
Accuracy of Non-Invasive Prenatal Testing for Duchenne Muscular Dystrophy in Families at Risk: A Systematic Review

Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Search Strategy
2.2. Eligibility Criteria
2.3. Screening Process and Critical Appraisal
2.4. Data Collection Process and Risk of Bias Assessment
3. Results
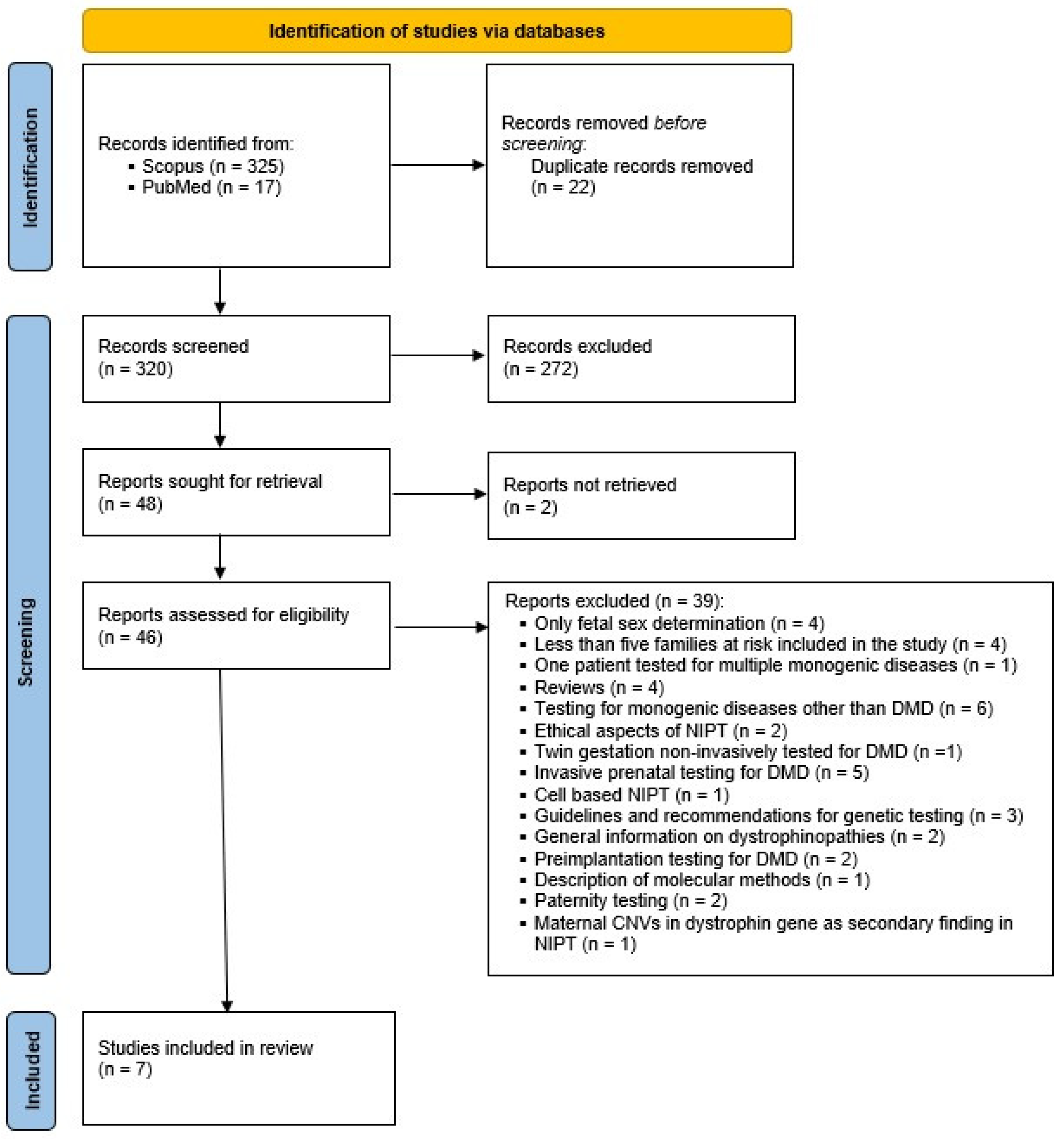
3.1. Search Results
3.2. Patient Characteristics and Acquisition of Samples
3.3. Types of Dystrophin Gene Mutations
3.4. Molecular Techniques Used for the Analysis
3.5. Confirmatory Testing
3.6. Study Outcomes
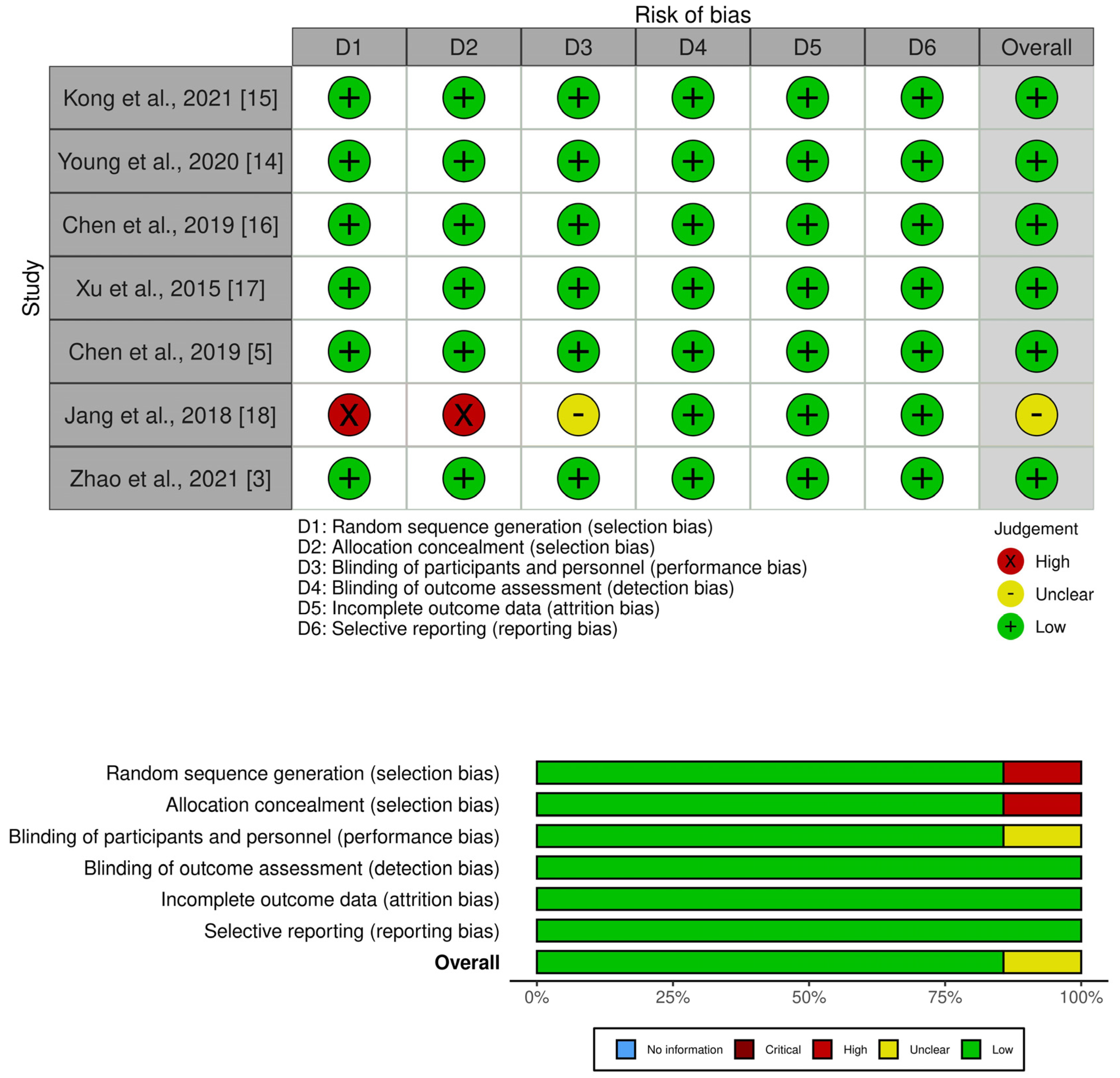
3.7. Risk of Bias in Primary Studies
3.8. Limiting Factors
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Allen, S.; Young, E.; Bowns, B. Noninvasive Prenatal Diagnosis for Single Gene Disorders. Curr. Opin. Obstet. Gynecol. 2017, 29, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.; Hill, M.; Chitty, L.S. Non-invasive prenatal diagnosis for single gene disorders: Experience of patients. Clin. Genet. 2014, 85, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Wang, X.; Liu, L.; Dai, P.; Kong, X. Noninvasive Prenatal Diagnosis of Duchenne Muscular Dystrophy in Five Chinese Families Based on Relative Mutation Dosage Approach. BMC Med. Genom. 2021, 14, 275. [Google Scholar] [CrossRef] [PubMed]
- Muscular Dystrophy, Duchenne Type; DMD. Available online: https://www.omim.org/entry/310200 (accessed on 24 November 2022).
