
Best Practices in Nuclear Imaging for the Diagnosis of Transthyretin Amyloid Cardiomyopathy (ATTR-CM) in KSA: The Eagle Eyes of Local Experts

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. ATTR-CM Presentation
3.2. Clinical Suspicion
3.3. Diagnosis
Nuclear Scintigraphy for Diagnosing ATTR-CM
4. Best Practices Recommended by the Expert Panel
4.1. Suspicion of ATTR-CM
- Considering the multi-organ involvement of ATTR-CM, an interdisciplinary strategy is required involving collaboration between nuclear medicine specialists, cardiologists, and other healthcare professionals is recommended for assessment and diagnosis.
- The patient evaluation should start with an assessment of medical history, including symptoms and risk factors for ATTR-CM. Further, the patient evaluation should include a thorough physical examination to assess for signs of cardiac involvement, such as heart murmurs, abnormal heart sounds, and signs of fluid retention.
- Patients should be screened for any clinical features suggestive of the phenotype of ATTR-CM, particularly those with a constellation of cardiac, neurological, and musculoskeletal manifestations.
- A transthoracic echocardiogram (TTE) should be conducted evaluating cardiac structure and function. One should look for signs of cardiac hypertrophy, thickened myocardium, and abnormal diastolic function, which are common findings in ATTR-CM. TTE can often help differentiate between restrictive and hypertrophic cardiomyopathies.
- Patients exhibiting an enlarged ventricular wall thickness (≥12 mm), along with at least one of the following indicators or warning signs, should be evaluated for ATTR-CM: an apical sparing longitudinal strain pattern, orthopedic symptoms (such as bilateral carpal tunnel syndrome, lumbar spinal stenosis, or a rupture of the bicep tendon), consistently elevated troponin levels, atrioventricular conduction block, or peripheral or autonomic neuropathy.
- In instances where the TTE is of subpar quality or yields indeterminate results, CMR becomes necessary. Diagnostic characteristics of CMR encompass an enlargement of the extra-cellular volume, irregular kinetics of gadolinium contrast (imperfect nulling), and widespread late gadolinium enhancement.
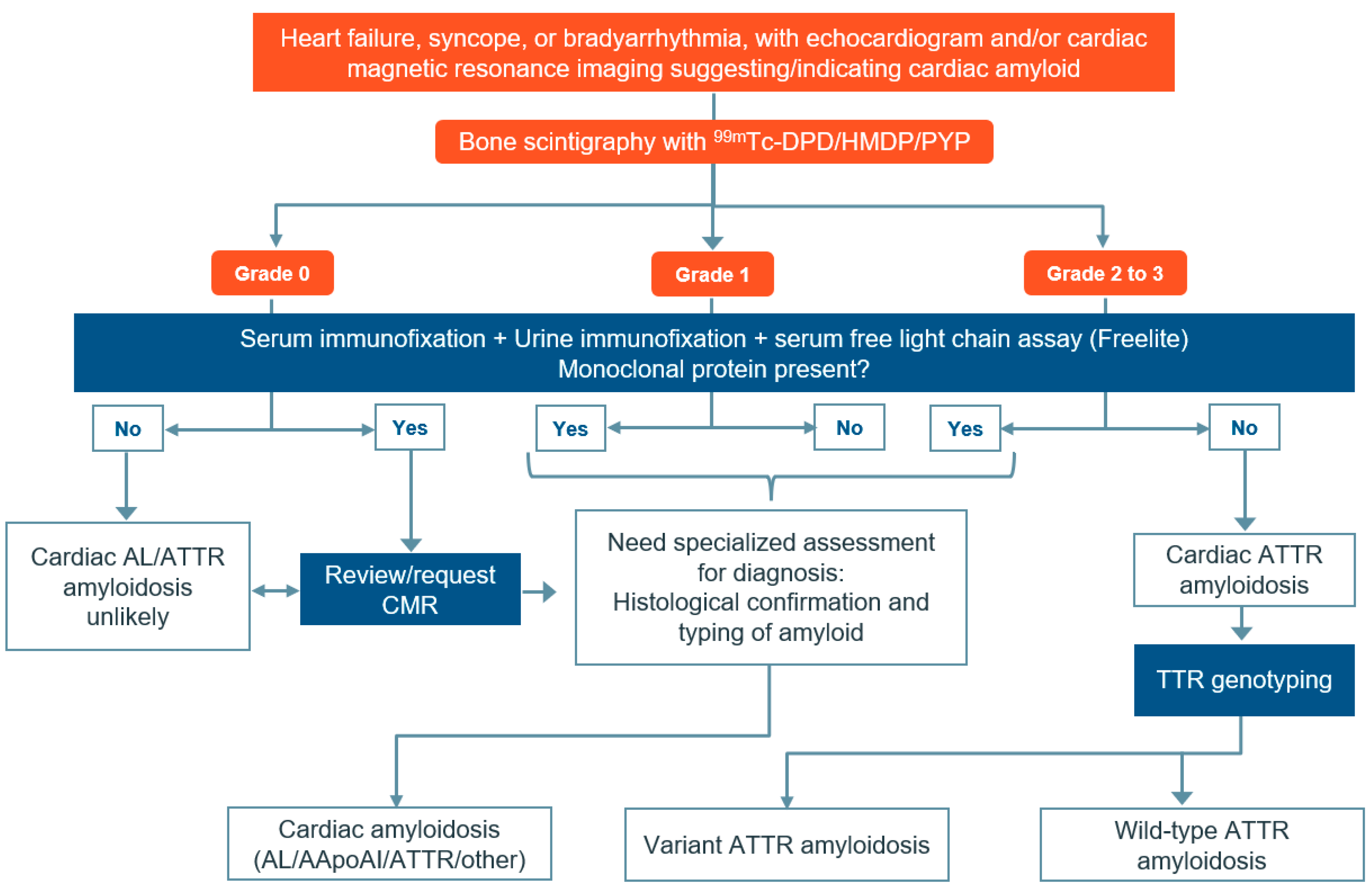
