
Emphasis on Early Prenatal Diagnosis and Perinatal Outcomes Analysis of Apert Syndrome



Abstract
1. Introduction
2. Literature Review
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hehr, U.; Muenke, M. Craniosynostosis Syndromes: From Genes to Premature Fusion of Skull Bones. Mol. Genet. Metab. 1999, 68, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, A.O.M.; Byren, J.C.; Hurst, J.A.; Jayamohan, J.; Johnson, D.; Knight, S.J.L.; Lester, T.; Richards, P.G.; Twigg, S.R.F.; Wall, S.A. Prevalence and Complications of Single-Gene and Chromosomal Disorders in Craniosynostosis. Pediatrics 2010, 126, e391–e400. [Google Scholar] [CrossRef] [PubMed]
- Arroyo Carrera, I.; Martínez-Frías, M.L.; Marco Pérez, J.J.; Paisán Grisolía, L.; Cárdenes Rodríguez, A.; Nieto Conde, C.; Félix Rodríguez, V.; Egüés Jimeno, J.J.; Morales Fernández, M.C.; Gómez-Ullate Vergara, J.; et al. Apert syndrome: Clinico-epidemiological analysis of a series of consecutive cases in Spain. An. Esp. Pediatr. 1999, 51, 667–672. [Google Scholar] [PubMed]
- Das, S.; Munshi, A. Research Advances in Apert Syndrome. J. Oral Biol. Craniofac. Res. 2018, 8, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.; Wilkie, A.O.M. Craniosynostosis. Eur. J. Hum. Genet. EJHG 2011, 19, 369–376. [Google Scholar] [CrossRef] [PubMed]
- López-Estudillo, A.-S.; Rosales-Bérber, M.-A.; Ruiz-Rodríguez, S.; Pozos-Guillén, A.; Noyola-Frías, M.-Á.; Garrocho-Rangel, A. Dental Approach for Apert Syndrome in Children: A Systematic Review. Med. Oral Patol. Oral Cirugia Bucal 2017, 22, e660–e668. [Google Scholar] [CrossRef] [PubMed]
- Vieira, C.; Teixeira, N.; Cadilhe, A.; Reis, I. Apert Syndrome: Prenatal Diagnosis Challenge. BMJ Case Rep. 2019, 12, e231982. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.M.; Kreiborg, S.; Lammer, E.J.; Cordero, J.F.; Mastroiacovo, P.; Erickson, J.D.; Roeper, P.; Martínez-Frías, M.L. Birth Prevalence Study of the Apert Syndrome. Am. J. Med. Genet. 1992, 42, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Tolarova, M.M.; Harris, J.A.; Ordway, D.E.; Vargervik, K. Birth Prevalence, Mutation Rate, Sex Ratio, Parents’ Age, and Ethnicity in Apert Syndrome. Am. J. Med. Genet. 1997, 72, 394–398. [Google Scholar] [CrossRef]
- Conrady, C.D.; Patel, B.C.; Sharma, S. Apert Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- David, A.L.; Turnbull, C.; Scott, R.; Freeman, J.; Bilardo, C.M.; van Maarle, M.; Chitty, L.S. Diagnosis of Apert Syndrome in the Second-Trimester Using 2D and 3D Ultrasound. Prenat. Diagn. 2007, 27, 629–632. [Google Scholar] [CrossRef]
- Werner, H.; Castro, P.; Daltro, P.; Lopes, J.; Ribeiro, G.; Araujo Júnior, E. Prenatal Diagnosis of Apert Syndrome Using Ultrasound, Magnetic Resonance Imaging, and Three-Dimensional Virtual/Physical Models: Three Case Series and Literature Review. Childs Nerv. Syst. ChNS Off. J. Int. Soc. Pediatr. Neurosurg. 2018, 34, 1563–1571. [Google Scholar] [CrossRef]
- Overview of Craniosynostosis—UpToDate. Available online: https://www.uptodate.com/contents/overview-of-craniosynostosis?search=Craniosynostosis%20syndromes&source=search_result&selectedTitle=2~150&usage_type=default&display_rank=2 (accessed on 24 January 2024).
- Craniosynostosis Syndromes—UpToDate. Available online: https://www.uptodate.com/contents/craniosynostosis-syndromes?search=Craniosynostosis%20syndromes&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1 (accessed on 24 January 2024).
- Bouaré, F.; Noureldine, M.H.A.; Hajhouji, F.; Ghannane, H.; Jallo, G.I.; Ait Benali, S. Complex Craniosynostosis in the Context of Carpenter’s Syndrome. Childs Nerv. Syst. ChNS Off. J. Int. Soc. Pediatr. Neurosurg. 2022, 38, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Xue, A.S.; Buchanan, E.P.; Hollier, L.H.J. Update in Management of Craniosynostosis. Plast. Reconstr. Surg. 2022, 149, 1209e. [Google Scholar] [CrossRef] [PubMed]
- Apert Syndrome: Acrocephalosyndactyly. Available online: https://thefetus.net/content/apert-syndrome-acrocephalosyndactyly (accessed on 24 January 2024).
