
Improving CNV Detection Performance in Microarray Data Using a Machine Learning-Based Approach

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects and Sample Preparation
2.2. Subjects and Sample Preparation
2.3. Preparation of Positive CNV Control Samples for Analytical Validation
2.4. Data Processing and CNV Analysis
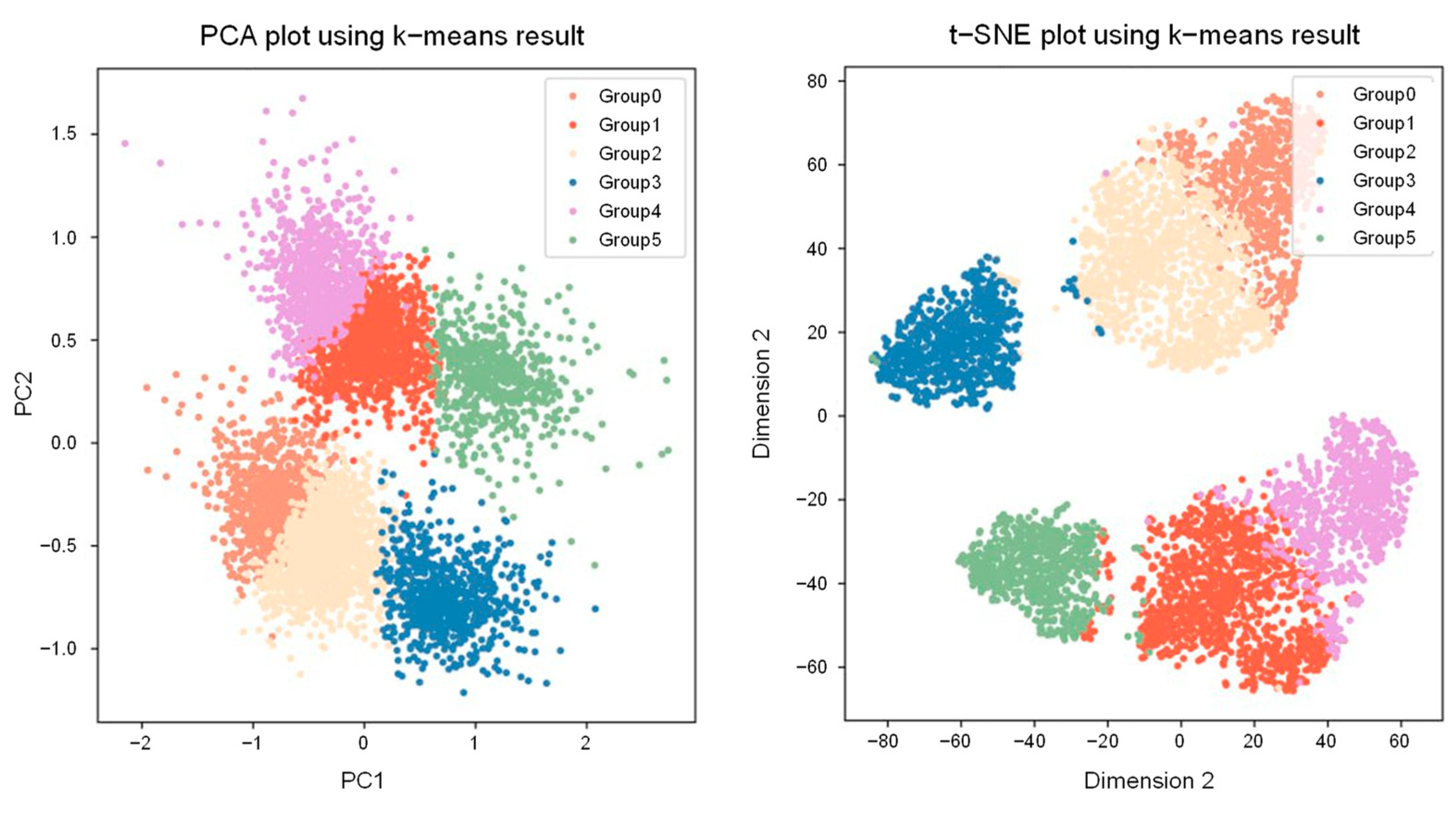
2.5. Clustering Genomic Waves from 5399 Clinical Samples Using GSA
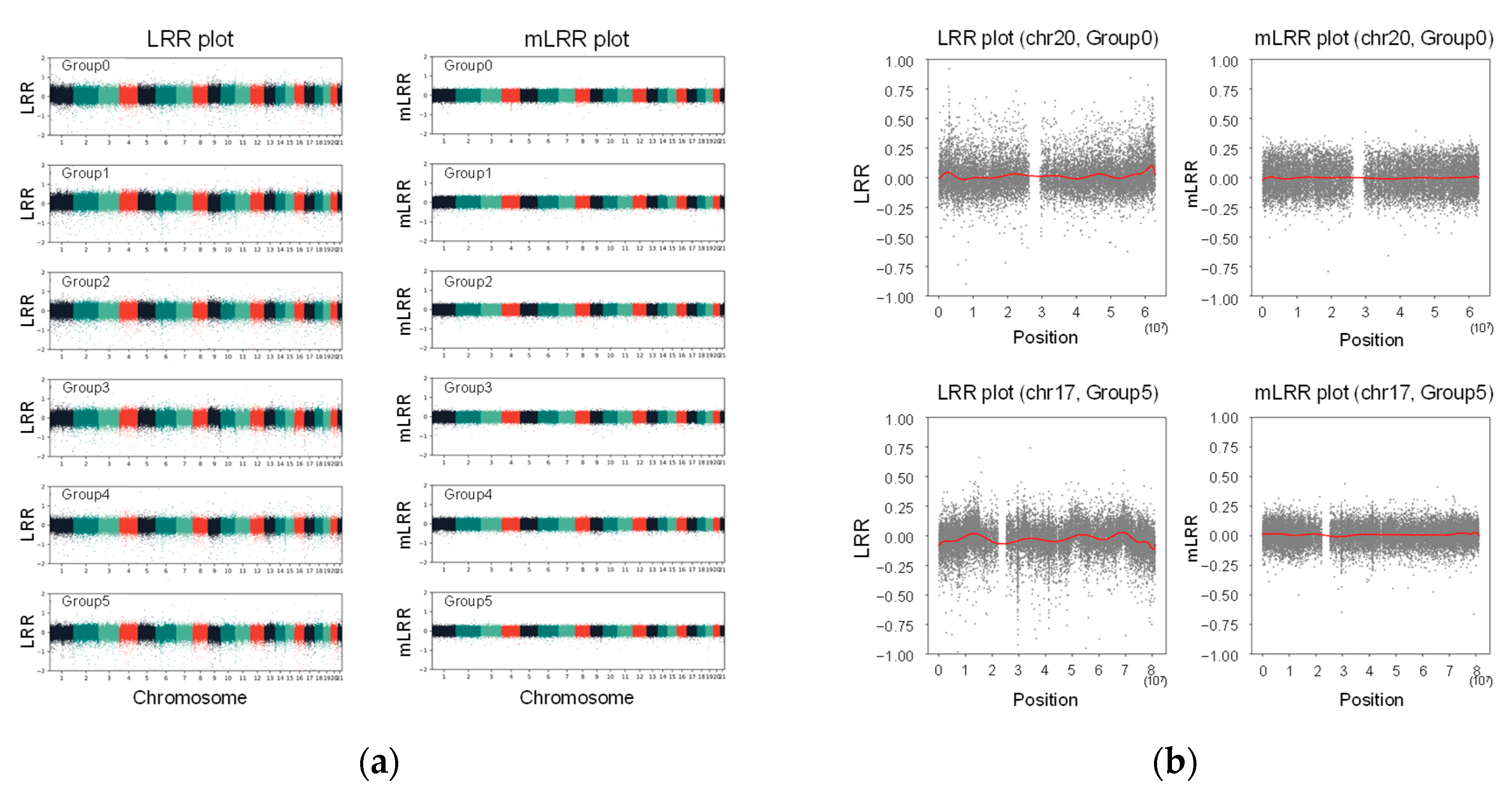
2.6. Cluster Matching of Analytical Samples and Calculation of Modified LRR Values
2.7. NGS Sequencing for Accuracy Validation
3. Results
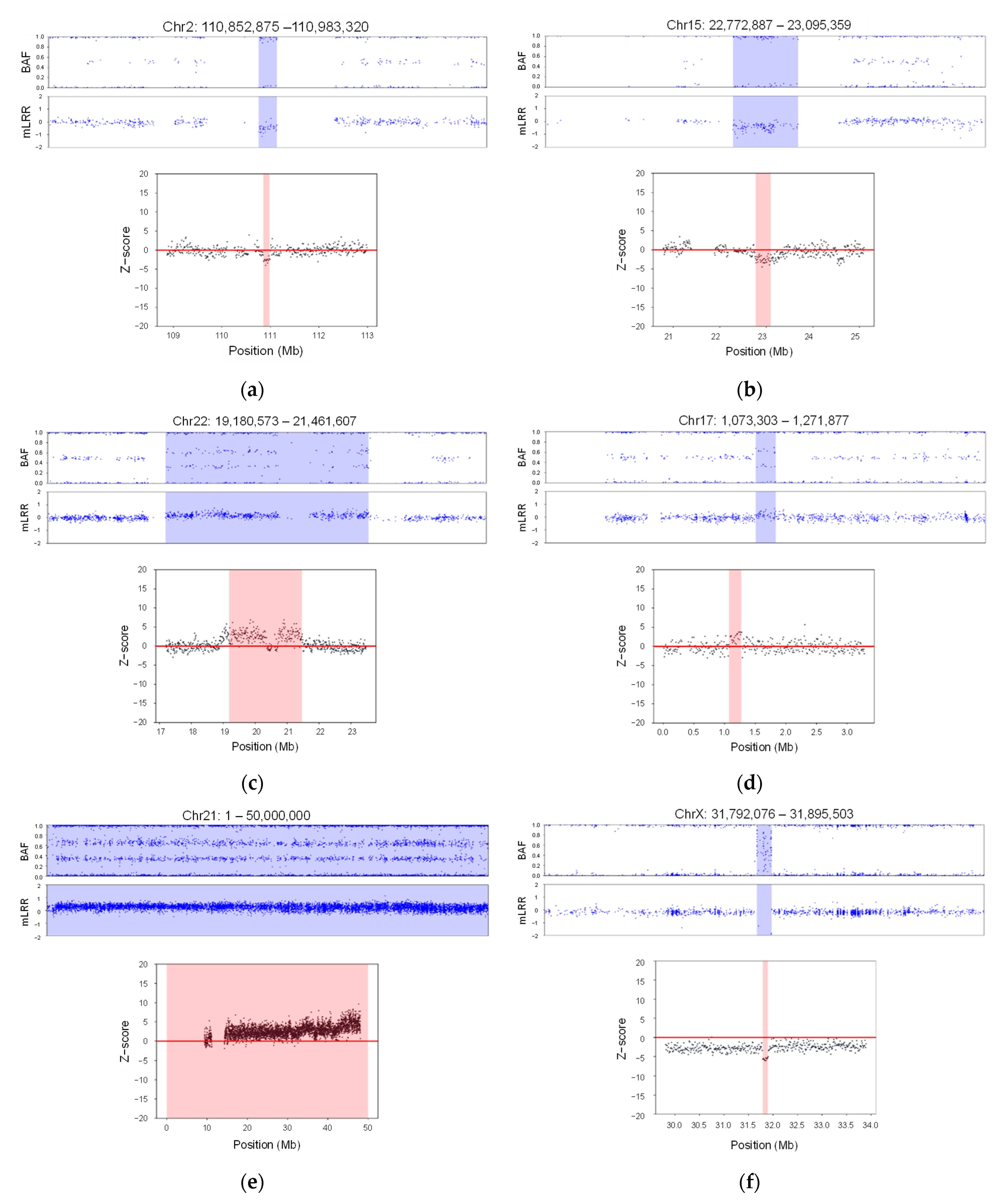
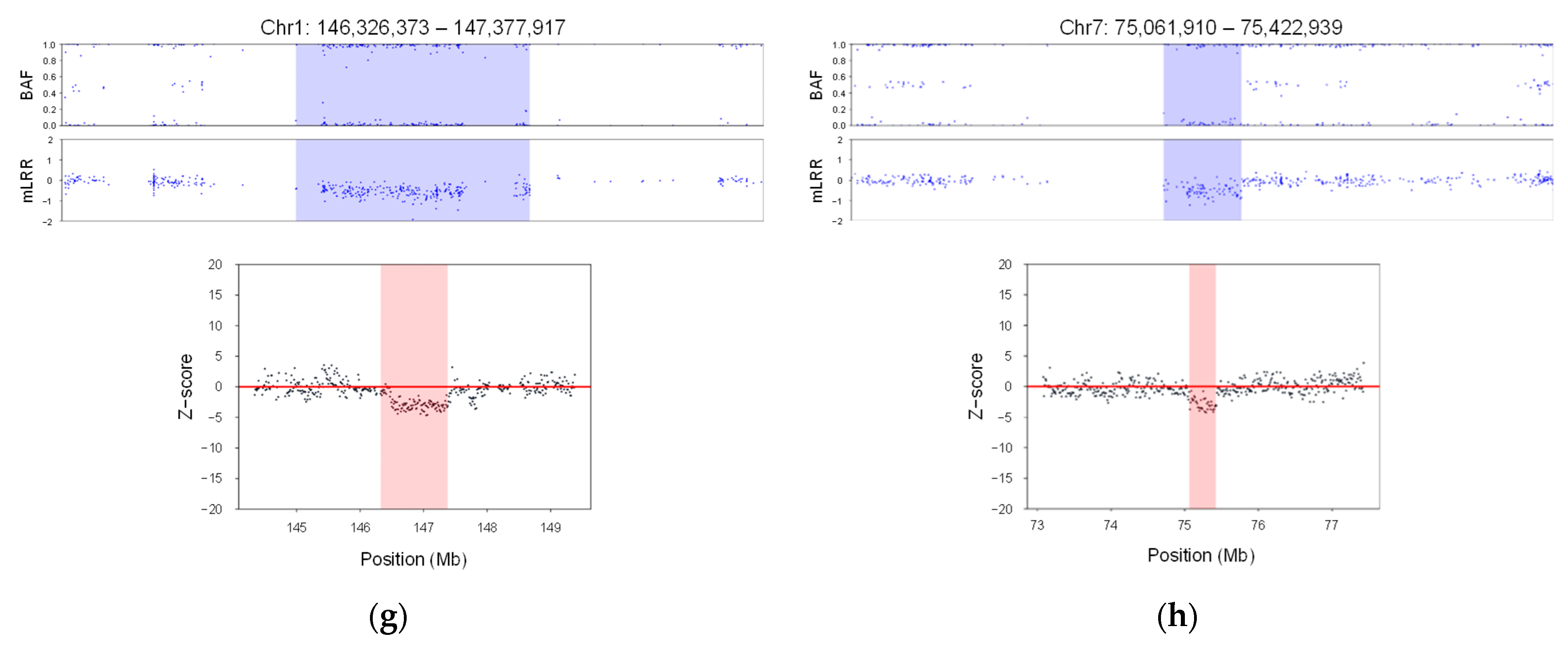
3.1. Enhancing CNV Analysis Accuracy through Customized Machine Learning Model
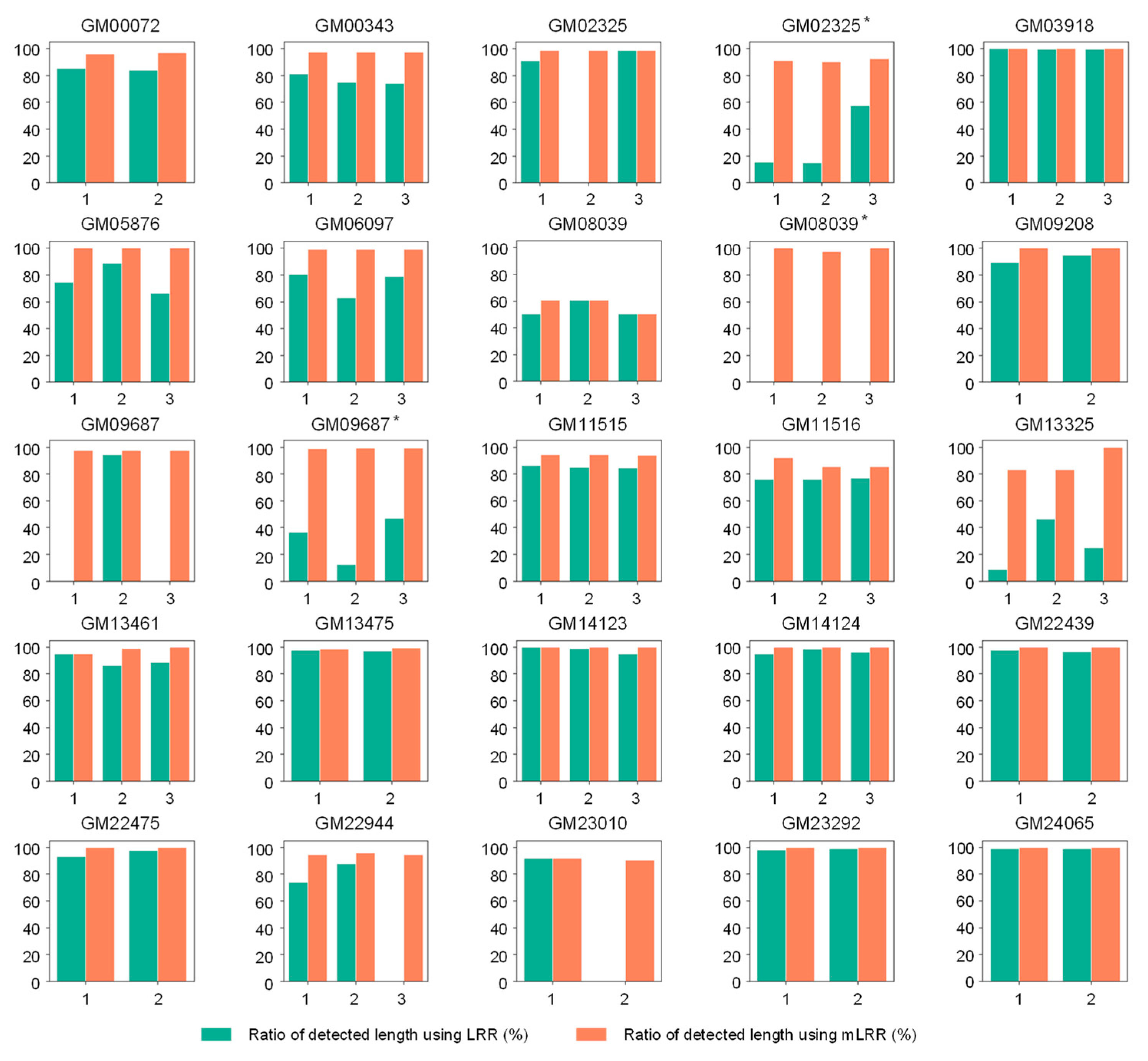
3.2. Analytical Validation Using Known Positive CNV Control DNA Samples
3.3. CNV Analysis Using 16,046 Neonate Samples from South Korea
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Zarrei, M.; MacDonald, J.R.; Merico, D.; Scherer, S.W. A copy number variation map of the human genome. Nat. Rev. Genet. 2015, 16, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Zaninović, L.; Bašković, M.; Ježek, D.; Bojanac, A.K. Validity and utility of non-invasive prenatal testing for copy number variations and microdeletions: A systematic review. J. Clin. Med. 2022, 11, 3350. [Google Scholar] [CrossRef] [PubMed]
- Pös, O.; Radvanszky, J.; Styk, J.; Pös, Z.; Buglyó, G.; Kajsik, M.; Budis, J.; Nagy, B.; Szemes, T. Copy number nariation: Methods and clinical applications. Appl. Sci. 2021, 11, 819. [Google Scholar] [CrossRef]
- Weiss, L.A.; Shen, Y.; Korn, J.M.; Arking, D.E.; Miller, D.T.; Fossdal, R.; Saemundsen, E.; Stefansson, H.; Ferreira, M.A.; Green, T.; et al. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 2008, 358, 667–675. [Google Scholar] [CrossRef]
- Wang, L.; Wang, B.; Wu, C.; Wang, J.; Sun, M. Autism spectrum disorder: Neurodevelopmental risk factors, biological mechanism, and precision therapy. Int. J. Mol. Sci. 2023, 24, 1819. [Google Scholar] [CrossRef]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef]
- Malhotra, D.; Sebat, J. CNVs: Harbingers of a rare variant revolution in psychiatric genetics. Cell 2012, 148, 1223–1241. [Google Scholar] [CrossRef]
