
Insights into the Value of Lyso-Gb1 as a Predictive Biomarker in Treatment-Naïve Patients with Gaucher Disease Type 1 in the LYSO-PROOF Study
 , , ,
, , ,  , ,
, ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Analysis
2.2. Statistical Analysis
3. Results
3.1. Study Cohort
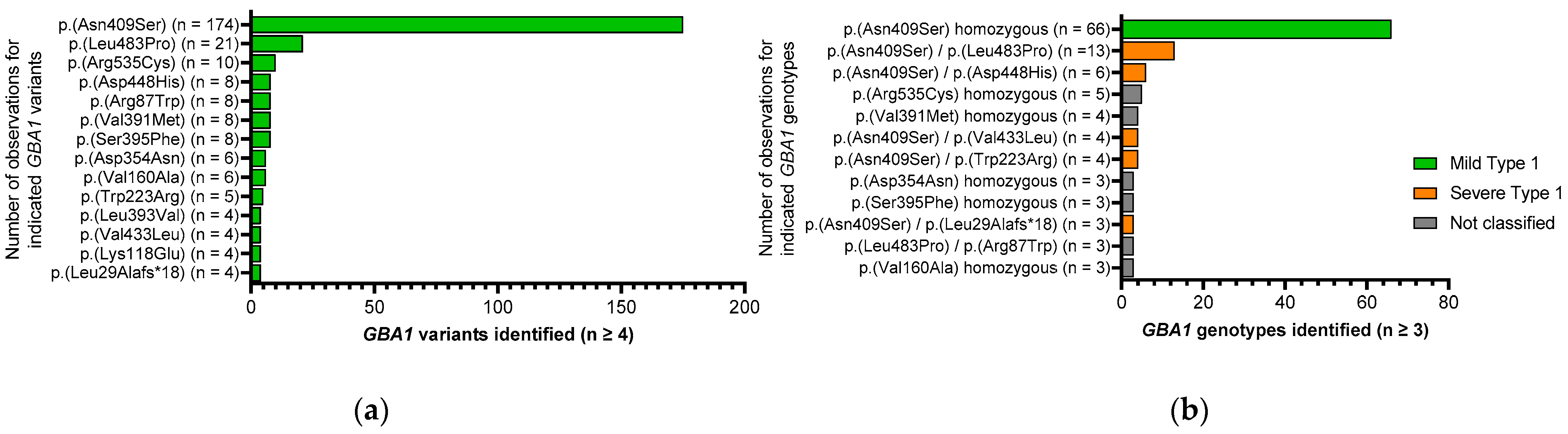
3.2. Genetic Characterization of the Cohort
3.3. Distribution of Disease Onset for Several Genotypes
3.4. Presence of Clinical Symptoms
3.5. Lyso-Gb1 Levels in GD Patients
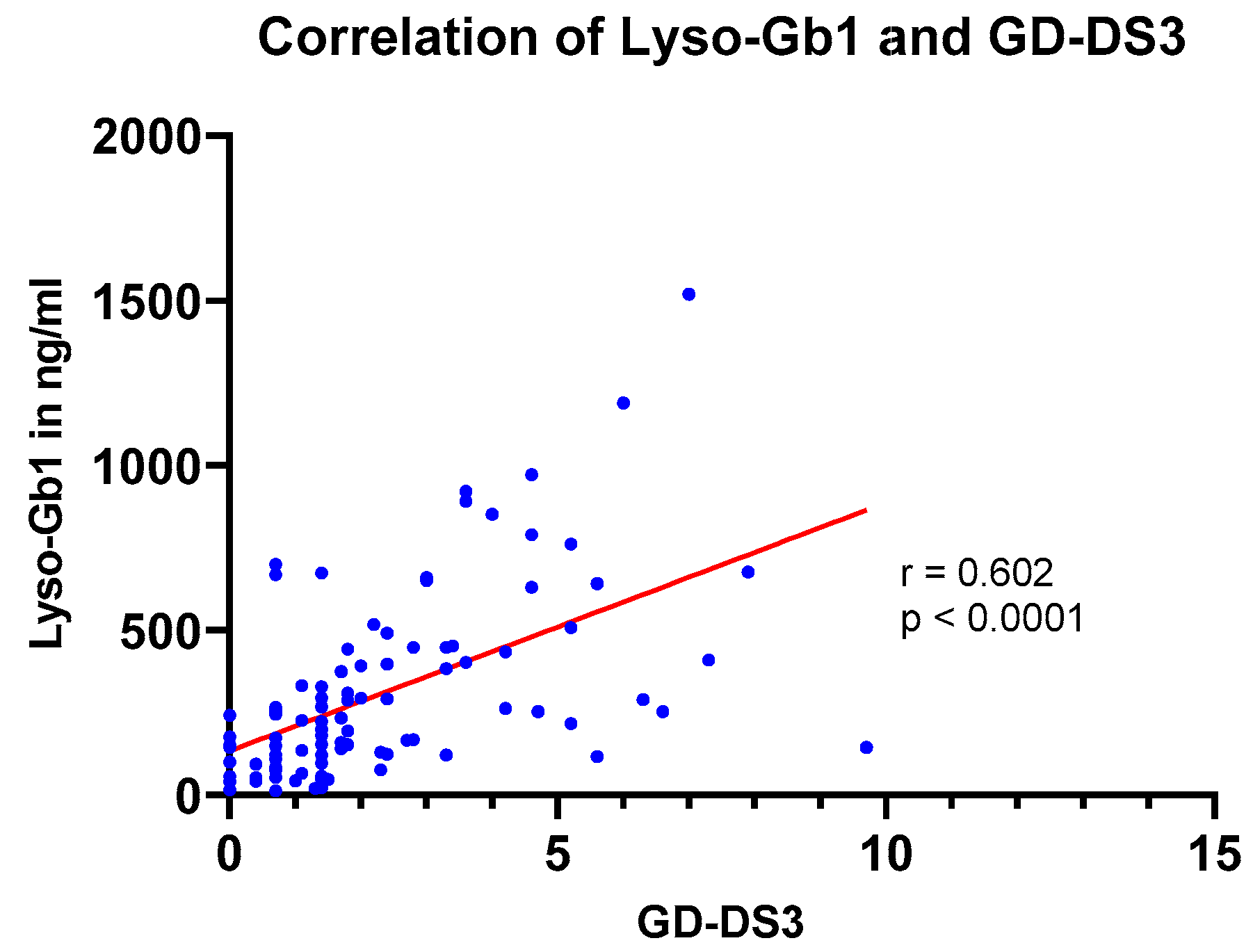
3.6. Correlation of lyso-Gb1 with Disease Severity (GD-DS3)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marques, A.R.A.; Saftig, P. Lysosomal storage disorders—Challenges, concepts and avenues for therapy: Beyond rare diseases. J. Cell Sci. 2019, 132, jcs221739. [Google Scholar] [CrossRef] [PubMed]
- Pastores, G.M.; Hughes, D.A. Gaucher Disease. In GeneReviews((R)); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; 2000 July (updated 2023 March); University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Zuckerman, S.; Lahad, A.; Shmueli, A.; Zimran, A.; Peleg, L.; Orr-Urtreger, A.; Levy-Lahad, E.; Sagi, M. Carrier screening for Gaucher disease: Lessons for low-penetrance, treatable diseases. JAMA 2007, 298, 1281–1290. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sidransky, E. Gaucher disease: Complexity in a “simple” disorder. Mol. Genet. Metab. 2004, 83, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, N.J.; Cappellini, M.D.; Cox, T.M.; Giannini, E.H.; Grabowski, G.A.; Hwu, W.L.; Mankin, H.; Martins, A.M.; Sawyer, C.; vom Dahl, S.; et al. A validated disease severity scoring system for adults with type 1 Gaucher disease. Genet. Med. 2010, 12, 44–51. [Google Scholar] [CrossRef]