- Red flags, ECG, and ECHO can be employed to raise suspicions of amyloidosis. Nuclear scintigraphy may be considered to differentiate ATTR-CM and AL.
- Hematologic tests should be carried out to rule out AL amyloidosis either before or at the time of ordering PYP imaging. An abnormal hematologic test should trigger a hematology consultation.
4.2. Diagnosing ATTR-CM
- Imaging procedures:
- ○
- No special preparation or fasting is required for the patient undergoing bone scintigraphy.
- ○
- Rest scan is preferred using the radio tracer, 99mTc-PYP at a10–20 mCi/370–740 MBq (intravenous) dose; HMDP can be used in case of shortages of PYP.
- ○
- Time between injection and acquisition:
- ▪
- Depending on the patient’s uptake and various other factors, 2 h or 3 h is recommended. One hour interval (only with 99mTc-PYP) is optional. If any excess blood pool activity is observed on the 1 h images, then imaging after a 3 h interval is recommended.
- ▪
- For elderly or intolerant patients, if results are conclusive in 1 h, consider stopping imaging instead of proceeding for 3 h.
- ○
- When available, SPECT/CT imaging is preferred for radio-tracer localization.
- Imaging parameters:
- ○
- Suggested cardiac or chest field of view with SPECT and planar imaging in supine position.
- ○
- Suggested other imaging parameters include:
- ▪
- Energy window: 140 keV, 15–20%.
- ▪
- Collimators: Low energy, high resolution.
- ▪
- Matrix: Planar: 256 by 256; at least 64 by 64 is required. SPECT: 128 by 128; at least 64 by 64 is required.
- ▪
- Pixel: 2.3–6.5 mm.
- Planar imaging parameters:
- ○
- Number of views: anterior, lateral, and left anterior oblique.
- ○
- Detector configuration: 90 degrees.
- ○
- Image duration (count based): 750,000 counts.
- ○
- Magnification: 1.46.
- SPECT imaging parameters:
- ○
- Angular range: Recommended: 180 degrees; Optional: 360 degrees.
- ○
- Detector configuration: Recommended 90 degrees; Optional 180 degrees.
- ○
- ECG gating: Off; non-gated imaging.
- ○
- Number of views/detectors: 40.
- ○
- Time per stop: 20 s.
- ○
- Magnification: 1.0.
- Interpretation:
- ○
- Visual interpretation:
- ▪
- To confirm diffuse radio-tracer uptake in the myocardium, planar and SPECT or SPECT/CT images can be evaluated.