- Sharp, A.J.; Mefford, H.C.; Li, K.; Baker, C.; Skinner, C.; E Stevenson, R.; Schroer, R.J.; Novara, F.; De Gregori, M.; Ciccone, R.; et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat. Genet. 2008, 40, 322–328. [Google Scholar] [CrossRef]
- Gozzetti, A.; Le Beau, M.M. Fluorescence in situ hybridization: Uses and limitations. Semin. Hematol. 2000, 37, 320–333. [Google Scholar] [CrossRef]
- Kozlowski, P.; Jasinska, A.J.; Kwiatkowski, D.J. New applications and developments in the use of multiplex ligation-dependent probe amplification. Electrophoresis 2008, 29, 4627–4636. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.; Wapner, R. Prenatal diagnosis by chromosomal microarray analysis. Fertil. Steril. 2018, 109, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Valenzuela, N.J.; Peixoto, A.B.; Júnior, E.A. Prenatal diagnosis of congenital heart disease: A review of current knowledge. Indian Heart J. 2018, 70, 150–164. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, M.J.; Puck, J.M. Newborn screening for severe combined immunodeficiency in the United States: Lessons learned. Immunol. Allergy Clin. N. Am. 2019, 39, 1–11. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, Q.; Wang, Q.; Jia, P.; Zhao, Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: Features and perspectives. BMC Bioinform. 2013, 14 (Suppl. S1), S1. [Google Scholar] [CrossRef]
- Bharadwaj, S.; Dwivedi, V.D.; Kirtipal, N. Application of whole genome sequencing (WGS) approach against identification of foodborne bacteria. In Microbial Genomics in Sustainable Agroecosystems; Tripathi, V., Kumar, P., Tripathi, P., Kishore, A., Eds.; Springer: Singapore, 2019; Volume 1, pp. 131–148. [Google Scholar] [CrossRef]
- Henderson, L.B.; Applegate, C.D.; Wohler, E.; Sheridan, M.B.; Hoover-Fong, J.; Batista, D.A. The impact of chromosomal microarray on clinical management: A retrospective analysis. Anesth. Analg. 2014, 16, 657–664. [Google Scholar] [CrossRef]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Werling, A.M.; Grünblatt, E.; Oneda, B.; Bobrowski, E.; Gundelfinger, R.; Taurines, R.; Romanos, M.; Rauch, A.; Walitza, S. High-resolution chromosomal microarray analysis for copy-number variations in high-functioning autism reveals large aberration typical for intellectual disability. J. Neural Transm. 2020, 127, 81–94. [Google Scholar] [CrossRef]
