
Pathomorphological Diagnostic Criteria for Focal Cortical Dysplasias and Other Common Epileptogenic Lesions—Review of the Literature

 , , and
, , and 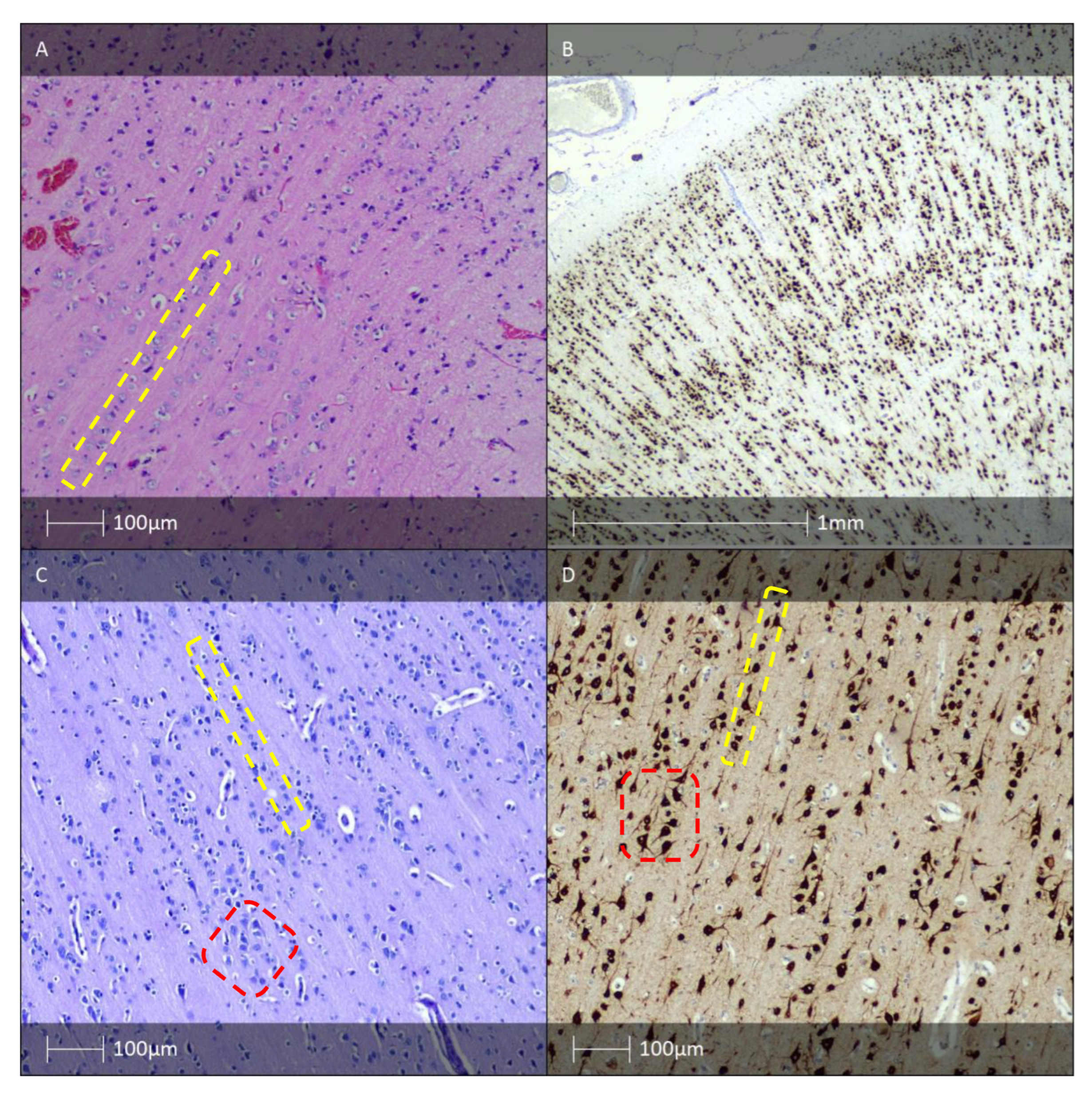{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
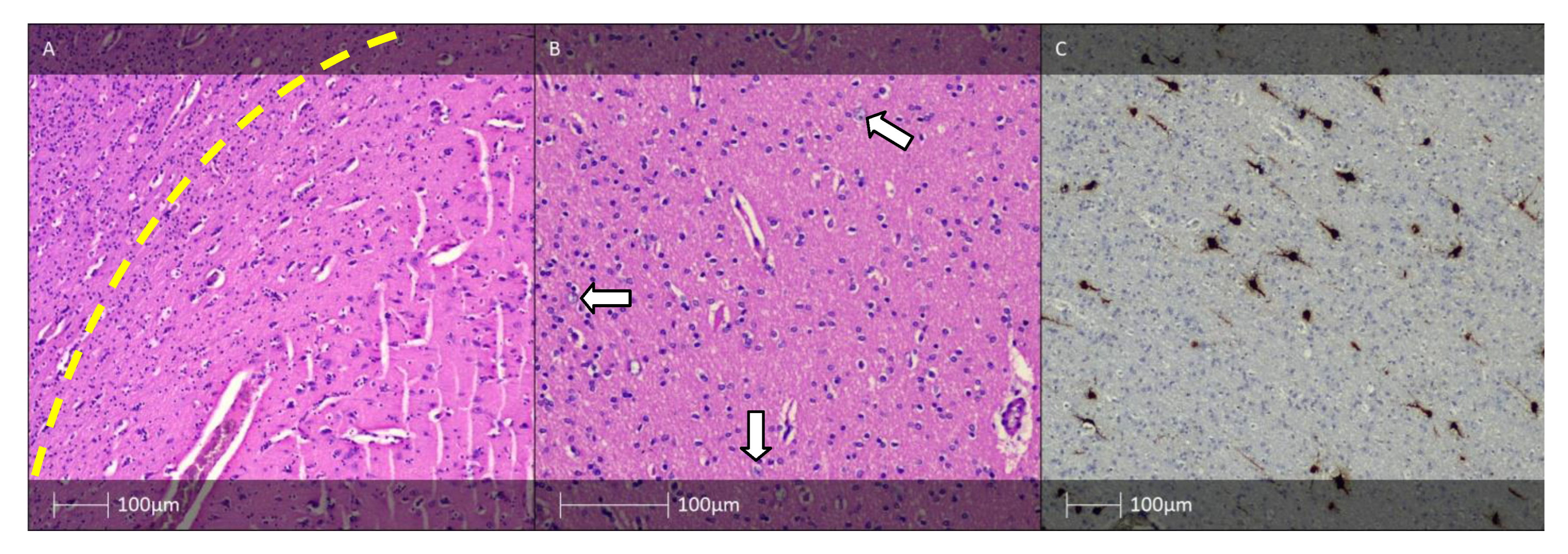{kind=link}
Abstract
1. Introduction
2. Brief History of Focal Cortical Dysplasia
3. Isolated Forms of FCD with Vertical, Horizontal, or a Combination of the Two Types of Cortical Dyslamination (FCD Type I)
4. Isolated Forms of FCD with Cortical Dyslamination and Dysmorphic Neurones with or without Balloon Cells (FCD Type II)
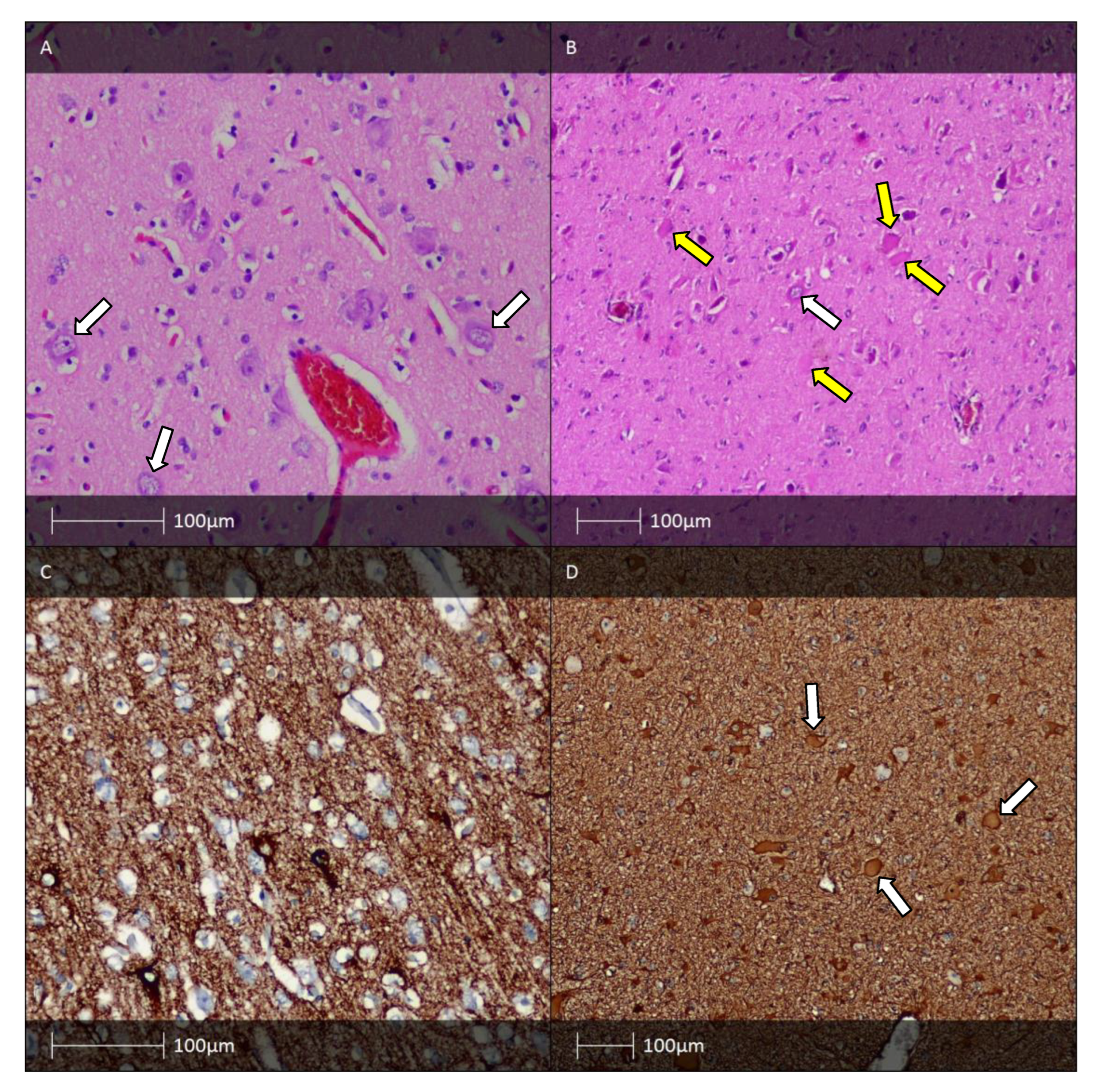
5. Hippocampal Sclerosis and FCD Associated with Hippocampal Sclerosis (FCD IIIa)
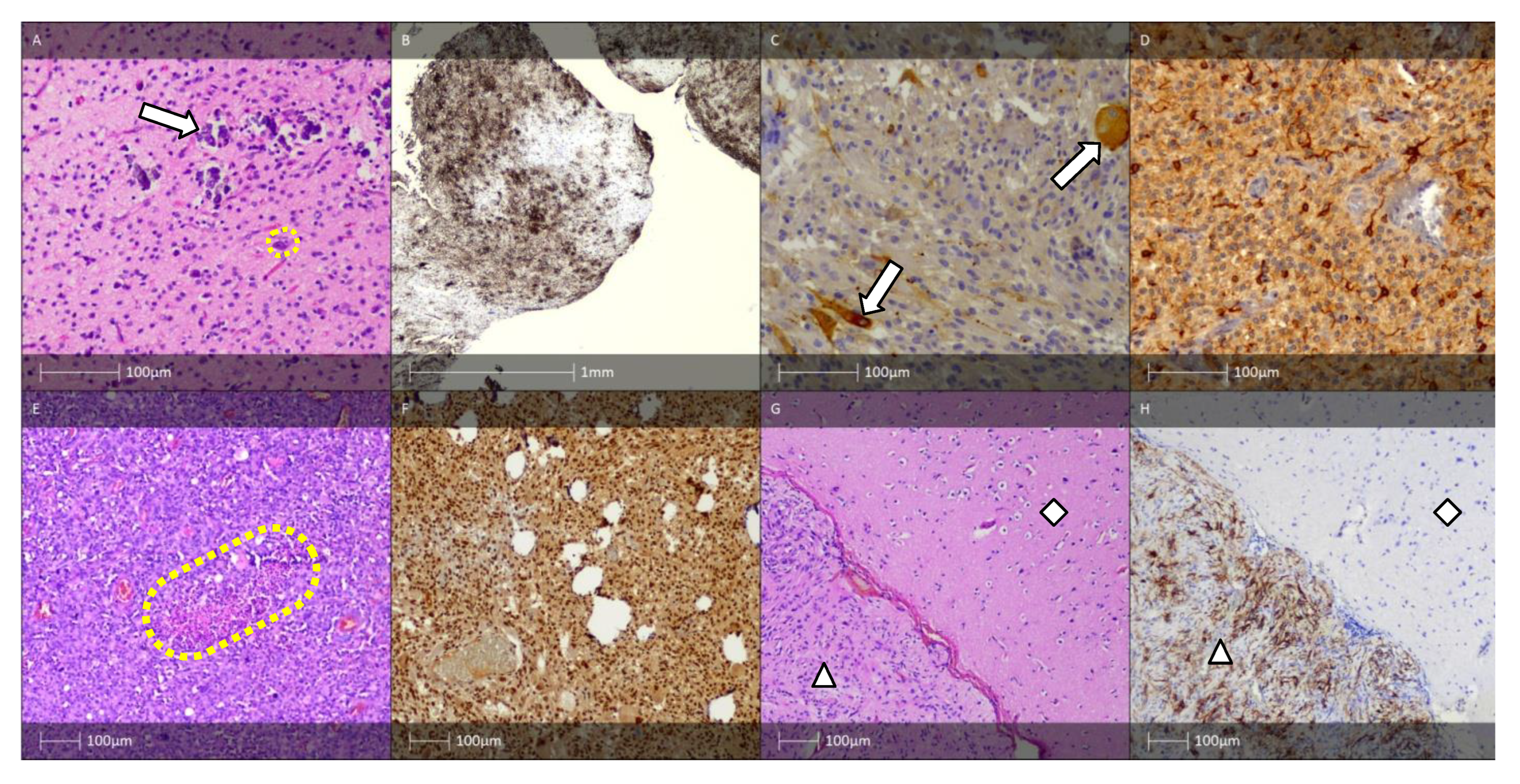
6. Long-Term Epilepsy-Associated Tumours (LEAT). FCD Associated with a Tumour Process (FCD Type IIIb)
6.1. Gangliocytoma, Ganglioglioma, Anaplastic Ganglioglioma, and Desmoplastic Ganglioglioma
6.2. Dysembryoplastic Neuroepithelial Tumour
6.3. Pleomorphic Xanthoastrocytoma
6.4. Rosette-Forming Glioneuronal Tumour
6.5. Papillary Glioneuronal Tumour
6.6. Hypothalamic Hamartoma
6.7. Angiocentric Glioma
6.8. Isomorphic Diffuse Astrocytoma
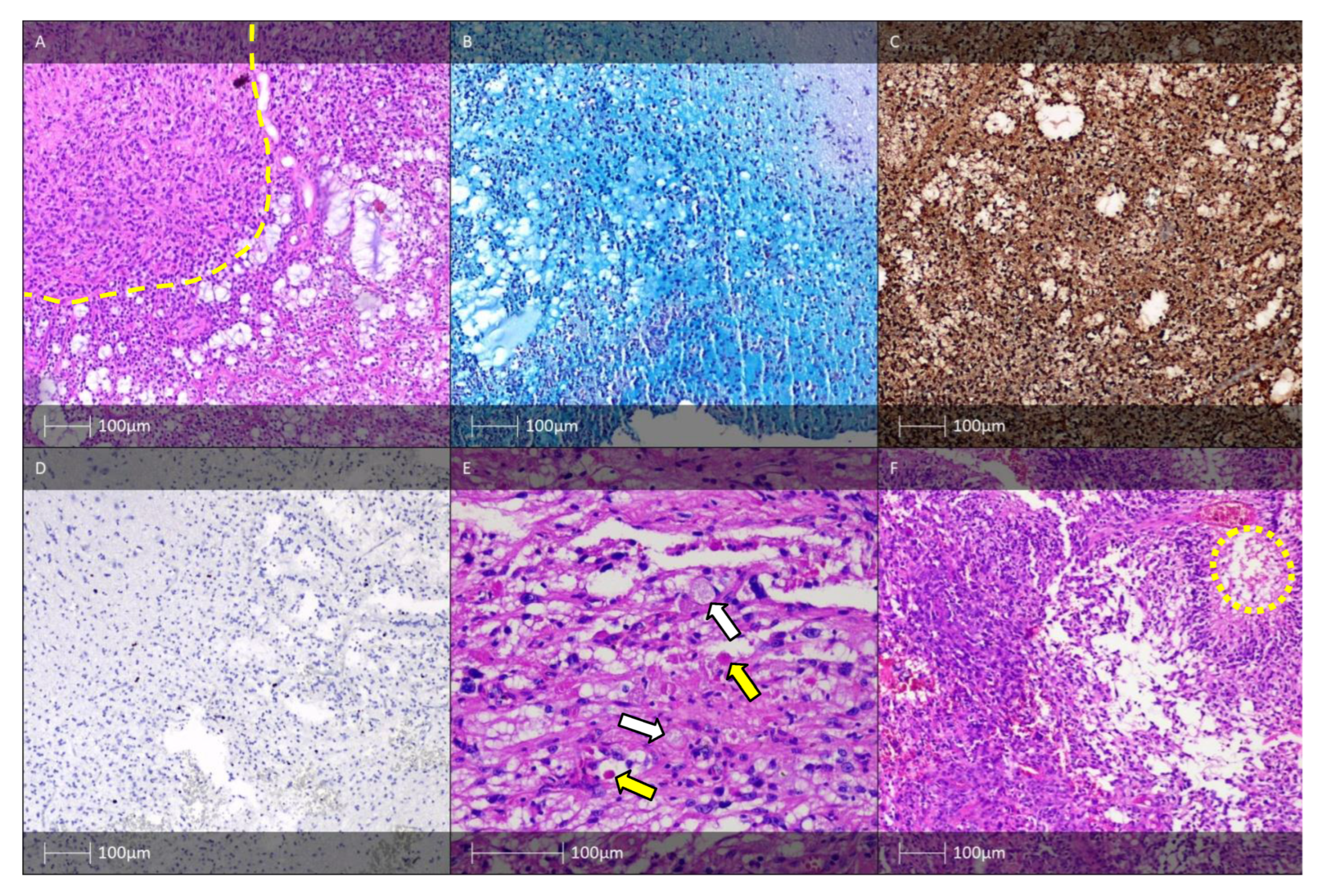
7. Epileptogenic Vascular Malformations, including in the Context of FCD IIIc, Meningioangiomatosis
7.1. Cavernous Hemangioma (Cavernoma)
7.2. Arteriovenous Malformation (AVM)
7.3. Sturge–Weber Syndrome
7.4. Meningioangiomatisis
8. Epileptogenic Encephalitides, Focal Cortical Dysplasia in Combination with Another Epileptogenic Lesion, Acquired Earlier in Life (Traumatic Injuries, Ischemic Injuries and Encephalitis)—FCD Type IIId
9. Epileptogenic Lesions with Predilection for White Matter Involvement
10. “Dual” and “Double Pathology”
11. The Integrated Approach for the Diagnosis of FCD
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Milanov, I.; Bozhinova, V. National Consensus on the Diagnosis and Treatment of Epilepsy; Bulgarian Society of Neurology: Golden Sands, Bulgaria, 2022; p. 6. [Google Scholar]
- Kwan, P.; Arzimanoglou, A.; Berg, A.T.; Brodie, M.J.; Hauser, W.A.; Mathern, G.; Moshé, S.L.; Perucca, E.; Wiebe, S.; French, J. Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010, 51, 1069–1077. [Google Scholar] [CrossRef]
- Minkin, K. Surgical Treatment Options for Patients with Symptomatic Pharmacoresistant Epilepsy. Ph.D. Thesis, Medical University-Sofia, Sofia, Bulgaria, 2009. [Google Scholar]
- Najm, I.; Lal, D.; Alonso Vanegas, M.; Cendes, F.; Lopes-Cendes, I.; Palmini, A.; Paglioli, E.; Sarnat, H.B.; Walsh, C.A.; Wiebe, S.; et al. The ILAE consensus classification of Focal Cortical Dysplasia (FCD): An update proposed by a hoc Task Force of ILAE Diagnostic Methods commission. Epilepsia 2022, 63, 1899–1919. [Google Scholar] [CrossRef]
- Schurr, J.; Coras, R.; Rössler, K.; Pieper, T.; Kudernatsch, M.; Holthausen, H.; Winkler, P.; Woermann, F.; Bien, C.G.; Polster, T.; et al. Mild malformation of cortical development with oligodendroglial hyperplasia in frontal lobe epilepsy: A new clinic-pathological entity. Brain Pathol. 2017, 27, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Prayson, R.; Spreafico, R.; Vinters, H. Pathologic characteristics of the cortical dysplasias. Neurosurg. Clin. N. Am. 2002, 13, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Blümcke, I.; Thom, M.; Aronica, E.; Armstrong, D.D.; Vinters, H.V.; Palmini, A.; Jacques, T.S.; Avanzini, G.; Barkovich, A.J.; Battaglia, G.; et al. The clinicopathologic spectrum of focal cortical dysplasias: A consensus classification proposed by an ad Task Force ot the ILAE Diagnostic Methods Commission. Epilepsia 2011, 52, 158–174. [Google Scholar] [CrossRef] [PubMed]
- Palmini, A.; Najm, I.; Avanzini, G.; Babb, T.; Guerrini, R.; Foldvary-Schaefer, N.; Jackson, G.; Luders, H.O.; Prayson, R.; Spreafico, R.; et al. Terminology and classification of the cortical dysplasias. Neurology 2004, 62, S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Falconer, M.; Bruton, C.; Corsellis, J. Focal dysplasia of the cerebral cortex in epilepsy. J. Neurol. Neurosurg. Psychiatry 1971, 34, 369–387. [Google Scholar] [CrossRef]
