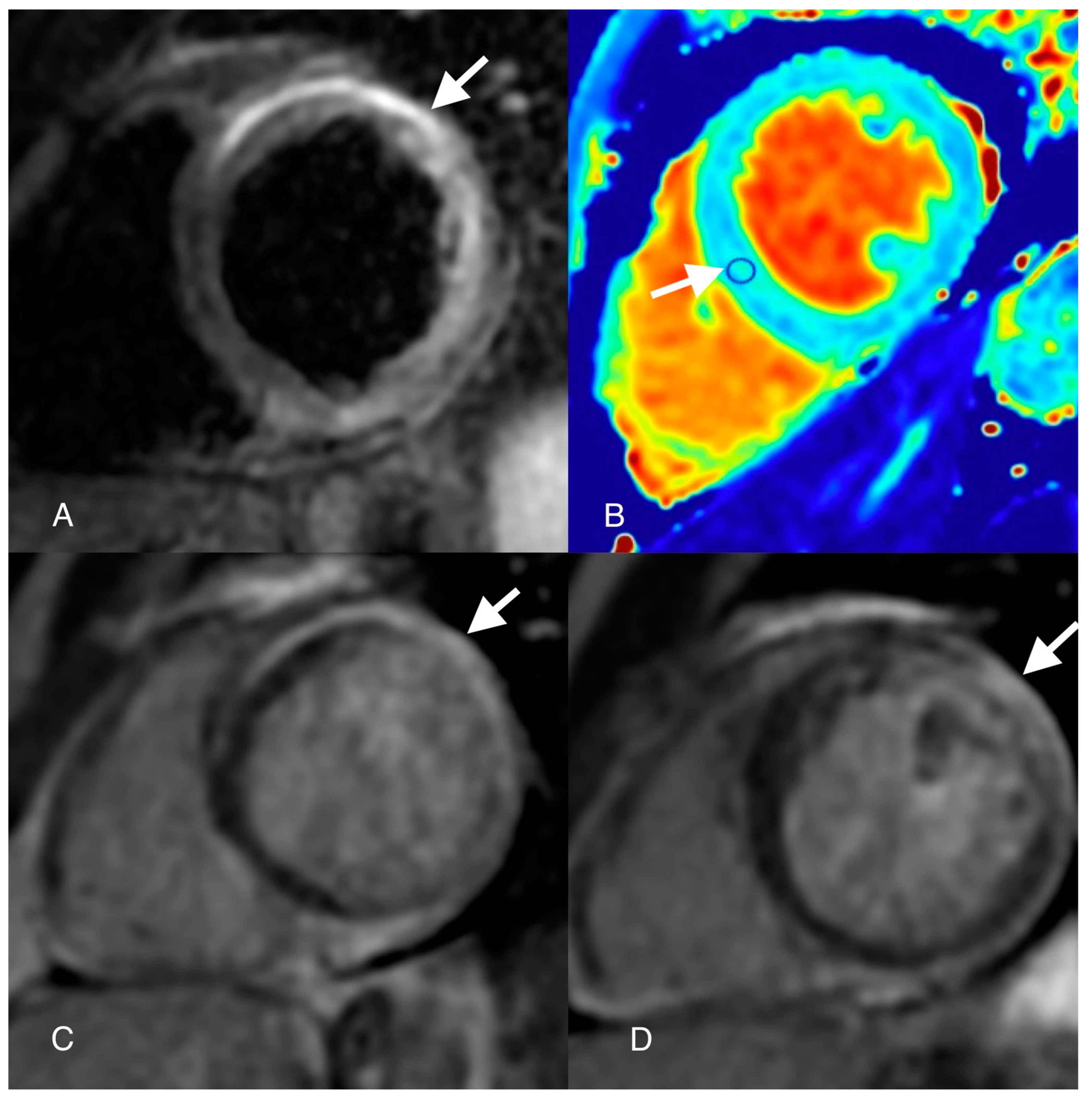{kind=link}
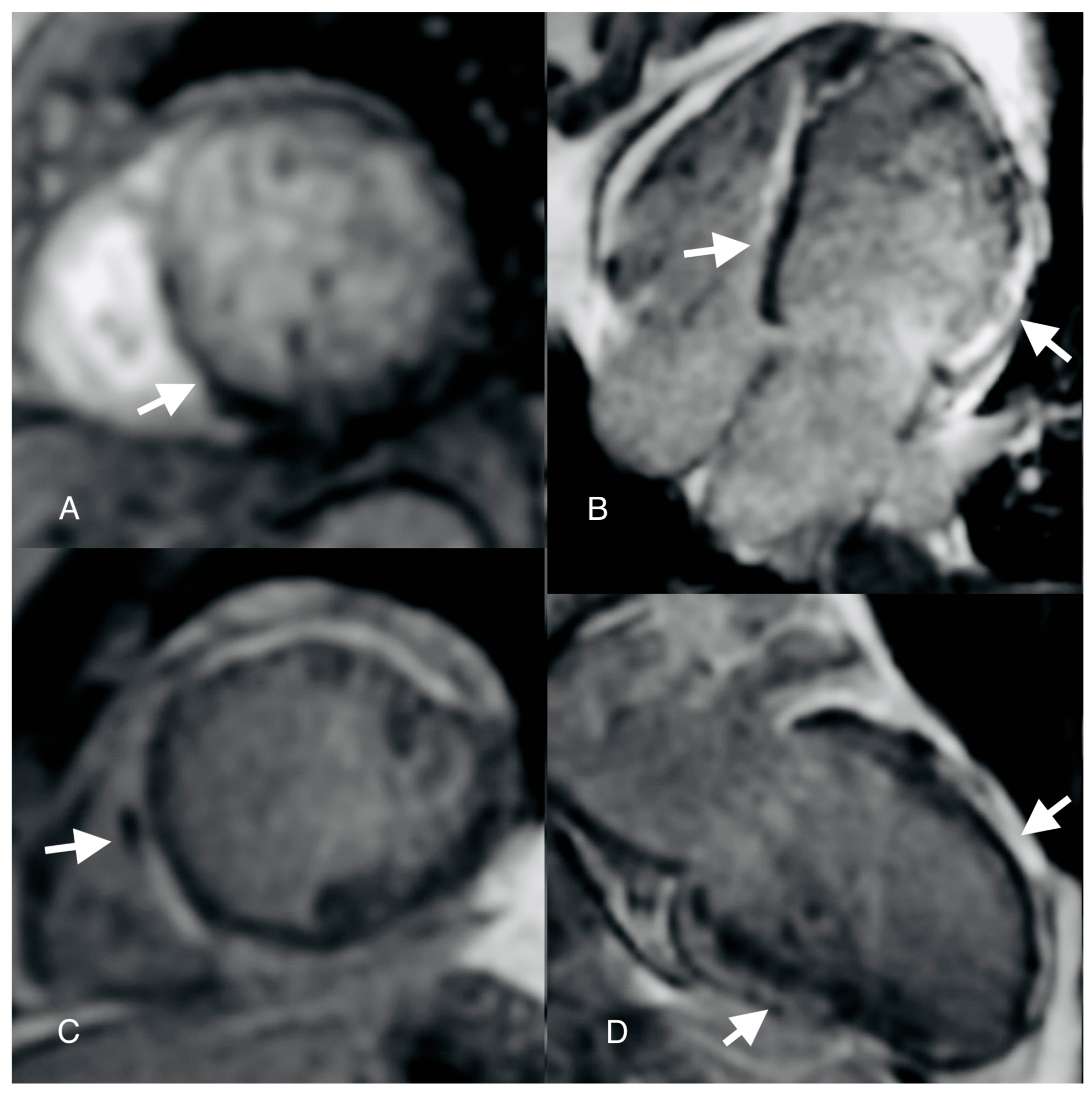{kind=link}
Abstract
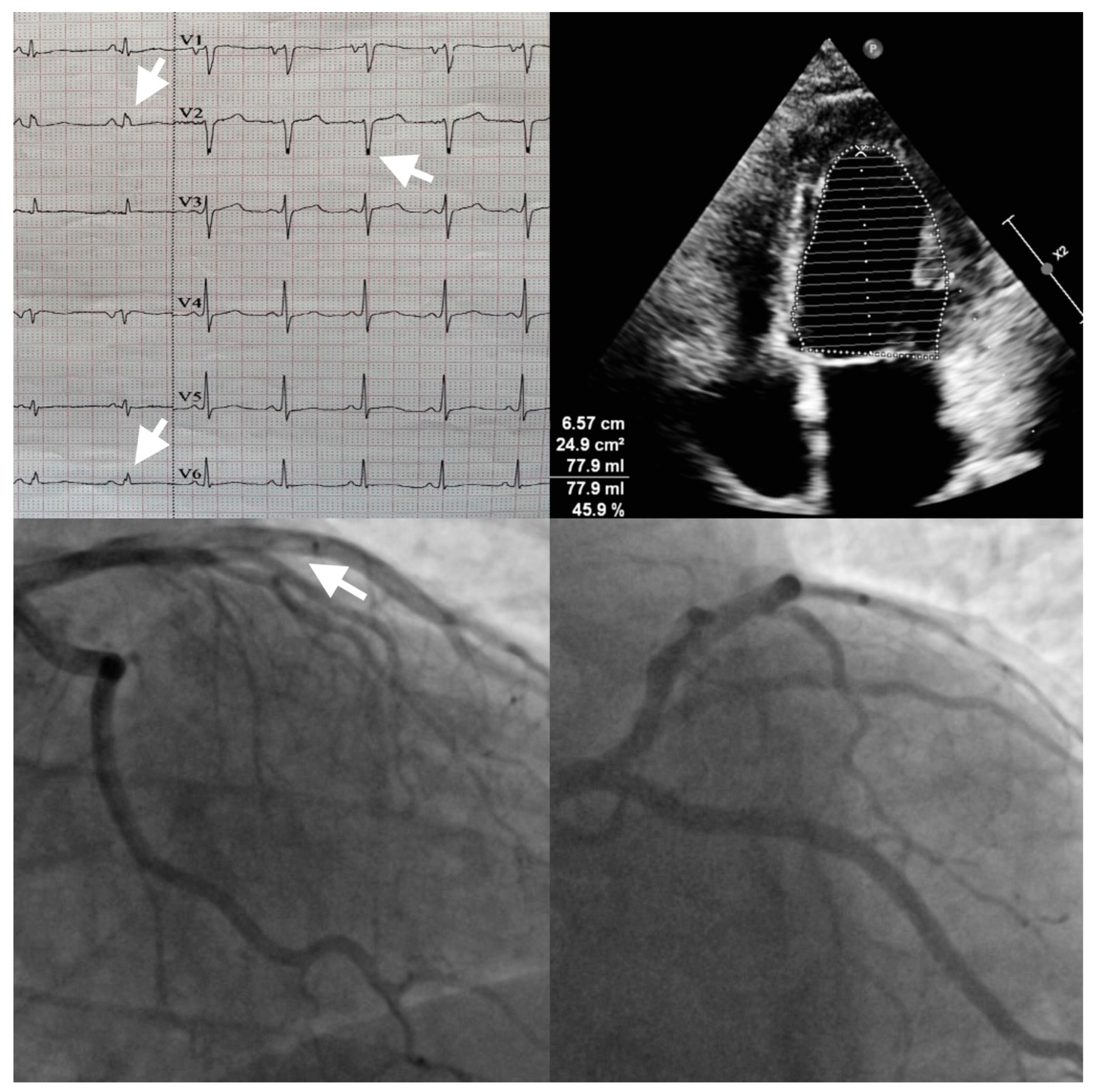
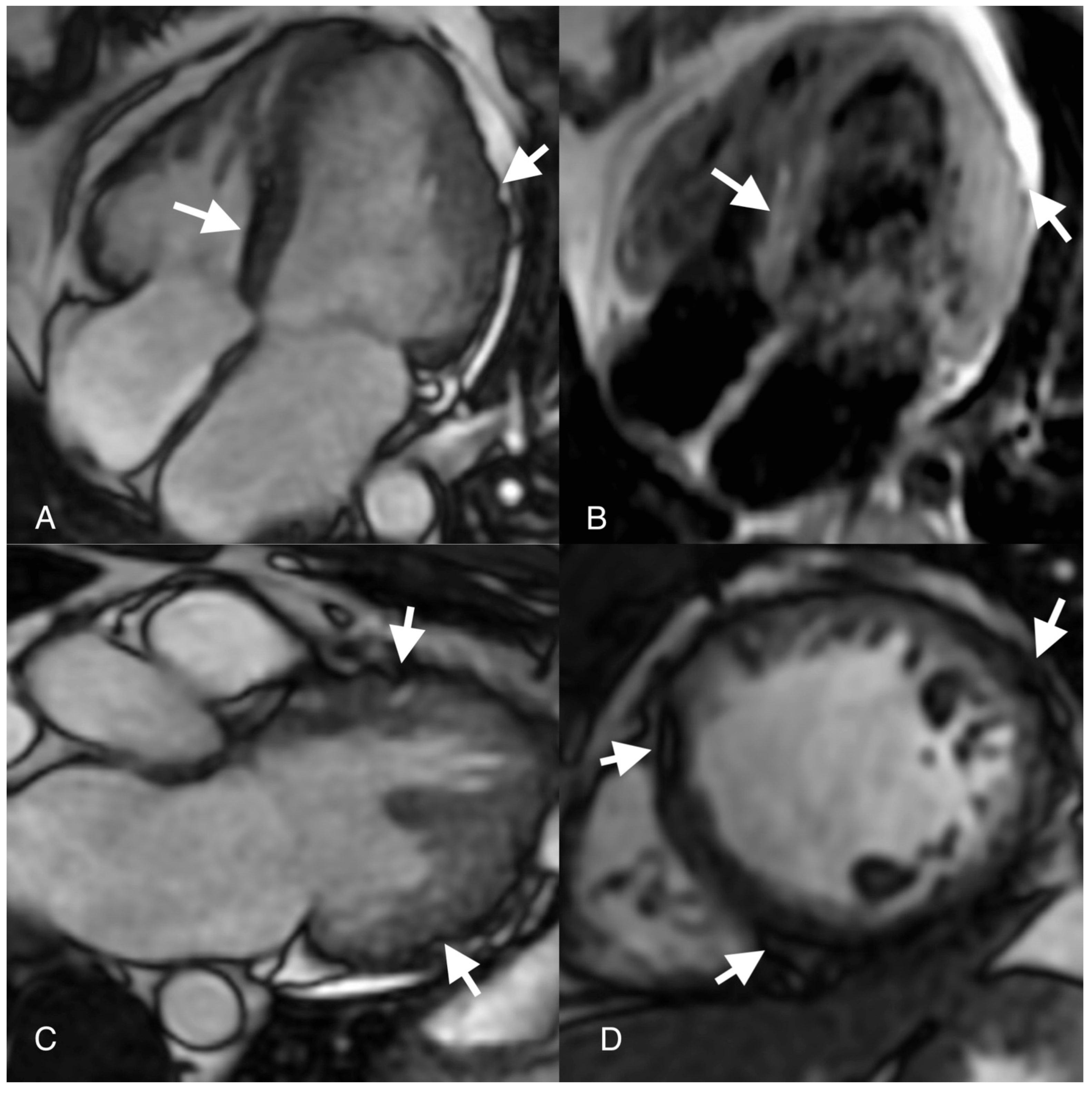
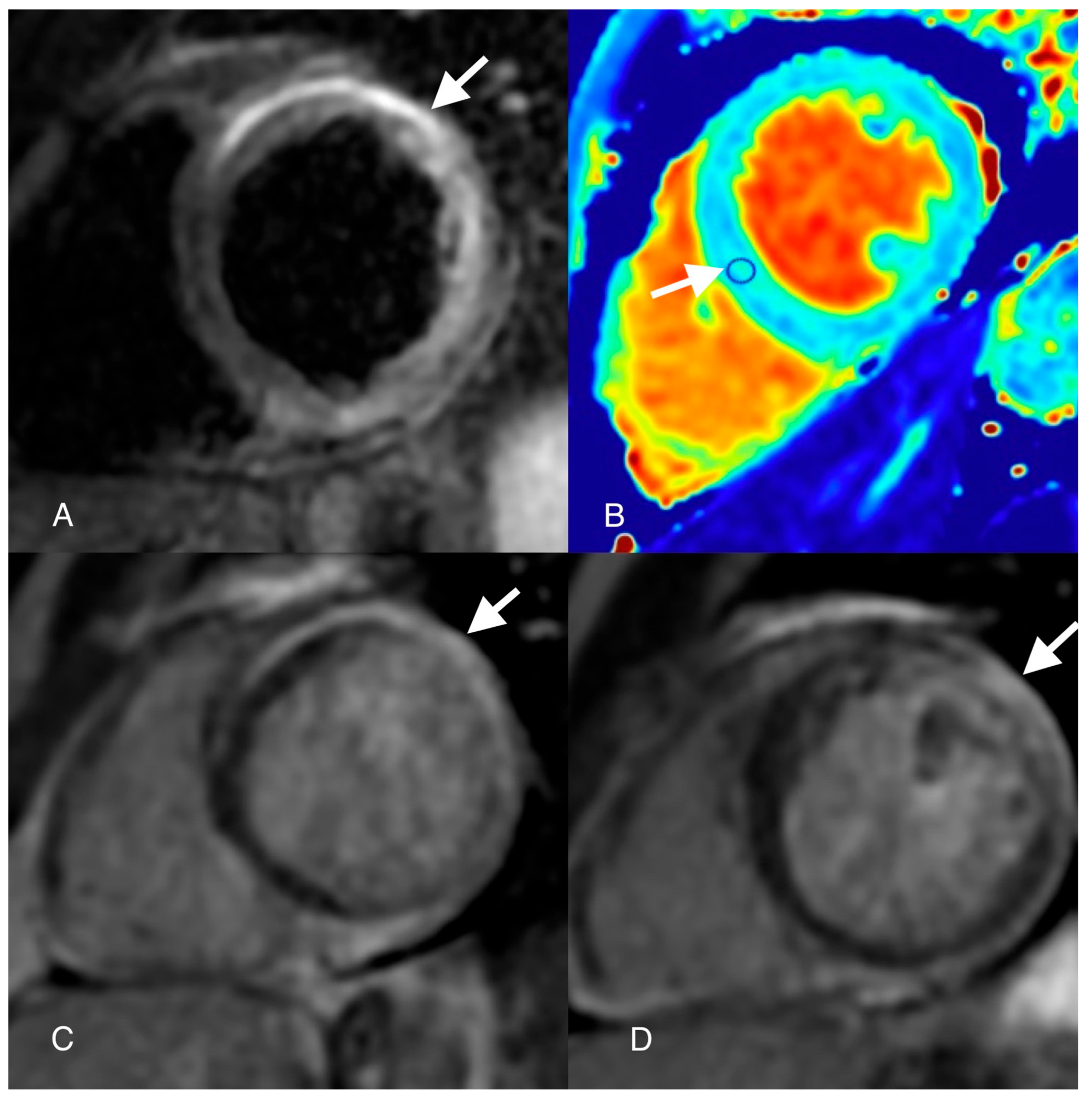
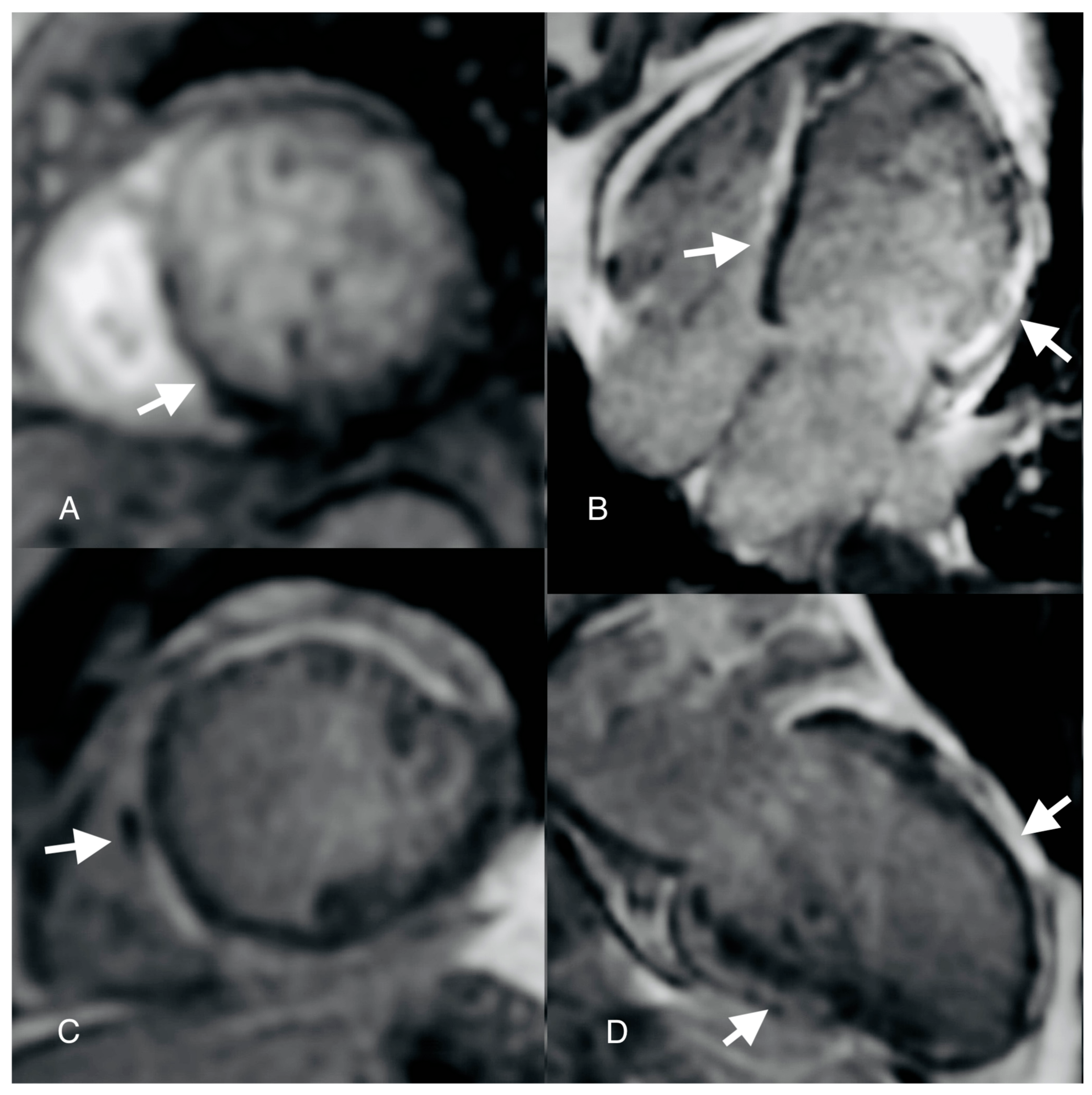
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weisberg, I.S.; Jacques, P.F.; Selhud, J.; Bostom, A.G.; Chen, Z.; Ellison, R.C.; Eckfeldt, J.H.; Rozen, R. The1298A → C polymorphism in methylenetetrahydrofolate reductase (MTHFR): In vitro expression and association with homocysteine. Atherosclerosis 2001, 156, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Lievers, K.J.A.; Boers, G.H.J.; Verhoef, P.; Heijer, M.; Kluijtmans, L.A.; Put, N.M.; Trijbels, F.J.; Blom, H.J. A second common variant in the methylenetetrahydrofolate reductase (MTHFR) gene and its relationship