- Hu, T.; Zhang, Z.; Wang, J.; Li, Q.; Zhu, H.; Lai, Y.; Wang, H.; Liu, S. Chromosomal aberrations in pediatric patients with developmental delay/intellectual disability: A single-center clinical investigation. BioMed Res. Int. 2019, 2019, 9352581. [Google Scholar] [CrossRef]
- Wu, X.-L.; Li, R.; Fu, F.; Pan, M.; Han, J.; Yang, X.; Zhang, Y.-L.; Li, F.-T.; Liao, C. Chromosome microarray analysis in the investigation of children with congenital heart disease. BMC Pediatr. 2017, 17, 117. [Google Scholar] [CrossRef]
- Tozzi, V.; Rosenberger, A.; Kube, D.; Bickeböller, H. Global, pathway and gene coverage of three Illumina arrays with respect to inflammatory and immune-related pathways. Eur. J. Hum. Genet. 2019, 27, 1716–1723. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Bucan, M. Copy number variation detection via high-density SNP genotyping. Cold Spring Harb. Protoc. 2008, 2008, pdb.top46. [Google Scholar] [CrossRef] [PubMed]
- Diskin, S.J.; Li, M.; Hou, C.; Yang, S.; Glessner, J.; Hakonarson, H.; Bucan, M.; Maris, J.M.; Wang, K. Adjustment of genomic waves in signal intensities from whole-genome SNP genotyping platforms. Nucleic Acids Res. 2008, 36, e126. [Google Scholar] [CrossRef] [PubMed]
- Ginsbach, P.; Chen, B.; Jiang, Y.; Engelter, S.T.; Grond-Ginsbach, C. Copy number studies in noisy samples. BioTech 2013, 2, 284–303. [Google Scholar] [CrossRef] [PubMed]
- Marioni, J.C.; Thorne, N.P.; Valsesia, A.; Fitzgerald, T.; Redon, R.; Fiegler, H.; Andrews, T.D.; Stranger, B.E.; Lynch, A.G.; Dermitzakis, E.T.; et al. Breaking the waves: Improved detection of copy number variation from microarray-based comparative genomic hybridization. Genome Biol. 2007, 8, R228. [Google Scholar] [CrossRef]
- Aboukhalil, A.; Bulyk, M.L. LOESS correction for length variation in gene set-based genomic sequence analysis. Bioinformatics 2012, 28, 1446–1454. [Google Scholar] [CrossRef]
- Komura, D.; Shen, F.; Ishikawa, S.; Fitch, K.R.; Chen, W.; Zhang, J.; Liu, G.; Ihara, S.; Nakamura, H.; Hurles, M.E.; et al. Genome-wide detection of human copy number variations using high-density DNA oligonucleotide arrays. Genome Res. 2006, 16, 1575–1584. [Google Scholar] [CrossRef]
- Hamerly, G.; Elkan, C. Learning the k in k-means. Adv. Neural Inf. Process. Syst. 2003, 16, 281–288. [Google Scholar]