- Crome, L. Infantile cerebral gliosis with giant nerve cells. J. Neurol. Neurosurg. Psychiatry 1957, 20, 117–124. [Google Scholar] [CrossRef]
- Barkovich, A.; Kuzniecky, R.; Bollen, A.; Grant, P. Focal transmantel dysplasia: A specific malformations of cortical development. Neurology 1997, 49, 1148–1152. [Google Scholar] [CrossRef]
- Blumcke, I.; Pieper, T.; Pauli, E.; Hildebrandt, M.; Kudernatsch, M.; Winkler, P.; Karlmeier, A.; Holthausen, H. A distinct variant of focal cortical dysplasia type I characterised by magnetic resonance imaging and neuropathological examination in children with severe epilepsies. Epileptic. Disord. 2010, 12, 172–180. [Google Scholar]
- Rakic, P.; Lombroso, P. Development of the cerebral cortex: I. Forming the cortical structure. J. Am. Acad. Child Adolesc. Psychiatry 1998, 37, 116–117. [Google Scholar] [CrossRef] [PubMed]
- Sarnat, H.; Flores-Sarnat, R. Radial microcolumnar cortical architecture: Maturational arrest or cortical dysplasia? Pediatr. Neurol. 2013, 48, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Leventer, R.; Jansen, F.; Mandelstam, S.; Ho, A.; Mohamed, I.; Sarnat, H.; Kato, M.; Fukasawa, T.; Saitsu, H.; Matsumoto, N.; et al. Is focal cortical dysplasia sporadic? Family evidence for genetic susceptibility. Epilepsia 2014, 55, e22–e26. [Google Scholar] [CrossRef] [PubMed]
- Blumcke, I.; Beck, H.; Nitsch, R.; Eickhoff, C.; Scheffler, B.; Celio, M.; Schramm, J.; Elger, C.; Wolf, H.; Wiestler, O. Preservation of calretinin-immunoreactive neurones in the hippocampus in epilepsy patients with Ammons horn sclerosis. J. Neuropathol. Exp. 1996, 55, 329–341. [Google Scholar]
- Blumcke, I.; Beck, H.; Suter, B.; Hoffmann, D.; Fodisch, H.; Wolf, H.; Schramm, J.; Elger, C.; Wiestler, O. An increase of hippocampal calretinin-immunoreactive neurones correlates with early febrile seizures in temporal lobe epilepsy. Acta Neuropathol. 1999, 97, 31–39. [Google Scholar]
- Martinian, L.; Boer, K.; Middeldorp, J.; Hol, E.; Sisodiya, S.; Squier, W.; Aronica, E.; Thom, M. Expression patterns of glial acidic fibrillary protein (GFAP)-delta in epilepsy-associated lesional pathologies. Neuropathol. Appl. Neurobiol. 2009, 35, 394–405. [Google Scholar] [CrossRef]
- Boer, K.; Spliet, W.; van Rijen, P.; Jansen, F.; Aronica, E. Expression patterns of AMOG in developing human cortex and malformations of cortical development. Epilepsy Res. 2010, 91, 84–93. [Google Scholar] [CrossRef]
- Poduri, A.; Evrony, G.; Cai, X.; Walsh, C. Somatic mutation, genomic variation, and neurological disease. Science 2013, 341, 1237758. [Google Scholar] [CrossRef]
- Mackenzie, I.; Neumann, M.; Cairns, N.; Munoz, D.; Isaacs, A. Novel types of frontotemporal lobar degeneration: Beyond tau and TDP-43. J. Mol. Neurosci. 2011, 45, 402–408. [Google Scholar] [CrossRef]
- Sarnat, H.; Flores-Sarnat, L.; Crino, P.; Hader, W.; Bello-Espinosa, L. Hemimegalencephaly: Foetal tauopathy with mTOR hyperactivation and neuronal lipidosis. Folia Neuropathol. 2012, 50, 330–345. [Google Scholar] [CrossRef]
- D’Gama, A.; Woodworth, M.; Hossain, A.; Bizzotto, S.; Hatem, N.; LaCoursiere, C.; Najm, I.; Ying, Z.; Yang, E.; Barkovich, A.; et al. Somatic mutations activating the mTOR pathway in dorsal telencephalic progenitors cause a continuum of cortical dysplasias. Cell Rep. 2017, 21, 3754–3766. [Google Scholar] [CrossRef] [PubMed]
- Baulac, S.; Ishida, S.; Marsan, E.; Miquel, C.; Biraben, A.; Nguyen, D.; Nordli, D.; Cossette, P.; Nguyen, S.; Lambrecq, V.; et al. Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Ann. Neurol. 2015, 77, 675–683. [Google Scholar] [CrossRef]
- Krueger, D.; Care, M.; Holland, K.; Agricola, K.; Tudor, C.; Mangeshkar, P.; Wilson, K.; Byars, A.; Sahmoud, T.; Franz, D. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N. Engl. J. Med. 2010, 363, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Blumcke, I.; Thom, M.; Wiestler, O. Ammon’s horn sclerosis: A maldevelopmental disorder. Brain Pathol. 2002, 12, 199–211. [Google Scholar] [PubMed]
- Blumcke, I.; Coras, R.; Miyata, H.; Ozkara, C. Defining clinico-neuropathological subtypes of mesial temporal lobe epilepsy with hippocampal sclerosis. Brain Pathol. 2012, 22, 402–411. [Google Scholar] [CrossRef]