- Weinreb, N.J.; Finegold, D.N.; Feingold, E.; Zeng, Z.; Rosenbloom, B.E.; Shankar, S.P.; Amato, D. Evaluation of disease burden and response to treatment in adults with type 1 Gaucher disease using a validated disease severity scoring system (DS3). Orphanet J. Rare Dis. 2015, 10, 64. [Google Scholar] [CrossRef]
- Ganz, M.L.; Stern, S.; Ward, A.; Nalysnyk, L.; Selzer, M.; Hamed, A.; Weinreb, N. A new framework for evaluating the health impacts of treatment for Gaucher disease type 1. Orphanet J. Rare Dis. 2017, 12, 38. [Google Scholar] [CrossRef]
- Mistry, P.K.; Lopez, G.; Schiffmann, R.; Barton, N.W.; Weinreb, N.J.; Sidransky, E. Gaucher disease: Progress and ongoing challenges. Mol. Genet. Metab. 2017, 120, 8–21. [Google Scholar] [CrossRef]
- Revel-Vilk, S.; Fuller, M.; Zimran, A. Value of Glucosylsphingosine (Lyso-Gb1) as a Biomarker in Gaucher Disease: A Systematic Literature Review. Int. J. Mol. Sci. 2020, 21, 7159. [Google Scholar] [CrossRef]
- Ferraz, M.J.; Kallemeijn, W.W.; Mirzaian, M.; Herrera Moro, D.; Marques, A.; Wisse, P.; Boot, R.G.; Willems, L.I.; Overkleeft, H.S.; Aerts, J.M. Gaucher disease and Fabry disease: New markers and insights in pathophysiology for two distinct glycosphingolipidoses. Biochim. Biophys. Acta 2014, 1841, 811–825. [Google Scholar] [CrossRef]
- Cabrera-Salazar, M.A.; O’Rourke, E.; Henderson, N.; Wessel, H.; Barranger, J.A. Correlation of surrogate markers of Gaucher disease. Implications for long-term follow up of enzyme replacement therapy. Clin. Chim. Acta 2004, 344, 101–107. [Google Scholar] [CrossRef]