- Krishna, K.; Murty, M.N. Genetic k-means algorithm. IEEE Trans. Syst. Man Cybern. Part B (Cybernetics) 1999, 29, 433–439. [Google Scholar] [CrossRef]
- Peterson, L.E. K-nearest neighbor. Scholarpedia 2009, 4, 1883. [Google Scholar] [CrossRef]
- Suguna, N.; Thanushkodi, K. An improved k-nearest neighbor classification using genetic algorithm. Int. J. Comput. Sci. Issues 2010, 7, 18–21. [Google Scholar]
- Wang, K.; Li, M.; Hadley, D.; Liu, R.; Glessner, J.; Grant, S.F.; Hakonarson, H.; Bucan, M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007, 17, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Colella, S.; Yau, C.; Taylor, J.M.; Mirza, G.; Butler, H.; Clouston, P.; Bassett, A.S.; Seller, A.; Holmes, C.C.; Ragoussis, J. QuantiSNP: An objective bayes hidden-Markov model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007, 35, 2013–2025. [Google Scholar] [CrossRef] [PubMed]
- Toyama, M.; Vargas, L.; Ticlihuanca, S.; Quispe, A.M. Regional clustering and waves patterns due to COVID-19 by the index virus and the lambda/gamma, and delta/omicron SARS-CoV-2 variants in Peru. Ann. Epidemiol. 2022, 6, 74. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Brancati, F.; Dallapiccola, B.; Valente, E.M. Joubert syndrome and related disorders. Orphanet J. Rare Dis. 2010, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.N.; Catalano, L.M.; Bernat, J.A.; Adams, S.D.; Martin, D.M.; Lalani, S.R.; Patel, A.; Burnside, R.D.; Innis, J.W.; Rudd, M.K. Recurrent deletions and duplications of chromosome 2q11.2 and 2q13 are associated with variable outcomes. Am. J. Med. Genet. Part A 2015, 167, 2664–2673. [Google Scholar] [CrossRef]
- Cox, D.M.; Butler, M.G. The 15q11.2 BP1–BP2 microdeletion syndrome: A review. Int. J. Mol. Sci. 2015, 16, 4068–4082. [Google Scholar] [CrossRef]
- Rafi, S.K.; Butler, M.G. The 15q11.2 BP1-BP2 microdeletion (Burnside–Butler) syndrome: In silico analyses of the four coding genes reveal functional associations with neurodevelopmental disorders. Int. J. Mol. Sci. 2020, 21, 3296. [Google Scholar] [CrossRef]
- Fischer, M.; Klopocki, E. Atypical 22q11.2 microduplication with “typical” signs and overgrowth. Cytogenet. Genome Res. 2020, 160, 659–663. [Google Scholar] [CrossRef]