- Blumcke, I.; Spreafico, R. Cause matters: A neuropathological challenge to human epilepsies. Brain Pathol. 2012, 22, 347–349. [Google Scholar] [CrossRef]
- Blumcke, I.; Suter, B.; Behle, K.; Kuhn, R.; Schramm, J.; Elger, C.; Wiestler, O. Loss of hilar mossy cells in Ammon’s horn sclerosis. Epilepsy 2000, 41 (Suppl. 6), S174–S180. [Google Scholar] [CrossRef]
- Sommer, W. Die Erkrankung des Ammonshorns als aetiologisches Moment der Epilepsie. Arch. Psychiat. Nervenkr. 1880, 308, 631–675. [Google Scholar] [CrossRef]
- Becker, A.; Chen., J.; Zien, A.; Sochivko, D.; Normann, S.; Schramm, J.; Elger, C.; Wiestler, O.; Blümcke, I. Correlates stage- and subfield-associated hippocampal gene expression patterns in experimental and human temporal lobe epilepsy. Eur. J. Neurosci. 2003, 18, 2792–2802. [Google Scholar] [CrossRef]
- Houser, C.R. Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res. 1990, 535, 195–204. [Google Scholar] [CrossRef]
- Marusic, P.; Tomasek, M.; Krsek, P.; Krijtová, H.; Zárubová, J.; Zámecník, J.; Mohapl, M.; Beneš, V.; Tichý, M.; Komárek, V. Clinical characteristics in patients with hippocampal sclerosis with or without cortical dysplasia. Epileptic. Disord. 2007, 9 (Suppl. 1), S75–S82. [Google Scholar]
- Metodiev, D.; Minkin, K.; Penkov, M.; Dimova, P.; Nachev, S. Histopathological Findings in Brain Tissue of Patients with Pharmacoresistant Focal Epilepsy. J. Neurol. Neurobiol. 2022, 8. [Google Scholar] [CrossRef]
- Blümcke, I.; Thom, M.; Aronica, E.; Armstrong, D.D.; Bartolomei, F.; Bernasconi, A.; Bernasconi, N.; Bien, C.G.; Cendes, F.; Coras, R.; et al. International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: A Task Force report from the ILAE Commission on Diagnostic Methods. Epilepsia 2013, 54, 1315–1329. [Google Scholar] [CrossRef]
- Coras, R.; Pauli, E.; Li, J.; Schwarz, M.; Rössler, K.; Buchfelder, M.; Hamer, H.; Stefan, H.; Blumcke, I. Differential influence of hippocampal subfields to memory formation: Insights from patients with tempiral lobe epilepsy. Brain 2014, 137, 1945–1957. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, G.; Kandratavicius, L.; Peixoto-Santos, J.; Monteiro, M.; Gargaro, A.; Geraldi, C.; Velasco, T.; Leite, J. Increased frequency of hippocampal sclerosis ILAE type 2 in patients with mesial temporal lobe epilepsy with normal episodic memory. Brain 2015, 138, e359. [Google Scholar] [CrossRef] [PubMed]
- Thom, M. Review: Hippocampal sclerosis in epilepsy: A neuropathology review. Neuropathol. Appl. Neurobiol. 2014, 40, 520–543. [Google Scholar] [CrossRef]
- Tassi, L.; Meroni, F.; Deleo, F.; Villani, F.; Mai, R.; Lo Russo, G.; Colombo, N.; Avanzini, G.; Falcone, C.; Bramerio, M.; et al. Temporal lobe epilepsy: Neuropathological and clinical correlations in 243 surgically treated patients. Epileptic. Disord. 2009, 4, 281–292. [Google Scholar] [CrossRef]
- Wolf, H.K.; Muller, M.B.; Spanle, M.; Zenter, J.; Schramm, J.; Wiestler, O.D. Ganglioglioma: A detailed histopathological and immunohistochemical analysis of 61 cases. Acta Neuropathol. 1994, 88, 166–173. [Google Scholar] [CrossRef]
- Blumcke, I. Neuropathology of focal epilepsies: A critical review. Epilepsy Behav. 2009, 15, 34–39. [Google Scholar] [CrossRef]
- Blumcke, I.; Speafico, R.; Haaker, G.; Coras, R.; Kobow, K.; Bien, C.; Pfäfflin, M.; Elger, C.; Widman, G.; Schramm, J.; et al. Histopathological findings in brain tissue obtained during epilepsy surgery. N. Engl. J. Med. 2017, 377, 1648–1656. [Google Scholar] [CrossRef]
- Urbach, H. High-grade magnetic resonance imaging of epilepsy. Neuroimaging Clin. N. Am. 2012, 22, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.; Brat, D. Practical Surgical Neuropathology. A Diagnostic Approach, 2nd ed.; Elsevier: Philadelphia, PA, USA, 2018; pp. 183–218. [Google Scholar]
- Russell, D.S.; Rubinstein, L.J. Pathology of Tumors of the Nervous System, 5th ed.; Williams and Wilkins: Baltimore, MD, USA, 1989; pp. 289–307. [Google Scholar]
- Johannson, J.H.; Rekate, H.L.; Roessmann, U. Gangliogliomas: Pathological and clinical correlation. J. Neurosurg. 1981, 54, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Prayson, R.; Khajavi, K.; Comair, Y. Cortical architectural abnormalities and MIB1 immunoreactivity in gangliogliomas: A study of 60 patients with intracranial tumors. J. Neuropathol. Exp. Neurol. 1995, 54, 513–520. [Google Scholar] [CrossRef] [PubMed]