- Lu, X.; Sawh-Martinez, R.; Jorge Forte, A.; Wu, R.; Cabrejo, R.; Wilson, A.; Steinbacher, D.M.; Alperovich, M.; Alonso, N.; Persing, J.A. Classification of Subtypes of Apert Syndrome, Based on the Type of Vault Suture Synostosis. Plast. Reconstr. Surg. Glob. Open 2019, 7, e2158. [Google Scholar] [CrossRef] [PubMed]
- Delahaye, S.; Bernard, J.P.; Rénier, D.; Ville, Y. Prenatal Ultrasound Diagnosis of Fetal Craniosynostosis. Ultrasound Obstet. Gynecol. Off. J. Int. Soc. Ultrasound Obstet. Gynecol. 2003, 21, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Miyashita, S.; Nagai, R.; Makino, S.; Murotsuki, J. Prenatal Sonographic Findings and Prognosis of Craniosynostosis Diagnosed during the Fetal and Neonatal Periods. Congenit. Anom. 2019, 59, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Helfer, T.M.; Peixoto, A.B.; Tonni, G.; Araujo Júnior, E. Craniosynostosis: Prenatal Diagnosis by 2D/3D Ultrasound, Magnetic Resonance Imaging and Computed Tomography. Med. Ultrason. 2016, 18, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.; Suchet, I. Postmortem Computed Tomography in a Case of Apert Syndrome: Correlation with Conventional Autopsy and Prenatal Ultrasound. Ultrasound Q. 2010, 26, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Apert Syndrome. Available online: https://thefetus.net/content/apert-syndrome-6 (accessed on 24 January 2024).
- Valentin, V.; Georgiana, B.; Dragos, G. Ultrasound and Clinical Correlations in Apert Syndrome. In Proceedings of the 5th Romanian Congress of Ultrasound in Obstetrics and Gynecology, 20–22 April 2017; Vlădăreanu, S., Mărginean, C., Vlădăreanu, R., Eds.; Editura Filodirrito: Bologna, Italy; Târgu Mureș, Romania; pp. 623–629. [Google Scholar]
- Franco, B.; Bruel, A.-L.; Thauvin-Robinet, C. Oral-Facial-Digital Syndrome Type I. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Thisse, B.; Thisse, C. Functions and Regulations of Fibroblast Growth Factor Signaling during Embryonic Development. Dev. Biol. 2005, 287, 390–402. [Google Scholar] [CrossRef]
- Conrady, C.D.; Patel, B.C. Crouzon Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Nguyen, Q.D.; Tran, T.N.A.; Nguyen, H.T. Crouzon Syndrome with Acanthosis Nigricans: A Case Report and Literature Review. Dermatol. Rep. 2023, 15, 9620. [Google Scholar] [CrossRef] [PubMed]
- Nørgaard, P.; Hagen, C.P.; Hove, H.; Dunø, M.; Nissen, K.R.; Kreiborg, S.; Jørgensen, F.S. Crouzon Syndrome Associated with Acanthosis Nigricans: Prenatal 2D and 3D Ultrasound Findings and Postnatal 3D CT Findings. Acta Radiol. Short Rep. 2012, 1, arsr.2012.110017. [Google Scholar] [CrossRef] [PubMed]
- M Das, J.; Winters, R. Pfeiffer Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Gallagher, E.R.; Ratisoontorn, C.; Cunningham, M.L. Saethre-Chotzen Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Kruszka, P.; Rolle, M.; Kahle, K.T.; Muenke, M. Muenke Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Heike, C.; Seto, M.; Hing, A.; Palidin, A.; Hu, F.Z.; Preston, R.A.; Ehrlich, G.D.; Cunningham, M. Century of Jackson-Weiss Syndrome: Further Definition of Clinical and Radiographic Findings in ?Lost? Descendants of the Original Kindred. Am. J. Med. Genet. 2001, 100, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Narayan, H.; Scott, I.V. Prenatal Ultrasound Diagnosis of Apert’s Syndrome. Prenat. Diagn. 1991, 11, 187–192. [Google Scholar] [CrossRef]