- Wenger, T.L.; Miller, J.S.; DePolo, L.M.; de Marchena, A.B.; Clements, C.C.; Emanuel, B.S.; Zackai, E.H.; McDonald-McGinn, D.M.; Schultz, R.T. 22q11.2 duplication syndrome: Elevated rate of autism spectrum disorder and need for medical screening. Mol. Autism 2016, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Armour, C.M.; E Bulman, D.; Jarinova, O.; Rogers, R.C.; Clarkson, K.B.; DuPont, B.R.; Dwivedi, A.; O Bartel, F.; McDonell, L.; Schwartz, C.E.; et al. 17p13.3 microduplications are associated with split-hand/foot malformation and long-bone deficiency (SHFLD). Eur. J. Hum. Genet. 2011, 19, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Petit, F.; Jourdain, A.; Andrieux, J.; Baujat, G.; Baumann, C.; Beneteau, C.; David, A.; Faivre, L.; Gaillard, D.; Gilbert-Dussardier, B.; et al. Split hand/foot malformation with long-bone deficiency and BHLHA9 duplication: Report of 13 new families. Clin. Genet. 2013, 85, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Merikangas, A.K.; Corvin, A.P.; Gallagher, L. Copy-number variants in neurodevelopmental disorders: Promises and challenges. Trends Genet. 2009, 25, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, R.; Mahjani, B.; Loos, R.J.F.; Sharp, A.J. Clinical characterization of copy number variants associated with neurodevelopmental disorders in a large-scale multiancestry biobank. JAMA Psychiatry 2022, 79, 250–259. [Google Scholar] [CrossRef]
- Lionel, A.C.; Crosbie, J.; Barbosa, N.; Goodale, T.; Thiruvahindrapuram, B.; Rickaby, J.; Gazzellone, M.; Carson, A.R.; Howe, J.L.; Wang, Z.; et al. Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci. Transl. Med. 2011, 3, 95ra75. [Google Scholar] [CrossRef]
- Glessner, J.T.; Wang, K.; Cai, G.; Korvatska, O.; Kim, C.E.; Wood, S.; Zhang, H.; Estes, A.; Brune, C.W.; Bradfield, J.P.; et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573. [Google Scholar] [CrossRef]
- Cooper, G.M.; Coe, B.P.; Girirajan, S.; A Rosenfeld, J.; Vu, T.H.; Baker, C.; Williams, C.; Stalker, H.; Hamid, R.; Hannig, V.; et al. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011, 43, 838–846. [Google Scholar] [CrossRef]
- Yamasaki, M.; Makino, T.; Khor, S.-S.; Toyoda, H.; Miyagawa, T.; Liu, X.; Kuwabara, H.; Kano, Y.; Shimada, T.; Sugiyama, T.; et al. Sensitivity to gene dosage and gene expression affects genes with copy number variants observed among neuropsychiatric diseases. BMC Med. Genom. 2020, 13, 55. [Google Scholar] [CrossRef]