- Chen, M.; Chen, C.; Huang, X.; Sun, J.; Jiang, L.; Li, Y.; Zhu, Y.; Tian, C.; Li, Y.; Lu, Z.; et al. Noninvasive Prenatal Diagnosis for Duchenne Muscular Dystrophy Based on the Direct Haplotype Phasing. Prenat. Diagn. 2020, 40, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Parks, M.; Court, S.; Bowns, B.; Cleary, S.; Clokie, S.; Hewitt, J.; Williams, D.; Cole, T.; Macdonald, F.; Griffiths, M.; et al. Non-Invasive Prenatal Diagnosis of Spinal Muscular Atrophy by Relative Haplotype Dosage. Eur. J. Hum. Genet. 2017, 25, 416–422. [Google Scholar] [CrossRef]
- Gerson, K.D.; O’Brien, B.M. Cell-Free DNA: Screening for Single-Gene Disorders and Determination of Fetal Rhesus D Genotype. Obstet. Gynecol. Clin. N. Am. 2018, 45, 27–39. [Google Scholar] [CrossRef]
- Chitty, L.S.; Mason, S.; Barrett, A.N.; Mckay, F.; Lench, N.; Daley, R.; Jenkins, L.A. Non-Invasive Prenatal Diagnosis of Achondroplasia and Thanatophoric Dysplasia: Next-Generation Sequencing Allows for a Safer, More Accurate, and Comprehensive Approach. Prenat. Diagn. 2015, 35, 656–662. [Google Scholar] [CrossRef]
- Korpi-Steiner, N.; Chiu, R.W.K.; Chandrasekharan, S.; Chitty, L.S.; Evans, M.I.; Jackson, J.A.; Palomaki, G.E. Emerging Considerations for Noninvasive Prenatal Testing. Clin. Chem. 2017, 63, 946–953. [Google Scholar] [CrossRef]
- Yoo, S.-K.; Lim, B.C.; Byeun, J.; Hwang, H.; Kim, K.J.; Hwang, Y.S.; Lee, J.; Park, J.S.; Lee, Y.-S.; Namkung, J.; et al. Noninvasive Prenatal Diagnosis of Duchenne Muscular Dystrophy: Comprehensive Genetic Diagnosis in Carrier, Proband, and Fetus. Clin. Chem. 2015, 61, 829–837. [Google Scholar] [CrossRef]
- Parks, M.; Court, S.; Cleary, S.; Clokie, S.; Hewitt, J.; Williams, D.; Cole, T.; Macdonald, F.; Griffiths, M.; Allen, S. Non-Invasive Prenatal Diagnosis of Duchenne and Becker Muscular Dystrophies by Relative Haplotype Dosage. Prenat. Diagn. 2016, 36, 312–320. [Google Scholar] [CrossRef]
- Jang, S.S.; Lim, B.C.; Yoo, S.-K.; Shin, J.-Y.; Seo, J.-S.; Hwang, D.; Yoo, K.-Y.; Chae, J.H.; Kim, J.-I. Development of a Common Platform for the Noninvasive Prenatal Diagnosis of X-Linked Diseases. Prenat. Diagn. 2018, 38, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lu, J.; Su, F.; Yang, J.; Ju, J.; Lin, Y.; Xu, J.; Qi, Y.; Hou, Y.; Wu, J.; et al. Non-Invasive Prenatal Diagnosis of Monogenic Disorders Through Bayesian- and Haplotype-Based Prediction of Fetal Genotype. Front. Genet. 2022, 13, 911369. [Google Scholar] [CrossRef] [PubMed]
- Young, E.; Bowns, B.; Gerrish, A.; Parks, M.; Court, S.; Clokie, S.; Mashayamombe-Wolfgarten, C.; Hewitt, J.; Williams, D.; Cole, T.; et al. Clinical Service Delivery of Noninvasive Prenatal Diagnosis by Relative Haplotype Dosage for Single-Gene Disorders. J. Mol. Diagn. 2020, 22, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Li, S.; Zhao, Z.; Feng, J.; Chen, G.; Liu, L.; Tang, W.; Li, S.; Li, F.; Han, X.; et al. Haplotype-Based Noninvasive Prenatal Diagnosis of 21 Families With Duchenne Muscular Dystrophy: Real-World Clinical Data in China. Front. Genet. 2021, 12, 791856. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chen, C.; Li, Y.; Yuan, Y.; Lai, Z.; Guo, F.; Wang, Y.; Huang, X.; Li, S.; Wu, R.; et al. Haplotype-Based Noninvasive Prenatal Diagnosis for Duchenne Muscular Dystrophy: A Pilot Study in South China. Eur. J. Obstet. Gynecol. Reprod. Biol. 2019, 240, 15–22. [Google Scholar] [CrossRef]