- ▪
- To differentiate myocardial radio-tracer uptake from residual blood pool activity, focal myocardial infarct, and overlapping bone (e.g., from rib hot spots from fractures) on SPECT images, planar and SPECT or SPECT/CT images can be evaluated, as per physician’s discretion and accessibility to the technique. If excess blood pool activity is noted on the 1 h SPECT images, repeating SPECT imaging at 3 h is recommended. If myocardial tracer uptake is visually present on SPECT, proceed to semi-quantitative visual grading. If no myocardial tracer uptake is present on SPECT, the visual grade is 0.
- ▪
- If myocardial tracer uptake is visually present on SPECT, semi-quantitative visual grading can be performed. If no myocardial tracer uptake is present on SPECT, the visual grade is 0.
- ○
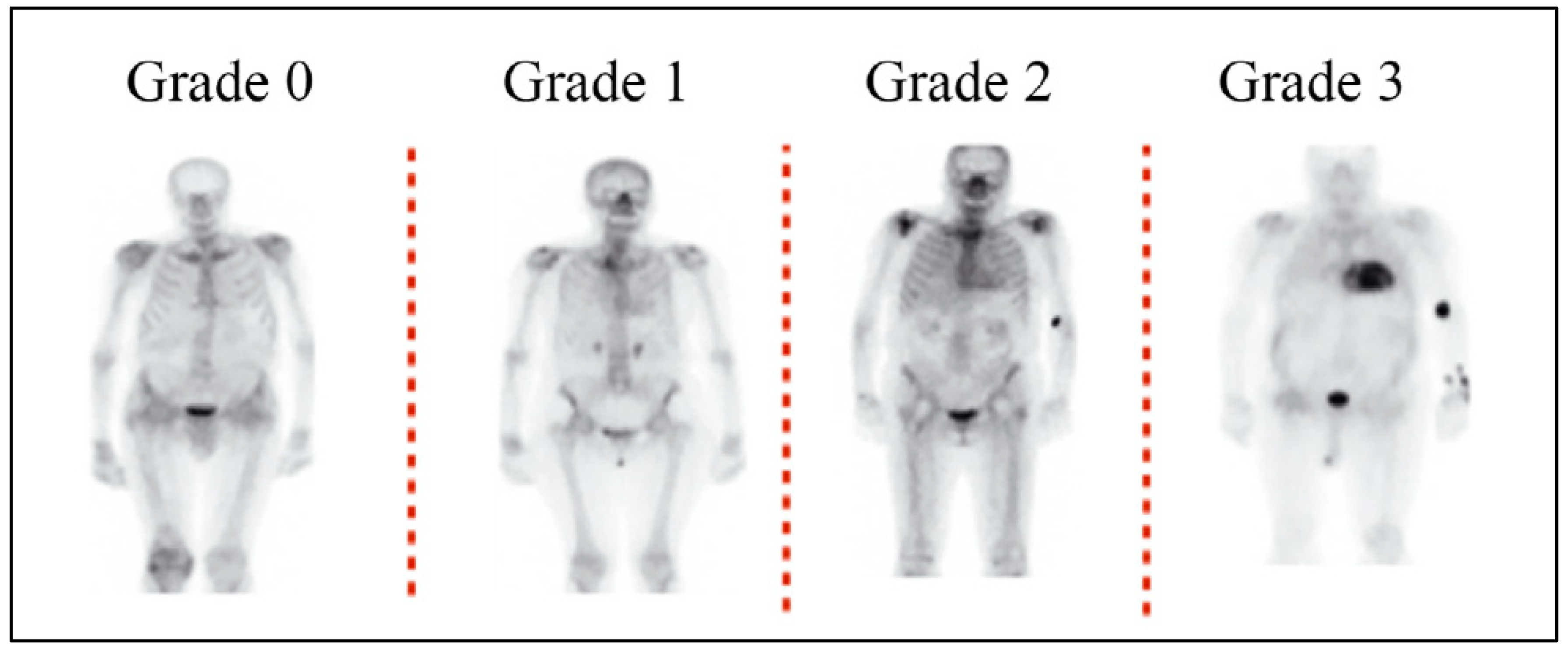
- To distinguish between AL and ATTR cardiac amyloidosis, semi-quantitative grading can be performed using planar and SPECT or SPECT/CT images. These images are then examined for any relative tracer uptake in the myocardium in comparison to the ribs. Based on these observations, the images are then graded using the following scale:
- ▪
- Grade 0: This grade is assigned when there is no observable myocardial uptake, and the bone uptake appears to be within normal limits.