- Ferreira, J.C.; Carter, S.M.; Bernstein, P.S.; Jabs, E.W.; Glickstein, J.S.; Marion, R.W.; Baergen, R.N.; Gross, S.J. Second-Trimester Molecular Prenatal Diagnosis of Sporadic Apert Syndrome Following Suspicious Ultrasound Findings. Ultrasound Obstet. Gynecol. 1999, 14, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Boog, G.; Le Vaillant, C.; Winer, N.; David, A.; Quere, M.P.; Nomballais, M.F. Contribution of Tridimensional Sonography and Magnetic Resonance Imaging to Prenatal Diagnosis of Apert Syndrome at Mid-Trimester. Fetal Diagn. Ther. 1999, 14, 20–23. [Google Scholar] [CrossRef]
- Lyu, K.J.; Ko, T.M. Prenatal Diagnosis of Apert Syndrome with Widely Separated Cranial Sutures. Prenat. Diagn. 2000, 20, 254–256. [Google Scholar] [CrossRef]
- Skidmore, D.L.; Pai, A.P.; Toi, A.; Steele, L.; Chitayat, D. Prenatal Diagnosis of Apert Syndrome: Report of Two Cases. Prenat. Diagn. 2003, 23, 1009–1013. [Google Scholar] [CrossRef] [PubMed]
- Hansen, W.F.; Rijhsinghani, A.; Grant, S.; Yankowitz, J. Prenatal Diagnosis of Apert Syndrome. Fetal Diagn. Ther. 2004, 19, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Esser, T.; Rogalla, P.; Bamberg, C.; Kalache, K.D. Application of the Three-Dimensional Maximum Mode in Prenatal Diagnosis of Apert Syndrome. Am. J. Obstet. Gynecol. 2005, 193, 1743–1745. [Google Scholar] [CrossRef]
- Lam, H.; Lo, T.-K.; Lau, E.; Chin, R.; Tang, L. The Use of 2- and 3-Dimensional Sonographic Scans in the Evaluation of Cranial Sutures: Prenatal Diagnosis of Apert Syndrome. J. Ultrasound Med. Off. J. Am. Inst. Ultrasound Med. 2006, 25, 1481–1484. [Google Scholar] [CrossRef] [PubMed]
- Quintero-Rivera, F.; Robson, C.D.; Reiss, R.E.; Levine, D.; Benson, C.; Mulliken, J.B.; Kimonis, V.E. Apert Syndrome: What Prenatal Radiographic Findings Should Prompt Its Consideration? Prenat. Diagn. 2006, 26, 966–972. [Google Scholar] [CrossRef]
- Respondek-Liberska, M.; Smigiel, R.; Zielinski, A.; Sasiadek, M.M. Progressive Development of Sonographic Features in Prenatal Diagnosis of Apert Syndrome--Case Report and Literature Review. Ginekol. Pol. 2010, 81, 935–939. [Google Scholar] [PubMed]
- Chen, C.-P.; Su, Y.-N.; Hsu, C.-Y.; Ling, P.-Y.; Tsai, F.-J.; Chern, S.-R.; Wu, P.-C.; Chen, H.-E.C.; Wang, W. Second-Trimester Molecular Prenatal Diagnosis of Sporadic Apert Syndrome Following Sonographic Findings of Mild Ventriculomegaly and Clenched Hands Mimicking Trisomy 18. Taiwan. J. Obstet. Gynecol. 2010, 49, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, K.; Salmaso, R.; Manara, R.; Cosmi, E.; Baldi, M.; Rugge, M. Apert Syndrome with Fused Thalami. Fetal Pediatr. Pathol. 2012, 31, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Ercoli, G.; Bidondo, M.P.; Senra, B.C.; Groisman, B. Apert Syndrome with Omphalocele: A Case Report. Birt. Defects Res. A Clin. Mol. Teratol. 2014, 100, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Pi, G.; Zúñiga, A.; Cervera, J.; Ortiz, M. Prenatal diagnosis of Apert syndrome caused by de novo mutation in FGFR2 gene. An. Pediatr. Barc. Spain 2003 2014, 80, e104–e105. [Google Scholar] [CrossRef]
- Giancotti, A.; D’Ambrosio, V.; De Filippis, A.; Aliberti, C.; Pasquali, G.; Bernardo, S.; Manganaro, L.; PECRAM Study Group. Comparison of Ultrasound and Magnetic Resonance Imaging in the Prenatal Diagnosis of Apert Syndrome: Report of a Case. Childs Nerv. Syst. ChNS Off. J. Int. Soc. Pediatr. Neurosurg. 2014, 30, 1445–1448. [Google Scholar] [CrossRef] [PubMed]