to MTHFR enzyme activity, homocysteine, and cardiovascular disease risk. J. Mol. Med. 2001, 79, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Asatryan, B.; Asimaki, A.; Landstrom, A.P.; Khanji, M.Y.; Odening, K.E.; Cooper, L.T.; Marchlinski, F.E.; Gelzer, A.R.; Semsarian, C.; Reichlin, T.; et al. Inflammation and Immune Response in Arrhythmogenic Cardiomyopathy: State-of-the-Art Review. Circulation 2021, 144, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Thiene, G.; Corrado, D.; Angelini, A.; Nava, A.; Valente, M. Arrhythmo- genic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation 1996, 94, 983–991. [Google Scholar] [CrossRef]
- Reichl, K.; Kreykes, S.E.; Martin, C.M.; Shenoy, C. Desmoplakin variant-as- sociated arrhythmogenic cardiomyopathy presenting as acute myo- carditis. Circ. Genom. Precis. Med. 2018, 11, e002373. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Thiene, G.; McKenna, W.J.; Davies, M.J.; Fontaliran, F.; Nava, A.; Silvestri, F.; Blomstrom-Lundqvist, C.; Wlodarska, E.K.; et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J. Am. Coll. Cardiol. 1997, 30, 1512–1520. [Google Scholar] [CrossRef]
- Campuzano, O.; Alcalde, M.; Iglesias, A.; Barahona-Dussault, C.; Sarquella-Brugada, G.; Benito, B.; Arzamendi, D.; Flores, J.; Leung, T.K.; Talajic, M.; et al. Arrhythmogenic right ventricular cardiomyopathy: Severe structural alterations are associated with inflammation. J. Clin. Pathol. 2012, 65, 1077–1083. [Google Scholar] [CrossRef]
- Bariani, R.; Rigato, I.; Cipriani, A.; Bueno Marinas, M.; Celeghin, R.; Basso, C.; Corrado, D.; Pilichou, K.; Bauce, B. Myocarditis-like Episodes in Patients with Arrhythmogenic Cardiomyopathy: A Systematic Review on the So-Called Hot-Phase of the Disease. Biomolecules 2022, 12, 1324. [Google Scholar] [CrossRef]