- ▪
- Grade 1: This grade is given when the myocardial uptake is observed to be less than the uptake in the ribs.
- ▪
- Grade 2: This grade is assigned when the myocardial uptake is found to be equal to the rib uptake.
- ▪
- Grade 3: This grade is given when the myocardial uptake is greater than the rib uptake, and the rib uptake is either mild or not present at all.
- ○
- When applicable, heart-to-contralateral lung (H/CL) uptake ratio assessment can be performed to identify ATTR cardiac amyloidosis. H/CL ratios of ≥1.5 at 1 h can accurately identify ATTR cardiac amyloidosis if myocardial pyrophosphate (PYP) uptake is visually confirmed on SPECT and systemic AL amyloidosis is excluded. An H/CL ratio of ≥1.3 at 3 h can also identify ATTR cardiac amyloidosis.
- ○
- The detection of ATTR-CM cannot rely solely on H/CL ratio with PYP. H/CL ratio is not recommended if there is an absence of myocardial uptake on SPECT. If there is a discordant result or the visual grade is equivocal, the H/CL ratio may be helpful to classify it as positive or negative. Both H/CL and a semi-quantitative visual system are recommended for assessing cardiac amyloidosis via scintigraphy.
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2018: Recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2018, 25, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pavia, P.; Bengel, F.; Brito, D.; Damy, T.; Duca, F.; Dorbala, S.; Nativi-Nicolau, J.; Obici, L.; Rapezzi, C.; Sekijima, Y.; et al. Expert consensus on the monitoring of transthyretin amyloid cardiomyopathy. Eur. J. Heart Fail. 2021, 23, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Chattranukulchai, P.; Lee, A.P.; Lin, Y.H.; Yu, W.C.; Liew, H.B.; Oomman, A. Clinical recommendations to diagnose and monitor patients with transthyretin amyloid cardiomyopathy in Asia. Clin. Cardiol. 2022, 45, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.W.; Colon, W.; Lai, Z.; Lashuel, H.A.; McCulloch, J.; McCutchen, S.L.; Miroy, G.J.; Peterson, S.A. Transthyretin quaternary and tertiary structural changes facilitate misassembly into amyloid. Adv. Protein Chem. 1997, 50, 161–181. [Google Scholar] [CrossRef] [PubMed]
- Liz, M.A.; Mar, F.M.; Franquinho, F.; Sousa, M.M. Aboard transthyretin: From transport to cleavage. IUBMB Life 2010, 62, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Cornwell, G.G., 3rd; Sletten, K.; Johansson, B.; Westermark, P. Evidence that the amyloid fibril protein in senile systemic amyloidosis is derived from normal prealbumin. Biochem. Biophys. Res. Commun. 1988, 154, 648–653. [Google Scholar] [CrossRef]
- Kittleson, M.M.; Maurer, M.S.; Ambardekar, A.V.; Bullock-Palmer, R.P.; Chang, P.P.; Eisen, H.J.; Nair, A.P.; Nativi-Nicolau, J.; Ruberg, F.L. Cardiac Amyloidosis: Evolving Diagnosis and Management: A Scientific Statement From the American Heart Association. Circulation 2020, 142, e7–e22. [Google Scholar] [CrossRef]
- Jain, A.; Zahra, F. Transthyretin Amyloid Cardiomyopathy (ATTR-CM). In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Ruberg, F.L.; Grogan, M.; Hanna, M.; Kelly, J.W.; Maurer, M.S. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 2872–2891. [Google Scholar] [CrossRef]
- González-López, E.; Gallego-Delgado, M.; Guzzo-Merello, G.; de Haro-Del Moral, F.J.; Cobo-Marcos, M.; Robles, C.; Bornstein, B.; Salas, C.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur. Heart J. 2015, 36, 2585–2594. [Google Scholar] [CrossRef]