- WHO. Classification of Tumours of the Central Nervous System, 5th ed.; IARC: Lyon, France, 2021.
- Blumcke, I.; Giencke, K.; Wardelmann, E.; Beyenburg, S.; Kral, T.; Sarioglu, N.; Pietsch, T.; Wolf, H.; Schramm, J.; Elger, C.; et al. The CD34 epitope is expressed in neoplastic and malformative lesions associated with chronic, focal epilepsies. Acta Neuropathol. 1999, 97, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Koelsche, C.; Wohhrer, A.; Jeibman, A.; Schittenhelm, J.; Schindler, G.; Preusser, M.; Lasitschka, F.; von Deimling, A.; Capper, D. Mutant BRAF protein in ganglioglioma is predominantly expressed by neuronal tumor cells. Acta Neuropathol. 2013, 125, 891–900. [Google Scholar] [CrossRef]
- Blumcke, I.; Wiestler, O. Gangliogliomas: An intriguing tumor entity associated with focal epilepsies. J. Neuropathol. Exp. Neurol. 2002, 6, 575–584. [Google Scholar] [CrossRef]
- Luyken, C.; Blumcke, I.; Fimmers, R.; Urbach, H.; Wiestler, O.; Schramm, J. Supratentorial gangliogliomas: Histopayhologic grading and tumor recurrence in 184 patients with a medium follow-up of 8 years. Cancer 2004, 101, 146–155. [Google Scholar] [CrossRef]
- Kawataki, T.; Sato, E.; Sato, T.; Kinouchi, H. Anaplastic ganglioglioma with malignant features in both neuronal and glial komponents-case report. Neurol. Med. Chir. 2010, 50, 228–231. [Google Scholar] [CrossRef]
- Zhu, J.J.; Leon, S.P.; Folkerth, R.D.; Guo, S.Z.; Wu, J.K.; Black, P.M. Evidence for clonal origin of neoplastic neuronal and glial cells in gangliogliomas. Am. J. Pathol. 1997, 151, 565–571. [Google Scholar]
- Blumcke, I.; Lobach, M.; Wolf, H. Evidence for developmental precursor lesions in epilepsy-associated glioneuronal tumors. Microsc. Res. Tech. 1999, 46, 53–58. [Google Scholar] [CrossRef]
- Nair, V.; Suri, V.S.; Tatke, M.; Saran, R.K.; Mlhotra, V.; Singh, D. Gangliogliomas: A report of five cases. Indian J. Cancer 2004, 41, 41–46. [Google Scholar] [PubMed]
- Kurian, N.I.; Nair, S.; Radhakrishnan, V.V. Anaplastic ganglioglioma: Case report and review of the literature. Br. J. Neurosurg. 1998, 12, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Vinters, H.V. Ganglion cell tumors. Korean J. Pathol. 2002, 36, 167–174. [Google Scholar]
- Naydenov, E.; Tzekov, C.; Minkin, K.; Nachev, S. Malignant progression of anaplastic supratentorial ganglioglioma into Glioblastoma multiforme in a patient with Turner syndrome. J. Neurol. Surg. A Cent. Eur. Neurosurg. 2012, 73, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Vanderberg, S. Desmoplastic infantile ganglioglioma and desmoplastic cerebral astrocytoma of infancy. Brain Pathol. 1993, 3, 275–281. [Google Scholar] [CrossRef]
- Daumas-Duport, C.; Scheithauer, B.; Chodkiewiczd, J.; Laws, E., Jr.; Vedrenne, C. Dysembrioplastic neuroepithelial tumor: A surgically curable tumor of young patients with intractable partial seizures. Report of thirty-nine cases. Neurosurgery 1988, 23, 545–556. [Google Scholar] [CrossRef]
- Bilginer, B.; Yalnizoglu, D.; Soylemezoglu, F.; Turanlı, G.; Cila, A.; Topçu, M.; Akalan, N. Surgery for epilepsy in children with dysembryoplastic neuroepithelial tumor: Clinical spectrum, seizure outcome, neuroradiology, and pathology. Childs Nerv. Syst. 2009, 25, 485–491. [Google Scholar] [CrossRef]
- Cataltepe, O.; Marshall, P.; Smith, T. Dysembryoplastic neuroepithelial tumor located in pericallosal and intraventricular area in a child. Case report. J. Neurosurg. Pediatr. 2009, 3, 456–460. [Google Scholar] [CrossRef]
- Emmez, H.; Kale, A.; Egemen, E.; Eser, P.; Kaymaz, M.; Paşaoğlu, A. Intraventricular dysembryoplastic neuroepithelial tumour: Case report. Neurol. Neurochir. Pol. 2012, 46, 192–195. [Google Scholar] [CrossRef]
- Stoyanov, G.S.; Petkova, L.; Kondev, T.; Georgiev, R.; Enchev, Y. Posterior Fossa Dysembryoplastic Neuroepithelial Tumor: A Neuropathological Report. Cureus 2023, 15, e33525. [Google Scholar] [CrossRef]