- Martins, D.; Ovaert, C.; Khraiche, D.; Boddaert, N.; Bonnet, D.; Raimondi, F. Myocardial inflammation detected by cardiac MRI in Arrhythmogenic right ventricular cardiomyopathy: A paediatric case series. Int. J. Cardiol. 2018, 271, 81–86. [Google Scholar] [CrossRef]
- Bowles, N.E.; Ni, J.; Marcus, F.; Towbin, J.A. The detection of cardiotropic viruses in the myocardium of patients with arrhythmogenic right ventriculardysplasia/cardiomyopathy. J. Am. Coll. Cardiol. 2002, 39, 892–895. [Google Scholar] [CrossRef] [PubMed]
- Campian, M.E.; Verberne, H.J.; Hardziyenka, M.; de Groot, E.A.; van Moerkerken, A.F.; van Eck-Smit, B.L.; Tan, H.L. Assessment of inflammation in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 2079–2085. [Google Scholar] [CrossRef] [PubMed]
- Protonotarios, A.; Wicks, E.; Ashworth, M.; Stephenson, E.; Guttmann, O.; Savvatis, K.; Sekhri, N.; Mohiddin, S.A.; Syrris, P.; Menezes, L.; et al. Prevalence of 18F- fluorodeoxyglucose positron emission tomography abnormalities in pa- tients with arrhythmogenic right ventricular cardiomyopathy. Int. J. Cardiol. 2019, 284, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Poller, W.; Haas, J.; Klingel, K.; Kühnisch, J.; Gast, M.; Kaya, Z.; Escher, F.; Kayvanpour, E.; Degener, F.; Opgen-Rhein, B.; et al. Familial recurrent myocarditis triggered by exercise in patients with a truncating variant of the desmoplakin gene. J. Am. Heart Assoc. 2020, 9, e015289. [Google Scholar] [CrossRef] [PubMed]
- Mavrogeni, S.; Protonotarios, N.; Tsatsopoulou, A.; Papachristou, P.; Sfendouraki, E.; Papadopoulos, G. Naxos disease evolution mimicking acute myocarditis: The role of cardiovascular magnetic resonance imaging. Int. J. Cardiol. 