- Xu, Y.; Li, X.; Ge, H.-J.; Xiao, B.; Zhang, Y.-Y.; Ying, X.-M.; Pan, X.-Y.; Wang, L.; Xie, W.-W.; Ni, L.; et al. Haplotype-Based Approach for Noninvasive Prenatal Tests of Duchenne Muscular Dystrophy Using Cell-Free Fetal DNA in Maternal Plasma. Genet. Med. 2015, 17, 889–896. [Google Scholar] [CrossRef]
- Jang, S.S.; Lim, B.C.; Yoo, S.-K.; Shin, J.-Y.; Kim, K.-J.; Seo, J.-S.; Kim, J.-I.; Chae, J.H. Targeted Linked-Read Sequencing for Direct Haplotype Phasing of Maternal DMD Alleles: A Practical and Reliable Method for Noninvasive Prenatal Diagnosis. Sci. Rep. 2018, 8, 8678. [Google Scholar] [CrossRef]
- Dystrophin; DMD. Available online: https://www.omim.org/entry/300377 (accessed on 28 November 2022).
- Barrett, A.N.; Chitty, L.S. Developing Noninvasive Diagnosis for Single-Gene Disorders: The Role of Digital PCR. Methods Mol. Biol. 2014, 1160, 215–228. [Google Scholar]
- New, M.I.; Tong, Y.K.; Yuen, T.; Jiang, P.; Pina, C.; Chan, K.C.A.; Khattab, A.; Liao, G.J.W.; Yau, M.; Kim, S.M.; et al. Noninvasive Prenatal Diagnosis of Congenital Adrenal Hyperplasia Using Cell-Free Fetal DNA in Maternal Plasma. J. Clin. Endocrinol. Metab. 2014, 99, 1022–1030. [Google Scholar] [CrossRef]
- Lo, Y.M.D.; Chan, K.C.A.; Sun, H.; Chen, E.Z.; Jiang, P.; Lun, F.M.F.; Zheng, Y.W.; Leung, T.Y.; Lau, T.K.; Cantor, C.R.; et al. Maternal Plasma DNA Sequencing Reveals the Genome-Wide Genetic and Mutational Profile of the Fetus. Sci. Transl. Med. 2010, 2, 61ra91. [Google Scholar] [CrossRef]
- Verhoef, T.I.; Hill, M.; Drury, S.; Mason, S.; Jenkins, L.; Morris, S.; Chitty, L.S. Non-Invasive Prenatal Diagnosis (NIPD) for Single Gene Disorders: Cost Analysis of NIPD and Invasive Testing Pathways. Prenat. Diagn. 2016, 36, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Papasavva, T.; van Ijcken, W.F.J.; Kockx, C.E.M.; van den Hout, M.C.G.N.; Kountouris, P.; Kythreotis, L.; Kalogirou, E.; Grosveld, F.G.; Kleanthous, M. Next Generation Sequencing of SNPs for Non-Invasive Prenatal Diagnosis: Challenges and Feasibility as Illustrated by an Application to β-Thalassaemia. Eur. J. Hum. Genet. 2013, 21, 1403–1410. [Google Scholar] [CrossRef] [PubMed]
- Chiu, E.K.L.; Hui, W.W.I.; Chiu, R.W.K. CfDNA Screening and Diagnosis of Monogenic Disorders—Where Are We Heading? Prenat. Diagn. 2018, 38, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, L.A.; Deans, Z.C.; Lewis, C.; Allen, S. Delivering an Accredited Non-Invasive Prenatal Diagnosis Service for Monogenic Disorders and Recommendations for Best Practice. Prenat. Diagn. 2018, 38, 44–51. [Google Scholar] [CrossRef]
- Kong, L.; Li, S.; Zhao, Z.; Feng, J.; Liu, L.; Tang, W.; Zhang, H.; Wu, D.; Sun, L.; Kong, X. Noninvasive Prenatal Testing of Duchenne Muscular Dystrophy in a Twin Gestation. Prenat. Diagn. 2022, 42, 518–523. [Google Scholar] [CrossRef]