- Blumcke, I.; Aronica, E.; Urbach, H.; Alexopoulos, A.; Gonzalez-Martinez, J. A neuropathology-based approach to epilepsy surgery in brain tumors and proposal for a new terminology use for long-term epilepsy-associated tumors. Acta Neuropathol. 2014, 128, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Daumas-Duport, C. Dysembryoplastic neuroepithelial tumors. Brain Pathol. 1993, 3, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Koeller, K.; Dillon, W.P. Dysembryoplastic neuroepithelial tumors: MR appearance. Am. J. Neuroradiol. 1992, 13, 1319–1325. [Google Scholar] [PubMed]
- Prayson, R.A.; Napekoski, K. Composite ganglioglioma/dysembryoplastic neuroepithelial tumor: A clinicopathologic study of 8 cases. Hum. Pathol. 2012, 43, 1113–1118. [Google Scholar] [CrossRef]
- Qaddoumi, I.; Orisme, W.; Wen, J.; Santiago, T.; Gupta, K.; Dalton, J.; Tang, B.; Haupfear, K.; Punchihewa, C.; Easton, J.; et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MUB mutations occur at high frequency and align with morphology. Acta Neuropathol. 2016, 131, 833–845. [Google Scholar] [CrossRef]
- Taratuto, A.; Pomata, H.; Sevlever, G.; Gallo, G.; Monges, J. Dysembryoplastic neuroepithelial tumour: Morphological, immunohistochemical, and deoxyribonucleic acid analysis in a pediatric series. Neurosurgery 1995, 36, 474–481. [Google Scholar]
- Hirose, T.; Scheithauer, B.; Lopes, M.; VandenBerg, S. Dysembryoplastic neuroepithelial tumour (DNT): An immunohistochemical and ultrastructural study. J. Neuropathol. Exp. Neurol. 1994, 53, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Nolan, M.; Sakuta, R.; Chuang, N.; Otsubo, H.; Rutka, J.; Snead, O.; Hawkins, C.; Weiss, S. Dysembryoplastic neuroepithelial tumours in childhood: Long-term outcome and prognostic features. Neurology 2004, 62, 2270–2776. [Google Scholar] [CrossRef]
- Chao, L.; Tao, X.; Jun, Y.; Xia, H.; Wan, W.; Tao, Q. Recurrence and histological evolution of dysembryoplastic neuroepithelial tumor: A case report and review of the literature. Oncol. Lett. 2013, 6, 907–914. [Google Scholar] [CrossRef]
- Keppes, J. Pleomorphic xanthoastrocytoma: The birth of a diagnosis and a concept. Brain Pathol. 1993, 3, 269–274. [Google Scholar] [CrossRef]
- Keppes, J.; Rubinstein, L.; Eng, L. Pleomorphic xanthoastrogytoma: A distinctive meningocerebral glioma of young subjects with relatively favorable prognosis; a study of 12 cases. Cancer 1979, 44, 1839–1852. [Google Scholar] [CrossRef]
- Rippe, D.; Boyko, O.; Radi, M.; Worth, R.; Fuller, G. MRI of temporal lobe pleomorphic xantroastrocytoma. J. Comput. Assist. Tomogr. 1992, 16, 856–859. [Google Scholar] [CrossRef] [PubMed]
- Giannini, C.; Schethauer, B.; Burger, P.; Brat, D.; Wollan, P.; Lach, B.; O’Neill, B. Pleomorphic xanthoastrocytoma: What do we really know about it? Cancer 1999, 85, 2033–2045. [Google Scholar] [CrossRef]
- Marucci, G.; Morandi, L. Assessment of MGMT promoter methylation status in pleomorphic xanthoastrocytma. J. Neurooncol. 2011, 105, 397–400. [Google Scholar] [CrossRef]
- Chamberlain, M.C. Salvage therapy with BRAF inhibitors for recurrent pleomorphic xanthoastrocytoma: A retrospective case series. J. Neurooncol. 2013, 114, 237–240. [Google Scholar] [CrossRef]
- Schlamann, A.; von Bueren, A.; Hagel, C.; Zwiener, I.; Seidel, C.; Kortmann, R.; Müller, K. An invidual patient data meta-analysis on characteristics and outcome of patients with papillary glioneuronal tumor, rosette glioneuronal tumor with neuropil-like islands and rosette-forming glioneuronal tumor of the fourth ventricle. PLoS ONE 2014, 9, e101211. [Google Scholar] [CrossRef]
- Medhi, G.; Prasad, C.; Saini, J.; Pendharkar, H.; Bhat, M.; Pandey, P.; Muthane, Y. Imaging features of rosette-forming glioneuronal tumours (RGNTs): A series of seven cases. Eur. Radiol. 2016, 26, 262–270. [Google Scholar] [CrossRef]
- Komori, T.; Scheithauer, B.; Hirose, T. A rosette-forming glioneuronal tumor of the fourth ventricle: Infratentorial form of dysembryoplastic neuroepithelial tumor? Am. J. Surg. Pathol. 2002, 26, 582–591. [Google Scholar] [CrossRef]
- Allinson, K.; O’Donovan, D.; Jena, R.; Cross, J.; Santarius, T. Rosette-forming glioneuronal tumor with dissemination throughout the ventricular system: A case report. Clin. Neuropathol. 2015, 34, 64–69. [Google Scholar] [CrossRef]
- Komori, T.; Scheithauer, B.; Anthony, D.; Rosenblum, M.; McLendon, R.; Scott, R.; Okazaki, H.; Kobayashi, M. Papillary glioneuronal tumour: A new variant of mixed neuronal-glial neoplasm. Am. J. Surg. Pathol. 1998, 22, 1171–1183. [Google Scholar] [CrossRef]