- Stark, Z.; McGillivray, G.; Sampson, A.; Palma-Dias, R.; Edwards, A.; Said, J.M.; Whiteley, G.; Fink, A.M. Apert Syndrome: Temporal Lobe Abnormalities on Fetal Brain Imaging. Prenat. Diagn. 2015, 35, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Rubio, E.I.; Blask, A.; Bulas, D.I. Ultrasound and MR Imaging Findings in Prenatal Diagnosis of Craniosynostosis Syndromes. Pediatr. Radiol. 2016, 46, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Z.; Tsai, H.-D.; Hsieh, C.T.-C. Prenatal Diagnosis of a Sporadic Apert Syndrome by 3-D Ultrasound and 3-D Helical Computerized Tomography. Taiwan. J. Obstet. Gynecol. 2017, 56, 571–572. [Google Scholar] [CrossRef]
- Quintas-Neves, M.; Soares-Fernandes, J.P. Fetal Brain MRI in Apert Syndrome: Early in Vivo Detection of Temporal Lobe Malformation. Childs Nerv. Syst. ChNS Off. J. Int. Soc. Pediatr. Neurosurg. 2018, 34, 1617–1618. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Dai, R.; Wang, R.; Jing, J.; Yu, X.; Liu, R.; Liu, Y. A Novel FGFR2 (S137W) Mutation Resulting in Apert Syndrome: A Case Report. Medicine 2020, 99, e22340. [Google Scholar] [CrossRef] [PubMed]
- Partoune, S.; Masereel, M.C. Apert syndrome or acrocephalosyndactilia type I. Rev. Med. Liege 2021, 76, 715–718. [Google Scholar] [PubMed]
- Zhang, W.; Xue, H.; Huang, D.; Ye, Y.; Chen, X. Apert Syndrome: A Case Report of Prenatal Ultrasound, Postmortem Cranial CT, and Molecular Genetic Analysis. J. Clin. Ultrasound JCU 2021, 49, 250–253. [Google Scholar] [PubMed]
- Tonni, G.; Grisolia, G.; Baldi, M.; Bonasoni, M.; Ginocchi, V.; Rolo, L.C.; Araujo Júnior, E. Early Prenatal Ultrasound and Molecular Diagnosis of Apert Syndrome: Case Report with Postmortem CT-Scan and Chondral Plate Histology. Fetal Pediatr. Pathol. 2022, 41, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Kumari, K.; Saleh, I.; Taslim, S.; Ahmad, S.; Hussain, I.; Munir, Z.; Javed, T.; Virk, M.F.I.; Javed, S.; Bisharat, P.; et al. Unraveling the Complexity of Apert Syndrome: Genetics, Clinical Insights, and Future Frontiers. Cureus 2023, 15, e47281. [Google Scholar] [CrossRef]
- Anantheswar, Y.N.; Venkataramana, N.K. Pediatric Craniofacial Surgery for Craniosynostosis: Our Experience and Current Concepts: Part -1. J. Pediatr. Neurosci. 2009, 4, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Wenger, T.L.; Hing, A.V.; Evans, K.N. Apert Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
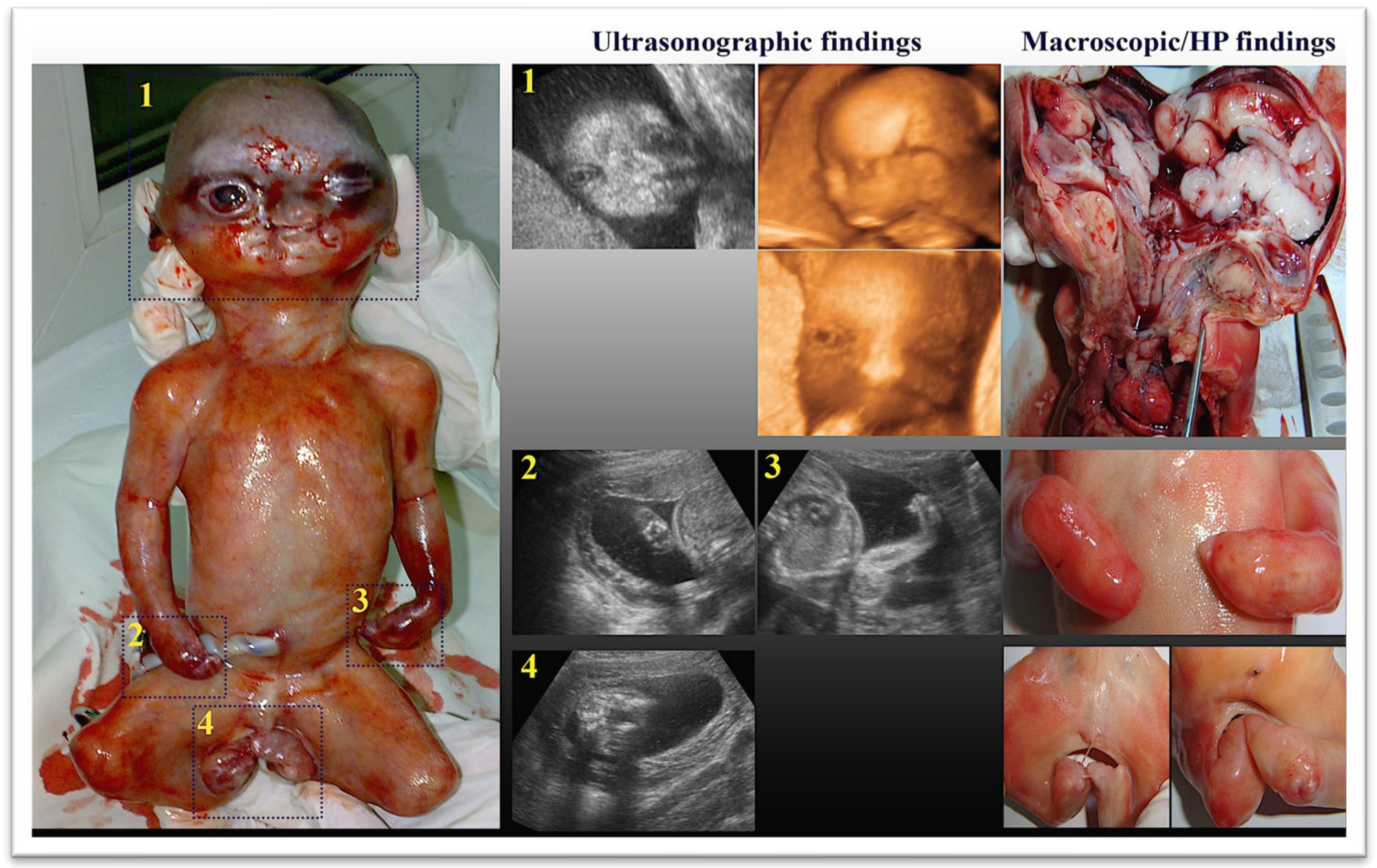

{kind=link}
| Syndrome | Incidence | Inheritance | Gene Mutations | Common Anomalies | Associated Anomalies | Outcome |
|---|---|---|---|---|---|---|
| Apert [7,8] | 6–15.5 in 1 million live births | Autosomal dominant | FGFR2 on chromosome 10q | Craniosynostosis, midface hypoplasia, and syndactyly in the hands and feet | Cardiovascular anomalies (atrial/ventricular septum defects, patent foramen ovale), cleft palate, central nervous system (agenesis corpus callosum, gyral anomalies), and urinary (hydronephrosis) | Cognitive impairment, respiratory complications, neurologic deficits, ocular anomalies |
| Crouzon [27] Crouzon + acanthosis nigricans [28,29] | 16 in 1 million live births | Autosomal dominant | FGFR2 | Compared to Apert, the degree of facial dysmorphism is milder, and the presence of cleft palate is rare. They usually have normal hands and feet | Cervical spine malformations | Strabismus, amblyopia, sleep apnea. Cognitive disabilities according to the severity of the cranial abnormality |
| 1 in 1 million live births | Autosomal dominant | FGFR3 | Craniosynostosis, midface hypoplasia, choanal atresia | Narrow sciatic notch, short vertebra, and broad metacarpals | ||