- Agarwal, K.; Alfirevic, Z. Pregnancy Loss after Chorionic Villus Sampling and Genetic Amniocentesis in Twin Pregnancies: A Systematic Review. Ultrasound Obstet. Gynecol. 2012, 40, 128–134. [Google Scholar] [CrossRef]
- Bakker, M.; Birnie, E.; Robles de Medina, P.; Sollie, K.M.; Pajkrt, E.; Bilardo, C.M. Total Pregnancy Loss after Chorionic Villus Sampling and Amniocentesis: A Cohort Study. Ultrasound Obstet. Gynecol. 2017, 49, 599–606. [Google Scholar] [CrossRef]
- Di Mascio, D.; Khalil, A.; Rizzo, G.; Buca, D.; Liberati, M.; Martellucci, C.A.; Flacco, M.E.; Manzoli, L.; D’Antonio, F. Risk of Fetal Loss Following Amniocentesis or Chorionic Villus Sampling in Twin Pregnancy: Systematic Review and Meta-Analysis. Ultrasound Obstet. Gynecol. 2020, 56, 647–655. [Google Scholar] [CrossRef]
- Lam, K.W.G.; Jiang, P.; Liao, G.J.W.; Chan, K.C.A.; Leung, T.Y.; Chiu, R.W.K.; Lo, Y.M.D. Noninvasive Prenatal Diagnosis of Monogenic Diseases by Targeted Massively Parallel Sequencing of Maternal Plasma: Application to β-Thalassemia. Clin. Chem. 2012, 58, 1467–1475. [Google Scholar] [CrossRef]
- Hudecova, I.; Jiang, P.; Davies, J.; Lo, Y.M.D.; Kadir, R.A.; Chiu, R.W.K. Noninvasive Detection of F8 Int22h-Related Inversions and Sequence Variants in Maternal Plasma of Hemophilia Carriers. Blood 2017, 130, 340–347. [Google Scholar] [CrossRef]
- Bermúdez-López, C.; García-de Teresa, B.; González-del Angel, A. Germinal Mosaicism in a Sample of Families with Duchenne/Becker Muscular Dystrophy with Partial Deletions in the DMD Gene. Genet. Test. Mol. Biomark. 2014, 18, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Oudet, C.; Hanauer, A.; Clemens, P.; Caskey, T.; Mandel, J.L. Two Hot Spots of Recombination in the DMD Gene Correlate with the Deletion Prone Regions. Hum. Mol. Genet. 1992, 1, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Nobile, C.; Galvagni, F.; Marchi, J.; Roberts, R.; Vitiello, L. Genomic Organization of the Human Dystrophin Gene across the Major Deletion Hot Spot and the 3’ Region. Genomics 1995, 28, 97–100. [Google Scholar] [CrossRef]
- Ferreiro, V.; Giliberto, F.; Francipane, L.; Szijan, I. The Role of Polymorphic Short Tandem (CA)n Repeat Loci Segregation Analysis in the Detection of Duchenne Muscular Dystrophy Carriers and Prenatal Diagnosis. Mol. Diagn. 2005, 9, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Giliberto, F.; Ferreiro, V.; Massot, F.; Ferrer, M.; Francipane, L.; Szijan, I. Prenatal Diagnosis of Duchenne/Becker Muscular Dystrophy by Short Tandem Repeat Segregation Analysis in Argentine Families. Muscle Nerve 2011, 43, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Lench, N.; Barrett, A.; Fielding, S.; Mckay, F.; Hill, M.; Jenkins, L.; White, H.; Chitty, L.S. The Clinical Implementation of Non-Invasive Prenatal Diagnosis for Single-Gene Disorders: Challenges and Progress Made. Prenat. Diagn. 2013, 33, 555–562. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Study | Country | Type of Study | Number of Families at Risk | Blood Samples | Fetal Fraction | Gestational Weeks | Molecular Method | Number of Informative SNPs | Coverage | Recombination Event Estimation | Confirmation Testing | Accuracy | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Maternal | Paternal | Proband’s | ||||||||||||