- Kashevarova, A.A.; Drozdov, G.V.; Fedotov, D.A.; Lebedev, I.N. Pleiotropy of copy number variation in human genome. Russ. J. Genet. 2022, 58, 1180–1192. [Google Scholar] [CrossRef]
- Park, J.; Jang, W.; Han, J.Y. Differing disease phenotypes of Duchenne muscular dystrophy and Moyamoya disease in female siblings of a Korean family. Mol. Genet. Genom. Med. 2019, 7, e862. [Google Scholar] [CrossRef] [PubMed]
- Szigeti, K.; Lupski, J.R. Charcot-Marie-Tooth disease. Eur. J. Hum. Genet. 2009, 17, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Helland, W.A.; Lundervold, A.J.; Heimann, M.; Posserud, M.-B. Stable associations between behavioral problems and language impairments across childhood—The importance of pragmatic language problems. Res. Dev. Disabil. 2014, 35, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Mitrakos, A.; Kattamis, A.; Katsibardi, K.; Papadhimitriou, S.; Kitsiou-Tzeli, S.; Kanavakis, E.; Tzetis, M. High resolution Chromosomal Microarray Analysis (CMA) enhances the genetic profile of pediatric B-cell acute lymphoblastic leukemia patients. Leuk. Res. 2019, 83, 106177. [Google Scholar] [CrossRef] [PubMed]
- Ronaghy, A.; Yang, R.K.; Khoury, J.D.; Kanagal-Shamanna, R. Clinical applications of chromosomal microarray testing in myeloid malignancies. Curr. Hematol. Malign. Rep. 2020, 15, 194–202. [Google Scholar] [CrossRef]
- Ganesamoorthy, D.; Bruno, D.; McGillivray, G.; Norris, F.; White, S.; Adroub, S.; Amor, D.; Yeung, A.; Oertel, R.; Pertile, M.D.; et al. Meeting the challenge of interpreting high-resolution single nucleotide polymorphism array data in prenatal diagnosis: Does increased diagnostic power outweigh the dilemma of rare variants? BJOG Int. J. Obstet. Gynaecol. 2013, 120, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Jing, W.; Samuels, D.C.; Sheng, Q.; Shyr, Y.; Guo, Y. Strategies for processing and quality control of Illumina genotyping arrays. Brief. Bioinform. 2018, 19, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Lü, Y.; Jiang, Y.; Zhou, X.; Hao, N.; Xu, C.; Guo, R.; Chang, J.; Li, M.; Zhang, H.; Zhou, J.; et al. Detection of mosaic absence of heterozygosity (AOH) using low-pass whole genome sequencing in prenatal diagnosis: A preliminary report. Diagnostics 2023, 13, 2895. [Google Scholar] [CrossRef]