- Koppe, T.; Doneda, D.; Siebert, M.; Paskulin, L.; Camargo, M.; Tirelli, K.M.; Vairo, F.; Daudt, L.; Schwartz, I.V. The prognostic value of the serum ferritin in a southern Brazilian cohort of patients with Gaucher disease. Genet. Mol. Biol. 2016, 39, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Hollak, C.E.; van Weely, S.; van Oers, M.H.; Aerts, J.M. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J. Clin. Investig. 1994, 93, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Schutyser, E.; Richmond, A.; Van Damme, J. Involvement of CC chemokine ligand 18 (CCL18) in normal and pathological processes. J. Leukoc. Biol. 2005, 78, 14–26. [Google Scholar] [CrossRef]
- Bussink, A.P.; Verhoek, M.; Vreede, J.; Ghauharali-van der Vlugt, K.; Donker-Koopman, W.E.; Sprenger, R.R.; Hollak, C.E.; Aerts, J.M.; Boot, R.G. Common G102S polymorphism in chitotriosidase differentially affects activity towards 4-methylumbelliferyl substrates. FEBS J. 2009, 276, 5678–5688. [Google Scholar] [CrossRef] [PubMed]
- Dekker, N.; van Dussen, L.; Hollak, C.E.; Overkleeft, H.; Scheij, S.; Ghauharali, K.; van Breemen, M.J.; Ferraz, M.J.; Groener, J.E.; Maas, M.; et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood 2011, 118, e118–e127. [Google Scholar] [CrossRef] [PubMed]
- Rolfs, A.; Giese, A.K.; Grittner, U.; Mascher, D.; Elstein, D.; Zimran, A.; Bottcher, T.; Lukas, J.; Hubner, R.; Golnitz, U.; et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS ONE 2013, 8, e79732. [Google Scholar] [CrossRef] [PubMed]
- Smid, B.E.; Ferraz, M.J.; Verhoek, M.; Mirzaian, M.; Wisse, P.; Overkleeft, H.S.; Hollak, C.E.; Aerts, J.M. Biochemical response to substrate reduction therapy versus enzyme replacement therapy in Gaucher disease type 1 patients. Orphanet J. Rare Dis. 2016, 11, 28. [Google Scholar] [CrossRef]