- Prayson, R. Papillary glioneuronal tumor. Arch. Pathol. Lab. Med. 2000, 124, 1820–1823. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.; Byeon, S.; Kim, B.; Suh, J.; Kim, S.; Park, C.; Chung, C.; Chang, K.; Park, S. Papillary glioneuronal tumors: A review of clinicopathologic and molecular genetic studies. Am. J. Surg. Pathol. 2011, 35, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Bridge, J.; Liu, X.; Sumegi, J.; Nelson, M.; Reyes, C.; Bruch, L.; Rosenblum, M.; Puccioni, M.; Bowdino, B.; McComb, R. Identification of a novel recurrent SLC44A1-PRKCA fusion in papillary glioneuronal tumor. Brain Pathol. 2013, 23, 121–128. [Google Scholar] [CrossRef]
- Fenoglio, K.; Wu, J.; Kim do, Y.; Simeone, T.; Coons, S.; Rekate, H.; Rho, J.; Kerrigan, J. Hypothalamic hamartomas: Basic mechanisms of intrinsic epileptogenesis. Semin. Pediatr. Neurol. 2007, 14, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Weissenberger, A.; Dell, M.; Liow, K.; Theodore, W.; Frattali, C.; Hernandez, D.; Zametkin, A. Aggression and psychiatric comorbidity in children with hypothalamic hamartomas and their unaffected siblings. J. Am. Acad. Child Adolesc. Psychiatry 2001, 40, 696–703. [Google Scholar] [CrossRef]
- Ng, Y.; Kerrigan, J.; Prenger, E.; White, W.; Rekate, H. Successful resection of a hypothalamic hamartoma and a Rathke cleft cyst. J. Neurosurg. 2005, 102, 78–80. [Google Scholar] [CrossRef]
- Coons, S.; Rekate, S.; Prenger, E.; Wang, N.; Drees, C.; Ng, Y.; Chung, S.; Kerrigan, J. The histopathology of hypothalamic hamartomas: Study of 57 cases. J. Neuropathol. Ex Neurol. 2007, 66, 131–141. [Google Scholar] [CrossRef]
- Harvey, A.; Freeman, J.; Berkovic, S.; Rosenfeld, J. Transcallosal resection of hypothalamic hamartomas in patient with intractable epilepsy. Epileptic. Disord. 2003, 5, 257–265. [Google Scholar]
- Ampie, L.; Choy, W.; DiDomenico, J.; Lamano, J.; Williams, C.; Kesavabhotla, K.; Mao, Q.; Bloch, O. Clinical attributes and surgical outcomes of angiocentric gliomas. J. Clin. Neurosci. 2016, 28, 117–122. [Google Scholar] [CrossRef]
- Marburger, T.; Prayson, R. Angiocentric glioma: A clinicopathologic review of 5 tumors with identification of associated cortical dysplasia. Arch. Pathol. Lab. Med. 2011, 135, 1037–1041. [Google Scholar] [CrossRef]
- Preusser, M.; Hoischen, M.; Novak, K.; Czech, T.; Prayer, D.; Hainfellner, J.; Baumgartner, C.; Woermann, F.; Tuxhorn, I.; Pannek, H.; et al. Angiocentric gliomas: Report of clinic-pathologic and genetic findings in 8 cases. Am. J. Surg. Pathol. 2007, 31, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Tihan, T.; Rojiani, M.; Bodhireddy, S.; Prayson, R.; Iacuone, J.; Alles, A.; Donahue, D.; Hessler, R.; Kim, J.; et al. Monomorphous angiocentric glioma: A distinctive epileptogenic neoplasm with features of infiltrating astrocytoma and ependymoma. J. Neuropathol. Exp. Neurol. 2005, 64, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Blumcke, I.; Luyken, C.; Urbach, H.; Schramm, J.; Wiestlerm, O.D. An isomorphic subtype of long-term epilepsy-associated astrocytomas associated with benign prognosis. Acta Neuropathol. 2004, 107, 381–388. [Google Scholar] [CrossRef]
- Schramm, J.; Luyken, C.; Urbach, H.; Fimmers, R.; Blumcke, I. Evidence for a clinically distinct new subtype of grade II astrocytomas in patients with long-term epilepsy. Neurosurgery 2004, 55, 340–347; discussion 347–348. [Google Scholar] [CrossRef] [PubMed]
- Wefers, A.K.; Stichel, D.; Schrimpf, D.; Coras, R.; Pages, M.; Tauziède-Espariat, A.; Varlet, P.; Schwarz, D.; Söylemezoglu, F.; Pohl, U.; et al. Isomorphic diffuse glioma is a morphologically and molecularly distinct tumour entity with recurrent gene fusions of MYBL1 or MYB and a benign disease course. Acta Neuropathol. 2020, 139, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Rammos, S.; Maina, R.; Lanzino, G. Developmental venous anomalies: Current concepts and implications for management. Neurosurgery 2009, 65, 20–29; discussion 29–30. [Google Scholar] [CrossRef] [PubMed]