- Wang, H.; Dong, Z.; Zhang, R.; Chau, M.H.K.; Yang, Z.; Tsang, K.Y.C.; Wong, H.K.; Gui, B.; Meng, Z.; Xiao, K.; et al. Low-pass genome sequencing versus chromosomal microarray analysis: Implementation in prenatal diagnosis. Anesth. Analg. 2020, 22, 500–510. [Google Scholar] [CrossRef]
- Chau, M.H.K.; Wang, H.; Lai, Y.; Zhang, Y.; Xu, F.; Tang, Y.; Wang, Y.; Chen, Z.; Leung, T.Y.; Chung, J.P.W.; et al. Low-pass genome sequencing: A validated method in clinical cytogenetics. Hum. Genet. 2020, 139, 1403–1415. [Google Scholar] [CrossRef]
- Chaubey, A.; Shenoy, S.; Mathur, A.; Ma, Z.; Valencia, C.A.; Nallamilli, B.R.R.; Szekeres, E.; Stansberry, L.; Liu, R.; Hegde, M.R. Low-pass genome sequencing: Validation and diagnostic utility from 409 clinical cases of low-pass genome sequencing for the detection of copy number variants to replace constitutional microarray. J. Mol. Diagn. 2020, 22, 823–840. [Google Scholar] [CrossRef]
- Singh, M.; Pujar, G.V.; Kumar, S.A.; Bhagyalalitha, M.; Akshatha, H.S.; Abuhaija, B.; Alsoud, A.R.; Abualigah, L.; Beeraka, N.M.; Gandomi, A.H. Evolution of machine learning in tuberculosis diagnosis: A review of deep learning-based medical applications. Electronics 2022, 11, 2634. [Google Scholar] [CrossRef]
- Senescau, A.; Kempowsky, T.; Bernard, E.; Messier, S.; Besse, P.; Fabre, R.; François, J.M. Innovative DendrisChips® technology for a syndromic approach of in vitro diagnosis: Application to the respiratory infectious diseases. Diagnostics 2018, 8, 77. [Google Scholar] [CrossRef]
- Kong, S.W.; Collins, C.D.; Shimizu-Motohashi, Y.; Holm, I.A.; Campbell, M.G.; Lee, I.-H.; Brewster, S.J.; Hanson, E.; Harris, H.K.; Lowe, K.R.; et al. Characteristics and predictive value of blood transcriptome signature in males with autism spectrum disorders. PLoS ONE 2012, 7, e49475. [Google Scholar] [CrossRef]
- Krishnan, A.; Zhang, R.; Yao, V.; Theesfeld, C.L.; Wong, A.K.; Tadych, A.; Volfovsky, N.; Packer, A.; Lash, A.; Troyanskaya, O.G. Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nat. Neurosci. 2016, 19, 1454–1462. [Google Scholar] [CrossRef]