| Pfeiffer [30] | 10 in 1 million live births | Autosomal dominant | FGFR1/FGFR2 | Compared to Apert, the degree of syndactyly is milder (partial syndactyly of the 2nd–3rd fingers as well as the 2nd–4th toes) | Imperforate anus, hydrocephalus, and radio-humeral synostosis | Obstructive sleep apnea, neurodevelopmental impairment, seizures, ocular complications. Type 1—better prognosis for intellectual outcome. Types 2 and 3—severe neurologic defects, death in childhood |
| Carpenter [15] | 1 in 1 million live births | Autosomal recessive | RAB23 (RAS-associated protein) | Coronal, sagittal, and lambdoid craniosynostosis, which cause prominence of the metopic suture and brachycephaly | Cardiovascular anomalies, hypogonadism, omphalocele, polydactyly, brachydactyly, and clinodactyly | Craniosynostosis surgery with satisfactory esthetics of the craniofacial bones. Psychomotor delay, potential blindness, decreased visual acuity |
| Saethre–Chotzen [31] | 20 in 1 million live births | Autosomal dominant | TWIST/FGFR2 | Compared to Apert, patients have a towering forehead, low-set hairline, facial asymmetry, ptosis of the upper eyelids | Cutaneous syndactyly is usually partial and involves the 2nd–3rd fingers and/or the 3rd–4th toes | Sleep disorder breathing, developmental delays. Dental malocclusion, swallowing difficulties. Hearing loss, amblyopia |
| Muenke [32] | 33.3 in 1 million live births | Autosomal dominant | FGFR3 | Coronal synostosis or pan synostosis | Anterior plagiocephaly, temporal bossing, fusions of carpal and/or tarsal bones; brachydactyly, broad toes and thumbs, clinodactyly | Behavioral issues, seizures. Hearing loss, strabismus. Developmental delays, intellectual disabilities |
| Jackson-Weiss [33] | Unknown | Autosomal dominant | FGFR2 on chromosome 10q26 | Craniosynostosis, mid-face hypoplasia, abnormally broad great toes, and/or malformation/fusion of bones from the feet. | - | Hearing impairment, normal intelligence, and a normal lifespan |
| Author/ Year | Maternal Age | GA at Diagnosis/Sex | Ultrasound/MRI Findings | Fetal Examination | FGFR2 Mutation | Outcome |
|---|---|---|---|---|---|---|
| Narayan 1991, [34] | 29, mother with AS | 19–20, F | The skull outline is very irregular and oval, “mitten” hands syndactyly. | Postmortem examination confirmed the features. | Not tested | Stillbirth, VD—34 wks |
| Ferreira 1999, [35] | 33 | 20, M | Turribrachycephaly, acrocephaly, frontal bossing, short nose, bilateral 2nd-to-5th-finger syndactyly of the hand (‘mitten type’), abnormal toes. | The pathological examination confirmed the prenatal diagnosis. | S252W | TOP |
| Boog 1999, [36] | 28 | 24, M | Turribrachycephaly, clover-leaf skull, midface hypoplasia, down slanting palpebral fissures, depressed nasal bridge, “mitten type” with 2nd-to-5th-finger total syndactyly of the hands, complete cutaneous fused toes with a broad big toe high forehead. MRI—brachycephaly and verticalization of the clivus with a flattened cranial base angle, hypertelorism. | The pathological examination confirmed the prenatal diagnosis. | Not tested | TOP |
| Lyu 2000, [37] | 35, mother with AS | 20, F | Frontal bossing, all cranial sutures widely separated, midface depression, syndactyly, fused toes. | The pathological examination confirmed the prenatal diagnosis, with both fontanels open. | Heterozygous 934C-G | TOP |
| Skidmore 2003, [38] | 34 | 17.3/19 + 5, F | Clover-leaf skull, proptosis, VM, low-set ears, syndactyly of the hands, and broad first toes. | Turribrachycephaly, proptosis, a narrow and high arched palate, bilateral bony syndactyly, 2nd-to-5th-finger hands and feet, mitten-thumb, coarctation of the aorta. | 755C → G, (S252W) | TOP |