- Bertalanffy, H.; Benes, L.; Miyazawa, T.; Alberti, O.; Siegel, A.; Sure, U. Cerebral cavernomas in the adult. Review of the literature and analysis of 72 surgically treated patients. Neurosurg Rev. 2002, 25, 1–53; discussion 54–55. [Google Scholar] [CrossRef] [PubMed]
- Costantino, A.; Vinters, H. A pathogenic correlate of the ‘steal’ phenomenon in a patient with cerebral arteriovenous malformation. Stroke 1986, 17, 103–106. [Google Scholar] [CrossRef]
- Scaravilli, F. Neuropathology of Epilepsy; World Scientific Publishing Co. Pte. Ltd.: Singapore; Hackensack, NJ, USA; London, UK; Hong Kong, China, 1998. [Google Scholar]
- Kossoff, E.; Hatfield, L.; Ball, K.; Comi, A. Comorbidity of epilepsy and headache in patients with Sturge-Weber syndrome. J. Child. Neurol. 2005, 20, 678–682. [Google Scholar]
- Aizpuru, R.N.; Quencer, R.M.; Norenberg, M.; Altman, N.; Smirniotopoulos, J. Meningioangiomatosis: Clinical, radiologic, and histopathologic correlation. Radiology 1991, 179, 819–821. [Google Scholar] [CrossRef]
- Kim, N.R.; Cho, S.J.; Suh, Y.L. Allelic loss on chromosomes 1p32, 9p21, 13q14, 16q22, 17p, and 22q12 in meningiomas associated with meningioangiomatosis and pure meningioangiomatosis. J. Neurooncol. 2009, 94, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, Y.; Amatya, V.J.; Nakayori, F.; Nakano, T.; Sugiyama, K.; Inai, K. Meningioangiomatosis occurring in a young male without neurofibromatosis: With special reference to its histogenesis and loss of heterozygosity in the NF2 gene region. Am. J. Surg. Pathol. 2002, 26, 125–129. [Google Scholar] [CrossRef]
- Cui, H.; Shi, H.; Chen, X.; Wang, W.; Lai, R.; Han, A. Clinicopathological features of meningioangiomatosis associated with meningioma: A case report with literature review. Case Rep Oncol Med. 2012, 2012, 296286. [Google Scholar] [CrossRef]
- Kim, N.R.; Choe, G.; Shin, S.H.; Wang, K.C.; Cho, B.K.; Choi, K.S.; Chi, J.G. Childhood meningiomas associated with meningioangiomatosis: Report of five cases and literature review. Neuropathol. Appl. Neurobiol. 2002, 28, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Jamil, O.; Ramkissoon, S.; Folkerth, R.; Smith, E. Multifocal meningioangiomatosis in a 3-year-old patient. J. Neurosurg. Pediatr. 2012, 10, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Miyata, H.; Kuwashige, H.; Hori, T.; Kubota, Y.; Pieper, T.; Coras, R.; Blumcke, I.; Yoshida, Y. Variable histopathology features of neuronal dyslamination in the cerebral neocortex adjacent to epilepsy-associated vascular malformations suggest complex pathogenesis of focal cortical dysplasia ILAE type IIIc. Brain Pathol. 2022, 32, e13052. [Google Scholar] [CrossRef]
- Bauer, J.; Vezzani, A.; Bien, C. Epileptic encephalitis: The role of the innate and adaptive immune system. Brain Pathol. 2012, 22, 412–421. [Google Scholar] [CrossRef]
- Bien, C.; Bauer, J.; Deckwerth, T.; Wiendl, H.; Deckert, M.; Wiestler, O.; Schramm, J.; Elger, C.; Lassmann, H. Destruction of neurons by cytotoxic T cells: A new pathogenic mechanisms in Rasmussen’s encephalitis. Ann. Neurol. 2002, 51, 311–318. [Google Scholar] [CrossRef]
- Marosso, M.; Balosso, S.; Ravizza, T.; Liu, J.; Aronica, E.; Iyer, A.; Rossetti, C.; Molteni, M.; Casalgrandi, M.; Manfredi, A.; et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 2010, 16, 413–419. [Google Scholar] [CrossRef]
- Rasmussen, T.; Olszewski, J.; Lloyd-Smith, D. Focal seizures due to chronic localized encephalitis. Neurology 1958, 8, 435–445. [Google Scholar] [CrossRef]
- Rogers, S.; Andrews, P.; Gahring, L.; Whisenand, T.; Cauley, K.; Crain, B.; Hughes, T.; Heinemann, S.; McNamara, J. Autoantibodies to glutamate receptor Glu3 in Rasmussen’s encephalitis. Science 1994, 265, 648–651. [Google Scholar] [CrossRef] [PubMed]
- Whitney, K.; Andrews, J.; McNamara, J. Immunoglobulin G and complement immunoreactivity in the cerebral cortex of patients with Rasmussen’s encephalitis. Neurology 1999, 53, 699–708. [Google Scholar] [CrossRef]
- Bien, C.; Elger, C.; Leitner, Y.; Gomori, M.; Ran, B.; Urbach, H.; Wilken, B.; Korn-Lubetzki, I. Slowly progressive hemiparesis in childhood as a consequence of Rasmussen encephalitis without or with delayed-onset seizures. Eur. J. Neurol. 2007, 14, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Korn-Lubetzki, I.; Bien, C.; Bauer, J.; Gomori, M.; Wiendl, H.; Trajo, L.; Ovadia, H.; Wilken, B.; Hans, V.; Elger, C.; et al. Rasmussen encephalitis with active inflammation and delayed seizures onset. Neurology 2004, 62, 984–986. [Google Scholar] [CrossRef]