- Castaño, A.; Narotsky, D.L.; Hamid, N.; Khalique, O.K.; Morgenstern, R.; DeLuca, A.; Rubin, J.; Chiuzan, C.; Nazif, T.; Vahl, T.; et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur. Heart J. 2017, 38, 2879–2887. [Google Scholar] [CrossRef]
- Damy, T.; Bourel, G.; Slama, M.; Algalarrondo, V.; Lairez, O.; Fournier, P.; Costa, J.; Pelcot, F.; Farrugia, A.; Zaleski, I.D. Incidence and survival of transthyretin amyloid cardiomyopathy from a French nationwide study of in-and out-patient databases. Arch. Cardiovasc. Dis. Suppl. 2023, 15, 47. [Google Scholar]
- Rozenbaum, M.H.; Large, S.; Bhambri, R.; Stewart, M.; Whelan, J.; van Doornewaard, A.; Dasgupta, N.; Masri, A.; Nativi-Nicolau, J. Impact of Delayed Diagnosis and Misdiagnosis for Patients with Transthyretin Amyloid Cardiomyopathy (ATTR-CM): A Targeted Literature Review. Cardiol. Ther. 2021, 10, 141–159. [Google Scholar] [CrossRef] [PubMed]
- Dang, D.; Fournier, P.; Cariou, E.; Huart, A.; Ribes, D.; Cintas, P.; Roussel, M.; Colombat, M.; Lavie-Badie, Y.; Carrié, D.; et al. Gateway and journey of patients with cardiac amyloidosis. ESC Heart Fail. 2020, 7, 2418–2430. [Google Scholar] [CrossRef] [PubMed]
- Al Badarin, F.; Al-Humood, K.; Bader, F.; Alsaid, S.; Sulaiman, K.; Alzadjali, M.; Sabbour, H.; Shehab, A.; Bazargani, N.; Perlini, S. Physician Knowledge and Awareness About Cardiac Amyloidosis in the Middle East and Gulf Region. JACC CardioOncol. 2022, 4, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Mohty, D.; Nasr, S.; Ragy, H.; Farhan, H.A.; Fadel, B.; Alayary, I.; Ghoubar, M. Cardiac amyloidosis: A survey of current awareness, diagnostic modalities, treatment practices, and clinical challenges among cardiologists in selected Middle Eastern countries. Clin. Cardiol. 2023, 46, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Dorbala, S.; Ando, Y.; Bokhari, S.; Dispenzieri, A.; Falk, R.H.; Ferrari, V.A.; Fontana, M.; Gheysens, O.; Gillmore, J.D.; Glaudemans, A.; et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI Expert Consensus Recommendations for Multimodality Imaging in Cardiac Amyloidosis: Part 1 of 2-Evidence Base and Standardized Methods of Imaging. Circ. Cardiovasc. Imaging 2021, 14, e000029. [Google Scholar] [CrossRef] [PubMed]
- Apostolou, E.A.; Fontrier, A.M.; Efthimiadis, G.K.; Kastritis, E.; Parissis, J.; Kanavos, P. The patient pathway in ATTR-CM in Greece and how to improve it: A multidisciplinary perspective. Hell. J. Cardiol. 2023, 73, 73–80. [Google Scholar] [CrossRef]
- Singh, V.; Falk, R.; Di Carli, M.F.; Kijewski, M.; Rapezzi, C.; Dorbala, S. State-of-the-art radionuclide imaging in cardiac transthyretin amyloidosis. J. Nucl. Cardiol. 2019, 26, 158–173. [Google Scholar] [CrossRef]
- Maurer, M.S.; Bokhari, S.; Damy, T.; Dorbala, S.; Drachman, B.M.; Fontana, M.; Grogan, M.; Kristen, A.V.; Lousada, I.; Nativi-Nicolau, J.; et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ. Heart Fail. 2019, 12, e006075. [Google Scholar] [CrossRef]
- Bokhari, S.; Castaño, A.; Pozniakoff, T.; Deslisle, S.; Latif, F.; Maurer, M.S. (99m)Tc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ. Cardiovasc. Imaging 2013, 6, 195–201. [Google Scholar] [CrossRef]
- Chander Mohan, J.; Dalal, J.; Chopra, V.K.; Narasimhan, C.; Kerkar, P.; Oomman, A.; Ray Fcsi, S.; Sharma, A.R.; Dougall, P.; Simon, S.; et al. Suspecting and diagnosing transthyretin amyloid cardiomyopathy (ATTR-CM) in India: An Indian expert consensus. Indian Heart J. 2022, 74, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Gillmore, J.D.; Maurer, M.S.; Falk, R.H.; Merlini, G.; Damy, T.; Dispenzieri, A.; Wechalekar, A.D.; Berk, J.L.; Quarta, C.C.; Grogan, M.; et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation 2016, 133, 2404–2412. [Google Scholar] [CrossRef] [PubMed]