| 33 | 21 | Clover-leaf skull, VM, hypoplastic left heart. | Turribrachycephaly, hypertelorism, flat occiput, proptosis, beaked nose, midface hypoplasia, syndactyly, hypoplastic left ventricle, and hypo-plastic ascending aorta proximal to the ductus arteriosus. | 755C → G, (S252W) | TOP | |
| Hansen 2004, [39] | 23 | 23, M | Abnormally shaped skull, syndactyly of the hands. | Craniosynostosis, craniofacial anomalies, bony and cutaneous syndactyly of the hands and feet. | Pro253Arg | VD at 32.5 wks |
| 26 | 21, F | Abnormally shaped skull, syndactyly, or an abnormal posturing of the hands. | N/A | Not tested | VD at 27 + 2 wks. Died at 4 m | |
| Esser 2005, [40] | 29 | 22 | Widely open metopic suture with craniosynostosis, 2nd-to-4th-finger hands syndactyly. | Postmortem CT confirmed the prenatal findings. No autopsy was performed. | Heterozygous Pro253Arg (C758G) | TOP |
| Lam 2006, [41] | 26 | 19 | Strawberry head shape, craniosynostosis, ventricular septal defect, overriding of the aorta, syndactyly of the hands and feet. | Widely opened metopic suture, craniosynostosis, midface hypoplasia, bilateral syndactyly of the hands and feet. No autopsy was performed. | Heterozygous C1347G— Ser242Trp | TOP |
| Quintero-Rivera 2006, [42] | 37 | 19, M | US—colpocephaly, ACC MRI—ACC, hypertelorism, oblong cranial shape. | Turricephaly, widely fontanelles and sagittal suture, fused toes, complex syndactyly, prominent downslanted eyes, hypertelorism, beaked nose, midface hypoplasia, depressed nasal bridge, mild tricuspid regurgitation. Postnatal 3D CT coronal craniosynostosis, wide midline calvarial defect, shallow orbits, midfacial hypoplasia. | S252W | CS at 39 wks |
| David 2007, [11] | N/A | 21 | Brachycephaly, VM, prominent forehead, proptosis, increased NT, absent nasal bone, abnormal DV, “mitten” hands, syndactyly of the hands and feet, arched foot, bilateral hydronephrosis. | The pathological examination confirmed the prenatal diagnosis. | S252W | TOP |
| N/A | 20 + 5 | Long head, craniosynostosis, pro-ptosis, “mitten” hands, syndactyly of the hands and feet, small feet, abnormal position. | Syndactyly of the hands and feet, proptosis, and abnormal head shape. No autopsy was performed. | S252W | TOP | |
| N/A | 22 + 5 | Long head, flat occiput, mild VM, syndactyly of the hands, feet abnormal position, clitoromegaly. | The pathological examination confirmed the prenatal diagnosis. | P253R | TOP | |
| Patel 2010, [22] | 31 | 22 + 3 | No significant brachycephaly, craniosynostosis, borderline VM, depressed nasal bridge, mild hypertelorism, cystic left orbit, or syndactyly of the hands and feet. | Postmortem 3D CT frontal bossing, wide metopic sutures with partial synostosis of coronal sutures, midface hypoplasia, hypertelorism, depressed nasal bridge. Autopsy—reasonable correlations of craniofacial anomalies. | Not tested | TOP |
| Respondek-Liberska 2010, [43] | 31 | 22, F | 22 wks—normal head shape, mild VM; 25 wks—colpocephaly, syndactyly of the hands and feet, mild hypertelorism; 28 wks—prominent forehead, craniosynostosis, midface hypoplasia, widely open metopic suture. | Typical facial appearance of AS, symmetrical, complex syndactyly of the hands 2nd-to-5th-fingers. The thumbs were not involved in the fusion. Feet syndactyly affected all toes. | S252W in exon 7 | CS at term |