- Murugesan, V.; Chuang, W.L.; Liu, J.; Lischuk, A.; Kacena, K.; Lin, H.; Pastores, G.M.; Yang, R.; Keutzer, J.; Zhang, K.; et al. Glucosylsphingosine is a key biomarker of Gaucher disease. Am. J. Hematol. 2016, 91, 1082–1089. [Google Scholar] [CrossRef]
- Elstein, D.; Mellgard, B.; Dinh, Q.; Lan, L.; Qiu, Y.; Cozma, C.; Eichler, S.; Bottcher, T.; Zimran, A. Reductions in glucosylsphingosine (lyso-Gb1) in treatment-naive and previously treated patients receiving velaglucerase alfa for type 1 Gaucher disease: Data from phase 3 clinical trials. Mol. Genet. Metab. 2017, 122, 113–120. [Google Scholar] [CrossRef]
- Arkadir, D.; Dinur, T.; Revel-Vilk, S.; Becker Cohen, M.; Cozma, C.; Hovakimyan, M.; Eichler, S.; Rolfs, A.; Zimran, A. Glucosylsphingosine is a reliable response biomarker in Gaucher disease. Am. J. Hematol. 2018, 93, E140–E142. [Google Scholar] [CrossRef]
- Kim, Y.M.; Yum, M.S.; Heo, S.H.; Kim, T.; Jin, H.K.; Bae, J.S.; Seo, G.H.; Oh, A.; Yoon, H.M.; Lim, H.T.; et al. Pharmacologic properties of high-dose ambroxol in four patients with Gaucher disease and myoclonic epilepsy. J. Med. Genet. 2020, 57, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Cozma, C.; Cullufi, P.; Kramp, G.; Hovakimyan, M.; Velmishi, V.; Gjikopulli, A.; Tomori, S.; Fischer, S.; Oppermann, S.; Grittner, U.; et al. Treatment Efficiency in Gaucher Patients Can Reliably Be Monitored by Quantification of Lyso-Gb1 Concentrations in Dried Blood Spots. Int. J. Mol. Sci. 2020, 21, 4577. [Google Scholar] [CrossRef]
- Dinur, T.; Bauer, P.; Beetz, C.; Kramp, G.; Cozma, C.; Iurascu, M.I.; Becker-Cohen, M.; Istaiti, M.; Rolfs, A.; Zimran, A.; et al. Gaucher Disease Diagnosis Using Lyso-Gb1 on Dry Blood Spot Samples: Time to Change the Paradigm? Int. J. Mol. Sci. 2022, 23, 1627. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A.; Zimran, A.; Ida, H. Gaucher disease types 1 and 3: Phenotypic characterization of large populations from the ICGG Gaucher Registry. Am. J. Hematol. 2015, 90 (Suppl. S1), S12–S18. [Google Scholar] [CrossRef] [PubMed]