2013, 166, e14–e15. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Belke, D.D.; Garnett, L.; Martens, K.; Abdelfatah, N.; Rodriguez, M.; Diao, C.; Chen, Y.-X.; Gordon, P.M.K.; Nygren, A.; et al. Transgenic Mice Overexpressing Desmocollin-2 (DSC2) Develop Cardiomyopathy Associated with MyocardialInflammation and Fibrotic Remodeling. PLoS ONE 2017, 12, e0174019. [Google Scholar] [CrossRef] [PubMed]
- Pilichou, K.; Remme, C.A.; Basso, C.; Campian, M.E.; Rizzo, S.; Barnett, P.; Scicluna, B.P.; Bauce, B.; Van Den Hoff, M.J.B.; De Bakker, J.M.T.; et al. Myocyte Necrosis Underlies Progressive Myocardial Dystrophy in Mouse Dsg2-Related Arrhythmogenic Right Ventricular Cardiomyopathy. J. Exp. Med. 2009, 206, 1787–1802. [Google Scholar] [CrossRef]
- Zhu, Q.; Wang, S.; Huang, X.; Zhao, C.; Wang, Y.; Li, X.; Jia, D.; Ma, C. Understanding the pathogenesis of coronary slow flow: Recent advances. Trends Cardiovasc. Med. 2022. [Google Scholar] [CrossRef]
- Woudstra, L.; Juffermans, L.J.M.; van Rossum, A.C.; Niessen, H.W.M.; Krijnen, P.A.J. Infectious myocarditis: The role of the cardiac vasculature. Heart Fail. Rev. 2018, 23, 583–595. [Google Scholar] [CrossRef]
- Paul, M.; Rahbar, K.; Gerss, J.; Kies, P.; Schober, O.; Schäfers, K.; Breithardt, G.; Schulze-Bahr, E.; Wichter, T.; Schäfers, M. Microvascular dysfunction in nonfailing arrhythmogenic right ventricular cardiomyopathy. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 416–420. [Google Scholar] [CrossRef]
- Gerull, B.; Brodehl, A. Insights Into Genetics and Pathophysiology of Arrhythmogenic Cardiomyopathy. Curr. Heart Fail. Rep. 2021, 18, 378–390. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lala, I.R.; Pop-Moldovan, A. Inflammation—A Possible Link between Myocarditis and Arrhythmogenic Cardiomyopathy. Diagnostics 2024, 14, 248. https://doi.org/10.3390/diagnostics14030248
Lala IR, Pop-Moldovan A. Inflammation—A Possible Link between Myocarditis and Arrhythmogenic Cardiomyopathy. Diagnostics. 2024; 14(3):248. https://doi.org/10.3390/diagnostics14030248
Chicago/Turabian StyleLala, Ioan Radu, and Adina Pop-Moldovan. 2024. "Inflammation—A Possible Link between Myocarditis and Arrhythmogenic Cardiomyopathy" Diagnostics 14, no. 3: 248. https://doi.org/10.3390/diagnostics14030248
APA StyleLala, I. R., & Pop-Moldovan, A. (2024). Inflammation—A Possible Link between Myocarditis and Arrhythmogenic Cardiomyopathy. Diagnostics, 14(3), 248. https://doi.org/10.3390/diagnostics14030248