- Graus, F.; Ribalta, T.; Campo, E.; Monforte, R.; Urbano, A.; Rozman, C. Immunohistochemical analysis of the immune reaction in the nervous system in paraneoplastic encephalomyelitis. Neurology 1990, 40, 219–222. [Google Scholar] [CrossRef]
- Dalmau, J.O.; Posner, J.B. Paraneoplastic syndromes. Neurol. Clin. 1999, 9, 919–936. [Google Scholar] [CrossRef]
- Wang, D.D.; Piao, Y.S.; Blumcke, I.; Coras, R.; Zhou, W.; Gui, Q.; Liu, C.; Hu, J.; Cao, L.; Zhang, G.; et al. A distinct clinicopathological variant of focal cortical dysplasia IIId characterized by loss of layer 4 in the occipital lobe in 12 children with remote hypoxic-ischemic injury. Epilepsia 2017, 58, 1697–1705. [Google Scholar] [CrossRef]
- Wang, D.; Blumcke, I.; Gui, Q.; Zhou, W.; Zuo, H.; Lin, J.; Luo, Y. Clinico-pathological investigations of Rasmussen encephalitis suggest multifocal disease progression and associated focal cortical dysplasia. Epileptic. Disord. 2013, 15, 32–43. [Google Scholar] [CrossRef]
- Blumcke, I.; Coras, R.; Busch, R.M.; Morita-Sherman, M.; Lal, D.; Prayson, R.; Cendes, F.; Lopes-Cendes, I.; Rogerio, F.; Almeida, V.; et al. Toward a better definition of focal cortical dysplasia: An iterative histopathological and genetic agreement trial. Epilepsia 2021, 62, 1416–1428. [Google Scholar] [CrossRef]
- Bonduelle, T.; Hartlieb, T.; Baldassari, S.; Sim, N.; Kim, S.; Kang, H.; Kobow, K.; Coras, R.; Chipaux, M.; Dorfmüller, G.; et al. Frequent SLC35A2 brain mosaicism in mild malformation of cortical development with oligodendroglial hyperplasia and epilepsy (MOGHE). Acta Neuropathol. Commun. 2021, 9, 3. [Google Scholar] [CrossRef]
- Bernasconi, A.; Cendes, F.; Theodore, W.; Gill, R.; Koepp, M.; Hogan, R.; Jackson, G.; Federico, P.; Labate, A.; Vaudanoet, A.; et al. Recommendations for the use of structural magnetic resonance imaging in the care of patients with epilepsy: A consensus report from the International League Against Epilepsy Neuroimaging Task Force. Epilepsia 2019, 60, 1054–1068. [Google Scholar] [CrossRef] [PubMed]
- Berkovic, S.; Jackson, G. ‘Idiopathic’ no more! Abnormal interaction of large-scale brain networks in generalized epilepsy. Brain 2014, 137, 2400–2402. [Google Scholar] [CrossRef] [PubMed]
- Opheim, G.; van der Kolk, A.; Bloch, K.; Colon, A.; Davis, K.; Henry, T.; Jansen, J.; Jones, S.; Pan, J.; Rössler, K.; et al. 7T Epilepsy Task Force Consensus Recommendations on the Use of 7T MRI in Clinical Practice. Neurology 2021, 96, 327–341. [Google Scholar] [CrossRef]
- Wang, Z.; Oh, S.; Lowe, M.; Larvie, M.; Ruggieri, P.; Hill, V.; Statsevych, V.; Moon, D.; Lee, J.; Emch, T.; et al. Radiological and Clinical Value of 7T MRI for Evaluating 3T-Visible Lesions in Pharmacoresistant Focal Epilepsies. Front. Neurol. 2021, 12, 591586. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Metodiev, D.; Minkin, K.; Ruseva, M.; Ganeva, R.; Parvanov, D.; Nachev, S. Pathomorphological Diagnostic Criteria for Focal Cortical Dysplasias and Other Common Epileptogenic Lesions—Review of the Literature. Diagnostics 2023, 13, 1311. https://doi.org/10.3390/diagnostics13071311
Metodiev D, Minkin K, Ruseva M, Ganeva R, Parvanov D, Nachev S. Pathomorphological Diagnostic Criteria for Focal Cortical Dysplasias and Other Common Epileptogenic Lesions—Review of the Literature. Diagnostics. 2023; 13(7):1311. https://doi.org/10.3390/diagnostics13071311
Chicago/Turabian StyleMetodiev, Dimitar, Krassimir Minkin, Margarita Ruseva, Rumiana Ganeva, Dimitar Parvanov, and Sevdalin Nachev. 2023. "Pathomorphological Diagnostic Criteria for Focal Cortical Dysplasias and Other Common Epileptogenic Lesions—Review of the Literature" Diagnostics 13, no. 7: 1311. https://doi.org/10.3390/diagnostics13071311
APA StyleMetodiev, D., Minkin, K., Ruseva, M., Ganeva, R., Parvanov, D., & Nachev, S. (2023). Pathomorphological Diagnostic Criteria for Focal Cortical Dysplasias and Other Common Epileptogenic Lesions—Review of the Literature. Diagnostics, 13(7), 1311. https://doi.org/10.3390/diagnostics13071311