- Dorbala, S.; Cuddy, S.; Falk, R.H. How to Image Cardiac Amyloidosis: A Practical Approach. JACC Cardiovasc. Imaging 2020, 13, 1368–1383. [Google Scholar] [CrossRef] [PubMed]
- Vergaro, G.; Aimo, A.; Barison, A.; Genovesi, D.; Buda, G.; Passino, C.; Emdin, M. Keys to early diagnosis of cardiac amyloidosis: Red flags from clinical, laboratory and imaging findings. Eur. J. Prev. Cardiol. 2020, 27, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Rauf, M.U.; Hawkins, P.N.; Cappelli, F.; Perfetto, F.; Zampieri, M.; Argiro, A.; Petrie, A.; Law, S.; Porcari, A.; Razvi, Y.; et al. Tc-99m labelled bone scintigraphy in suspected cardiac amyloidosis. Eur. Heart J. 2023, 44, 2187–2198. [Google Scholar] [CrossRef] [PubMed]
- Witteles, R.M.; Bokhari, S.; Damy, T.; Elliott, P.M.; Falk, R.H.; Fine, N.M.; Gospodinova, M.; Obici, L.; Rapezzi, C.; Garcia-Pavia, P. Screening for Transthyretin Amyloid Cardiomyopathy in Everyday Practice. JACC Heart Fail. 2019, 7, 709–716. [Google Scholar] [CrossRef]
- Sabbour, H.; Hasan, K.Y.; Al Badarin, F.; Alibazoglu, H.; Rivard, A.L.; Romany, I.; Perlini, S. From Clinical Clues to Final Diagnosis: The Return of Detective Work to Clinical Medicine in Cardiac Amyloidosis. Front. Cardiovasc. Med. 2021, 8, 644508. [Google Scholar] [CrossRef]
- Longhi, S.; Lorenzini, M.; Gagliardi, C.; Milandri, A.; Marzocchi, A.; Marrozzini, C.; Saia, F.; Ortolani, P.; Biagini, E.; Guidalotti, P.L.; et al. Coexistence of Degenerative Aortic Stenosis and Wild-Type Transthyretin-Related Cardiac Amyloidosis. JACC Cardiovasc. Imaging 2016, 9, 325–327. [Google Scholar] [CrossRef]
- Dorbala, S.; Ando, Y.; Bokhari, S.; Dispenzieri, A.; Falk, R.H.; Ferrari, V.A.; Fontana, M.; Gheysens, O.; Gillmore, J.D.; Glaudemans, A.; et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: Part 2 of 2-Diagnostic criteria and appropriate utilization. J. Nucl. Cardiol. 2020, 27, 659–673. [Google Scholar] [CrossRef]
- Brownrigg, J.; Lorenzini, M.; Lumley, M.; Elliott, P. Diagnostic performance of imaging investigations in detecting and differentiating cardiac amyloidosis: A systematic review and meta-analysis. ESC Heart Fail. 2019, 6, 1041–1051. [Google Scholar] [CrossRef]
- Kittleson, M.M.; Ruberg, F.L.; Ambardekar, A.V.; Brannagan, T.H.; Cheng, R.K.; Clarke, J.O.; Dember, L.M.; Frantz, J.G.; Hershberger, R.E.; Maurer, M.S.; et al. 2023 ACC Expert Consensus Decision Pathway on Comprehensive Multidisciplinary Care for the Patient With Cardiac Amyloidosis: A Report of the American College of Cardiology Solution Set Oversight Committee. J. Am. Coll. Cardiol. 2023, 81, 1076–1126. [Google Scholar] [CrossRef] [PubMed]
- Zeldenrust, S.R.; Benson, M.D. Familial and senile amyloidosis caused by transthyretin. In Protein Misfolding Diseases: Current and Emerging Principles and Therapies; Wiley: Hoboken, NJ, USA, 2010; pp. 795–815. [Google Scholar]