| Chen 2010, [44] | 30 | 24, M | Midface hypoplasia, low-set ears, mild VM, mitten hand, 2nd-to-4th-finger syndactyly and abroad proximally deviated thumb. | Prominent midface hypoplasia, low-set ears, bilateral mitten syndactyly of the hands and feet, broad and proximally displaced thumbs, and big toes. | Heterozygous c.755 C>G, Ser252Trp (S252W) | TOP |
| Ludwig 2012, [45] | 38 | 20 + 1, F | Turricephaly, frontal bossing, midface hypoplasia, low-set ears, hyperthelorism, and reduced facial movements. | Postmortem external examination—turribrachycephaly, prominent forehead, midface hypoplasia, proptosis, slightly down palpebral fissures, hypoplastic alae nasi, smooth palate cleft; hand—complete cutaneous syndactyly 2nd-to-4th-fingers II, III, IV with synonchia, Hitchhiker thumb; feet—broad halluces, complete cutaneous syndactyly toe 1st-to-4th-fingers, short V toe. Postmortem CT and MRI confirmed craniosynostosis, ACC, absence of the septum pellucidum, and complete fusion of the thalami. | Ser252Trp in exon 8 | TOP |
| Ercoli 2014, [46] | 30 | 23, M | Turricephaly, syndactyly, and omphalocele. | Turribrachycephaly, high arched palate, broad forehead, midface hypoplasia, hypertelorism, exophthalmos, syndactyly of hands and feet, bilateral cryptorchidism, omphalocele. Rx—craniosynostosis, synostosis between metacarpians and metatarsal bones. | Not tested | CS at 35 wks |
| Pi 2014, [47] | N/A | 18/20/21/22 + 4, F | 18 wks—cranial anomalies, syndactyly of the hands and feet; 20 wks—mild bilateral hydronephrosis, strawberry skull; 21 wks—craniosynostosis; 22 + 4 wks—craniosynostosis, low-set ears, frontal bulging, hypertelorism, syndactyly of the fingers of both hands. | Craniosynostosis, wide anterior fontanel, metopic suture dehiscence, synostosis of the lambdoid suture, ocular proptosis, low-set ears, medial palatal fissure, mild hypertelorism, the fingers and toes brachydactyly, syndactyly, 2–5 fingers and toes. | Heterozygous c.755C>G, p.Ser252Trp (NM 022970) | TOP |
| Giancotti 2014, [48] | 37 | 21, M | US—irregular head shape, dolicocephaly, prominent forehead, flat occiput, bilateral mild VM, partial ACC, hypertelorism, midface hypoplasia, depressed nasal bridge, feet abnormal position, and syndactyly. MRI—excluded ACC. | Fetus macroscopic analysis: syndactyly of the hands (“spoon hands”) and feet, midface hypoplasia, prominent forehead.No postmortem autopsy. | Heterozygous c.758C>G (p.Pro253Arg) | TOP |
| Stark 2015, [49] | N/A | 21 | US—craniosynostosis, syndactyly, coarctation of the aorta. MRI—turribrachycephaly, CCD, temporal over-expansion/over-sulcation, transverse temporal clefts | The pathological examination confirmed the prenatal diagnosis. | c.755C>G (p.Ser252Trp) | N/A |
| Rubio 2016, [50] | N/A | 22 | US/MRI—calvarial indentation, hypertelorism, proptosis, syndactyly of the hands and feet. | N/A | NS | TOP |
| N/A | 21/30 | US—calvarial indentation, “lamp-shade” calvarium, hypertelorism, syndactyly of the hands and feet, abnormally expanded lungs. MRI—plus proptosis, absent septum pellucidum. | N/A | NS | Survived | |
| Wang 2017, [51] | 27 | 23, F | Craniosynostosis, widely open metopic suture and anterior fontanel, midface hypoplasia, mitten hands, bony or cutaneous syndactyly of the hands and feet. | Fetal phenotype compatible with AS. No postmortem autopsy. | Not tested | TOP |
| Varlas 2017, [24] | 32 | 21 | Acrocephaly and brachycephaly, prominent forehead, nasal bone hypoplasia, exorbitism, flat midface, hypertelorism, low-set ears, cleft lip and palate, syndactyly of the hands and feet with lack of low extremities movements, vicious positions of the limbs. | The macroscopic examination confirmed the US diagnosis (craniosynostosis with acrocephaly and brachycephaly, flat midface, hypertelorism, cleft lip and palate, syndactyly of the hands and feet—the fusion of the bone and soft tissue components of all fingers). | Not tested | TOP |