- Cheng, L.; Wang, P.; Yang, S.; Yang, Y.; Zhang, Q.; Zhang, W.; Xiao, H.; Gao, H.; Zhang, Q. Identification of genes with a correlation between copy number and expression in gastric cancer. BMC Med. Genom. 2012, 5, 14. [Google Scholar] [CrossRef]
- Nogueira, A.; Ferreira, A.; Figueiredo, M. A Machine learning pipeline for cancer detection on microarray data: The role of feature discretization and feature selection. BioMedInformatics 2023, 3, 585–604. [Google Scholar] [CrossRef]
- Parisi, F.; Micsinai, M.; Strino, F.; Ariyan, S.; Narayan, D.; Bacchiocchi, A.; Cheng, E.; Xu, F.; Li, P.; Kluger, H.; et al. Integrated analysis of tumor samples sheds light on tumor heterogeneity. Yale J. Biol. Med. 2012, 85, 347–361. [Google Scholar]
- Joseph, S.M.; Sathidevi, P.S. An automated cDNA microarray image analysis for the determination of gene expression ratios. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 20, 136–150. [Google Scholar] [CrossRef]
- Belean, B.; Gutt, R.; Costea, C.; Balacescu, O. Microarray image analysis: From image processing methods to gene expression levels estimation. IEEE Access 2020, 8, 159196–159205. [Google Scholar] [CrossRef]


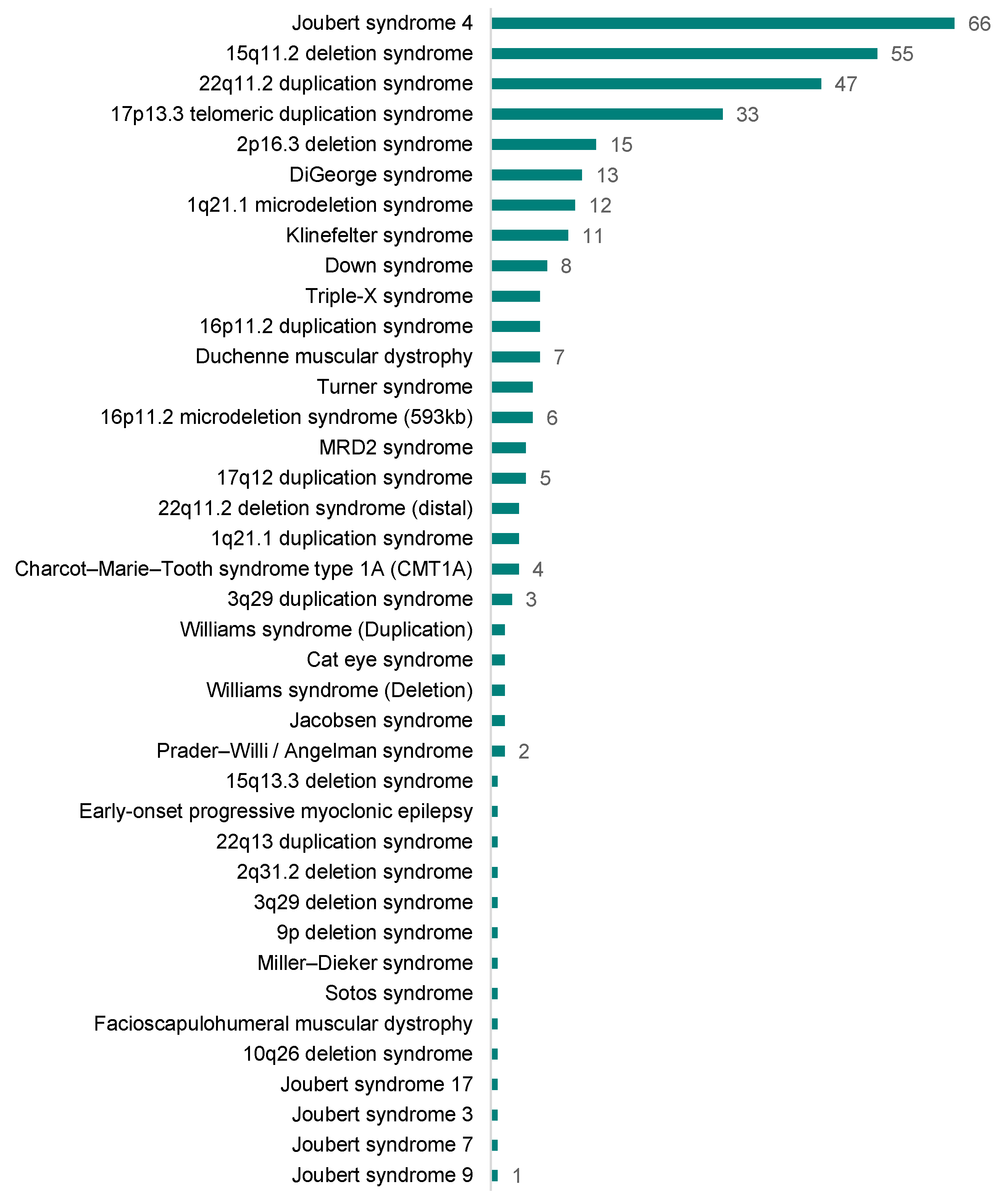
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goh, C.J.; Kwon, H.-J.; Kim, Y.; Jung, S.; Park, J.; Lee, I.K.; Park, B.-R.; Kim, M.-J.; Kim, M.-J.; Lee, M.-S. Improving CNV Detection Performance in Microarray Data Using a Machine Learning-Based Approach. Diagnostics 2024, 14, 84. https://doi.org/10.3390/diagnostics14010084
Goh CJ, Kwon H-J, Kim Y, Jung S, Park J, Lee IK, Park B-R, Kim M-J, Kim M-J, Lee M-S. Improving CNV Detection Performance in Microarray Data Using a Machine Learning-Based Approach. Diagnostics. 2024; 14(1):84. https://doi.org/10.3390/diagnostics14010084
Chicago/Turabian StyleGoh, Chul Jun, Hyuk-Jung Kwon, Yoonhee Kim, Seunghee Jung, Jiwoo Park, Isaac Kise Lee, Bo-Ram Park, Myeong-Ji Kim, Min-Jeong Kim, and Min-Seob Lee. 2024. "Improving CNV Detection Performance in Microarray Data Using a Machine Learning-Based Approach" Diagnostics 14, no. 1: 84. https://doi.org/10.3390/diagnostics14010084
APA StyleGoh, C. J., Kwon, H.-J., Kim, Y., Jung, S., Park, J., Lee, I. K., Park, B.-R., Kim, M.-J., Kim, M.-J., & Lee, M.-S. (2024). Improving CNV Detection Performance in Microarray Data Using a Machine Learning-Based Approach. Diagnostics, 14(1), 84. https://doi.org/10.3390/diagnostics14010084