- Hutt, D.F.; Fontana, M.; Burniston, M.; Quigley, A.M.; Petrie, A.; Ross, J.C.; Page, J.; Martinez-Naharro, A.; Wechalekar, A.D.; Lachmann, H.J.; et al. Prognostic utility of the Perugini grading of 99mTc-DPD scintigraphy in transthyretin (ATTR) amyloidosis and its relationship with skeletal muscle and soft tissue amyloid. Eur. Heart J. Cardiovasc. Imaging 2017, 18, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Perugini, E.; Guidalotti, P.L.; Salvi, F.; Cooke, R.M.; Pettinato, C.; Riva, L.; Leone, O.; Farsad, M.; Ciliberti, P.; Bacchi-Reggiani, L.; et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J. Am. Coll. Cardiol. 2005, 46, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Ren, J.; Tian, Z.; Du, Y.; Hao, Z.; Zhang, Z.; Fang, W.; Li, F.; Zhang, S.; Hsu, B.; et al. Assessment of cardiac amyloidosis with (99m)Tc-pyrophosphate (PYP) quantitative SPECT. EJNMMI Phys. 2021, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Elliott, P.; Comenzo, R.; Semigran, M.; Rapezzi, C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation 2017, 135, 1357–1377. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pavia, P.; Rapezzi, C.; Adler, Y.; Arad, M.; Basso, C.; Brucato, A.; Burazor, I.; Caforio, A.L.P.; Damy, T.; Eriksson, U.; et al. Diagnosis and treatment of cardiac amyloidosis: A position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2021, 42, 1554–1568. [Google Scholar] [CrossRef] [PubMed]
- Castano, A.; Haq, M.; Narotsky, D.L.; Goldsmith, J.; Weinberg, R.L.; Morgenstern, R.; Pozniakoff, T.; Ruberg, F.L.; Miller, E.J.; Berk, J.L.; et al. Multicenter Study of Planar Technetium 99m Pyrophosphate Cardiac Imaging: Predicting Survival for Patients With ATTR Cardiac Amyloidosis. JAMA Cardiol. 2016, 1, 880–889. [Google Scholar] [CrossRef]
- Stern, L.K.; Kittleson, M.M. Updates in Cardiac Amyloidosis Diagnosis and Treatment. Curr. Oncol. Rep. 2021, 23, 47. [Google Scholar] [CrossRef]
- Duran, J.M.; Borges-Neto, S. Bone scintigraphy imaging and transthyretin-related (ATTR) cardiac amyloidosis: New tricks from an old tool? J. Nucl. Cardiol. 2023, 30, 368–370. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Cardiac Signs | Extra-Cardiac Signs |
|---|---|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alqarni, A.; Aljizeeri, A.; Bakhsh, A.M.; El-Zeftawy, H.A.M.; Farghaly, H.R.; Alqadhi, M.A.M.; Algarni, M.; Asiri, Z.M.; Osman, A.; Haddadin, H.; et al. Best Practices in Nuclear Imaging for the Diagnosis of Transthyretin Amyloid Cardiomyopathy (ATTR-CM) in KSA: The Eagle Eyes of Local Experts. Diagnostics 2024, 14, 212. https://doi.org/10.3390/diagnostics14020212
Alqarni A, Aljizeeri A, Bakhsh AM, El-Zeftawy HAM, Farghaly HR, Alqadhi MAM, Algarni M, Asiri ZM, Osman A, Haddadin H, et al. Best Practices in Nuclear Imaging for the Diagnosis of Transthyretin Amyloid Cardiomyopathy (ATTR-CM) in KSA: The Eagle Eyes of Local Experts. Diagnostics. 2024; 14(2):212. https://doi.org/10.3390/diagnostics14020212
Chicago/Turabian StyleAlqarni, Abdullah, Ahmed Aljizeeri, Aquib Mohammadidrees Bakhsh, Hossam Ahmed Maher El-Zeftawy, Hussein R. Farghaly, Mukhtar Ahmed M. Alqadhi, Mushref Algarni, Zain Mohammed Asiri, Ahmed Osman, Haya Haddadin, and et al. 2024. "Best Practices in Nuclear Imaging for the Diagnosis of Transthyretin Amyloid Cardiomyopathy (ATTR-CM) in KSA: The Eagle Eyes of Local Experts" Diagnostics 14, no. 2: 212. https://doi.org/10.3390/diagnostics14020212
APA StyleAlqarni, A., Aljizeeri, A., Bakhsh, A. M., El-Zeftawy, H. A. M., Farghaly, H. R., Alqadhi, M. A. M., Algarni, M., Asiri, Z. M., Osman, A., Haddadin, H., Alayary, I., & Al-Mallah, M. H. (2024). Best Practices in Nuclear Imaging for the Diagnosis of Transthyretin Amyloid Cardiomyopathy (ATTR-CM) in KSA: The Eagle Eyes of Local Experts. Diagnostics, 14(2), 212. https://doi.org/10.3390/diagnostics14020212