| Quintas-Neves 2018, [52] | 33 | 22 + 2 | US—craniosynostosis, ocular proptosis, syndactyly of the hands and feet.MRI—turribrachycephaly, hypertelorism, over convolution, abnormal sulci in inferior and mesial temporal lobes. | N/A | c.755C>G (p.Ser252Trp) | TOP |
| Vieira 2019, [7] | 33 | 22, F | US—prominent forehead, hypertelorism, proptosis, mitten hands. MRI—hypertelorism, turribrachycephaly, cranial dysmorphism, cortical malformation. | Craniosynostosis, hypertelorism, low-set ears, proptosis, bilateral syndactyly of the 2nd-to-5th fingers in hands and all fingers in the feet. | c.755C>G (p.Ser252Trp) | TOP |
| Shi 2020, [53] | 32 | 23 + 5 | Clover-leaf skull, syndactyly from the 2nd-to-5th finger. | Acrocephalosyndactyly and hypertelorism. | c.410C>G, chr10:123279677 (p.S137W) | TOP |
| Partoune 2021, [54] | 39 | 23 + 5, M (twin) | Skull deformation, syndactyly—mitten hand. 27 wks—facial dysmorphism, small cerebellar vermis, syndactyly of the hands and feet, moderate VM, hypertelorism. | N/A | c.758C>G (p.Pro253Arg) | selective TOP at 33 + 4 wks |
| Zhang 2021, [55] | 36 | 23, F | Unusual head shape, craniosynostosis, mild left VM, slight frontal bossing, proptosis, hypertelorism, syndactyly. | The pathological examination confirmed the prenatal diagnosis syndactyly of type VI (known as mitten type). | c.755C>G (p.S252W) | TOP |
| Tonni 2022, [56] | 36 | 19, F | Turribrachicephaly, wide metopic suture, craniosynostosis, and syndactyly of the left hand. | Acrocephaly, broad forehead, mid-face hypoplasia, depressed nasal bridge, exophthalmos. The right hand—spade-shaped with thumb and index separated; the left hand—mitten-shaped with complete cutaneous syndactyly of fingers I-V, the feet—broad halluces and complete cutaneous syndactyly of all toes. | c.758C>Gp at 252 of the exon 8 | TOP |
| Anomalies Findings | Ultrasound/MRI Findings n (%) |
|---|---|
| Cranial | |
| Abnormal head shape | 31 (96.8%) |
| Suture morphology | 15 (46.8%) |
| Ventriculomegaly | 10 (31.2%) |
| ACC/DCC | 2/1 (9.3%) |
| Facial | |
| Hypertelorism | 14 (43.7%) |
| Proptosis | 7 (21.8%) |
| Midface hypoplasia | 8 (25%) |
| Prominent forehead | 5 (15.6%) |
| Low-set ears | 5 (15.6%) |
| Nasal deformity | 5 (15.6%) |
| Limbs | |
| Syndactyly | 28 (87.5%) |
| Abnormal toes | 3 (9.3%) |
| Cardiovascular | 3 (9.3%) |
| Renal | 2 (6.2%) |
| Digestive | 1 (3.1%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varlas, V.N.; Epistatu, D.; Varlas, R.G. Emphasis on Early Prenatal Diagnosis and Perinatal Outcomes Analysis of Apert Syndrome. Diagnostics 2024, 14, 1480. https://doi.org/10.3390/diagnostics14141480
Varlas VN, Epistatu D, Varlas RG. Emphasis on Early Prenatal Diagnosis and Perinatal Outcomes Analysis of Apert Syndrome. Diagnostics. 2024; 14(14):1480. https://doi.org/10.3390/diagnostics14141480
Chicago/Turabian StyleVarlas, Valentin Nicolae, Dragos Epistatu, and Roxana Georgiana Varlas. 2024. "Emphasis on Early Prenatal Diagnosis and Perinatal Outcomes Analysis of Apert Syndrome" Diagnostics 14, no. 14: 1480. https://doi.org/10.3390/diagnostics14141480
APA StyleVarlas, V. N., Epistatu, D., & Varlas, R. G. (2024). Emphasis on Early Prenatal Diagnosis and Perinatal Outcomes Analysis of Apert Syndrome. Diagnostics, 14(14), 1480. https://doi.org/10.3390/diagnostics14141480