| Kong et al., 2021 [15] | China | Prospective cohort | 21 | Yes | When available | Yes | 1.87–11.68% | 7+3–18+0 | Targeted MPS, RHDO (indirect) | 1511 (DMD region) 203 (X chromosome) 213 (autosomes) | 98×–563× (gDNA), 165×–490× (cfDNA) | Yes | CVS, amniocentesis (Sanger sequencing, MLPA) | 100% (21/21) |
| Young et al., 2020 [14] | United Kingdom | Retrospective cohort | 30 | Yes | Yes | When available | approximately 1–18% | >8 | Targeted MPS, RHDO (indirect) | - | >200× | Yes | CVS, cord blood, POC (Sanger sequencing, MLPA) | 86.67% (26/30) |
| Chen et al., 2019 [16] | China | Prospective cohort | 17 | Yes | Yes | Yes | 3.61–16.97% | 11+1–26+6 | Targeted MPS, RHDO (indirect) | 3965 (flanking region of DMD) | 152× (gDNA), 248× (cfDNA) | Yes | CVS, amniocentesis (targeted capture sequencing, MLPA) | 100% (17/17) |
| Xu et al., 2015 [17] | China | Prospective cohort | 8 | Yes | Yes | Yes | 3.52–22.67% | 17–22 | Targeted MPS, RHDO (indirect) | 1243 (DMD region) | 20× (gDNA) | Yes | Amniocentesis (microsatellites-based linkage analysis) | 100% (8/8) |
| Chen et al., 2019 [5] | China | Prospective cohort | 13 | Yes | No | No | 6.20–18.50% | 11+2–25+4 | Targeted linked-read sequencing, RHDO (direct) | 2261 (DMD region) 1704 (flanking region of DMD) | 329×–697× (maternal gDNA) | Yes | CVS (targeted capture sequencing) | 100% (13/13) |
| Jang et al., 2018 [18] | South Korea | Retrospective cohort | 5 | Yes | No | Yes | 4.10–9.25% | 6+5–17+1 | Targeted linked-read sequencing, RHDO (direct) | 700–1000 (DMD region) | 692× (maternal gDNA) | Yes | CVS, amniocentesis (Sanger sequencing) | 100% (5/5) |
| Zhao et al., 2021 [3] | China | Prospective cohort | 5 | Yes | No | Yes | 3.00–14.70% | 11–12 | RMD—based cfBEST | 109 | 695–3476 total unique reads | no | CVS (Sanger sequencing, MLPA) | 100% (5/5) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaninović, L.; Bašković, M.; Ježek, D.; Katušić Bojanac, A. Accuracy of Non-Invasive Prenatal Testing for Duchenne Muscular Dystrophy in Families at Risk: A Systematic Review. Diagnostics 2023, 13, 183. https://doi.org/10.3390/diagnostics13020183
Zaninović L, Bašković M, Ježek D, Katušić Bojanac A. Accuracy of Non-Invasive Prenatal Testing for Duchenne Muscular Dystrophy in Families at Risk: A Systematic Review. Diagnostics. 2023; 13(2):183. https://doi.org/10.3390/diagnostics13020183
Chicago/Turabian StyleZaninović, Luca, Marko Bašković, Davor Ježek, and Ana Katušić Bojanac. 2023. "Accuracy of Non-Invasive Prenatal Testing for Duchenne Muscular Dystrophy in Families at Risk: A Systematic Review" Diagnostics 13, no. 2: 183. https://doi.org/10.3390/diagnostics13020183
APA StyleZaninović, L., Bašković, M., Ježek, D., & Katušić Bojanac, A. (2023). Accuracy of Non-Invasive Prenatal Testing for Duchenne Muscular Dystrophy in Families at Risk: A Systematic Review. Diagnostics, 13(2), 183. https://doi.org/10.3390/diagnostics13020183