- Mozafari, H.; Tghikhani, M.; Rahimi, Z.; Vaisi Raygani, A.; Ansari, S.; Khatami, S.; Alaei, M.R.; Saghiri, R. Analysis of glucocerebrosidase (GBA) gene mutations in Iranian patients with Gaucher disease. Iran. J. Child Neurol. 2021, 15, 139–166. [Google Scholar] [CrossRef]
- Park, J.K.; Orvisky, E.; Tayebi, N.; Kaneski, C.; Lamarca, M.E.; Stubblefield, B.K.; Martin, B.M.; Schiffmann, R.; Sidransky, E. Myoclonic epilepsy in Gaucher disease: Genotype-phenotype insights from a rare patient subgroup. Pediatr. Res. 2003, 53, 387–395. [Google Scholar] [CrossRef]
- Pastores, G.M.; Patel, M.J.; Firooznia, H. Bone and joint complications related to Gaucher disease. Curr. Rheumatol. Rep. 2000, 2, 175–180. [Google Scholar] [CrossRef]
- Balwani, M.; Fuerstman, L.; Kornreich, R.; Edelmann, L.; Desnick, R.J. Type 1 Gaucher disease: Significant disease manifestations in “asymptomatic” homozygotes. Arch. Intern. Med. 2010, 170, 1463–1469. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
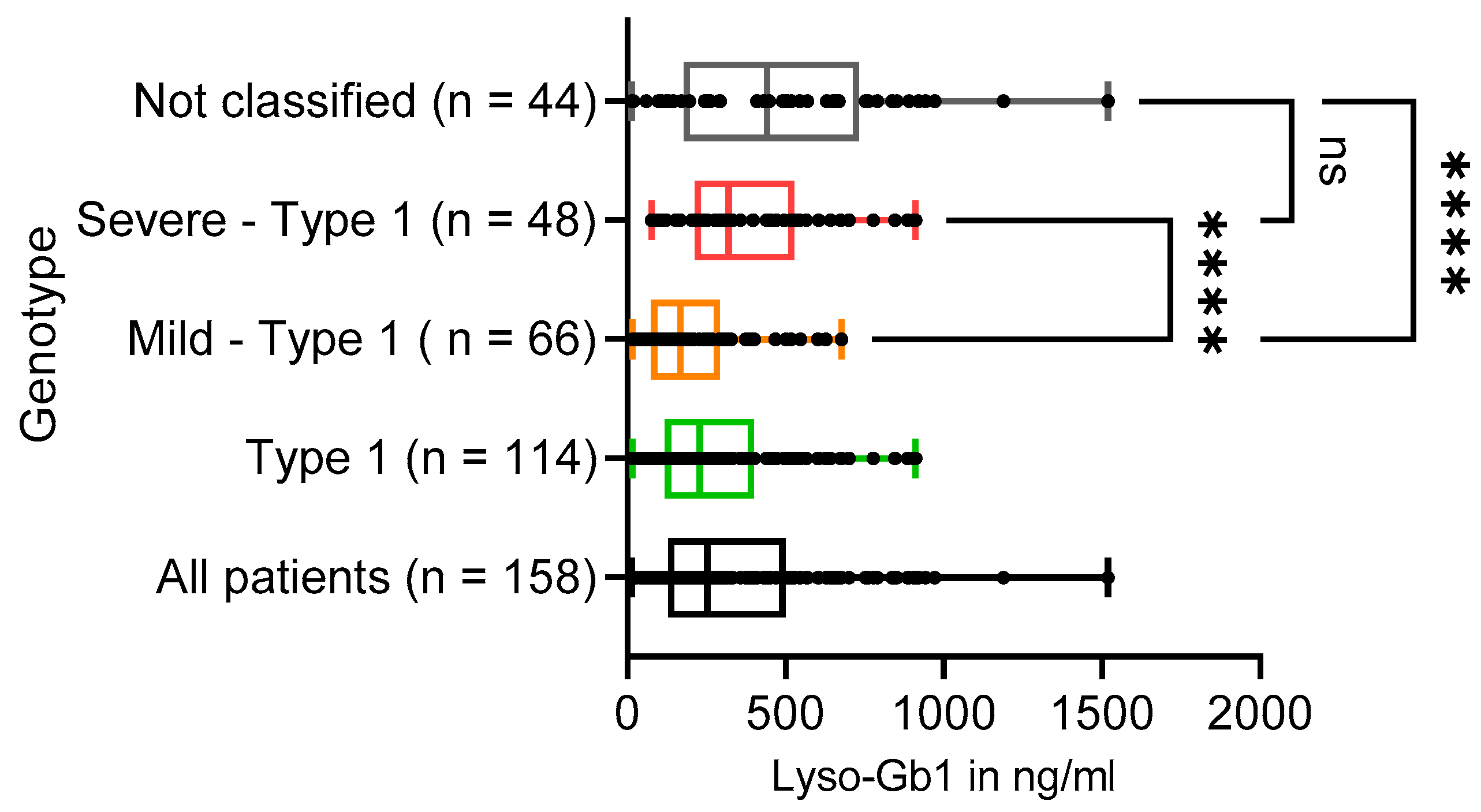{kind=link}
{kind=link}
| Parameter | All Patients N = 160 | Type 1 N = 114 | Mild—Type 1 N = 66 | Severe—Type 1 N = 48 | Not Classified N = 46 |
|---|---|---|---|---|---|
| Gender, n (%) | |||||
| Male | 78 (49%) | 61 (51%) | 29 (44%) | 30 (62%) | 19 (41%) |
| Female | 82 (51%) | 58 (49%) | 37 (56%) | 18 (38%) | 27 (59%) |
| Age at diagnosis (years) | |||||
| Mean (SD) | 23 (17) | 27 (17) | 29 (16) | 26 (18) | 10 (12) |
| Median [IQR] [range] | 20 [6–35] [1–81] | 25 [15–39] [2–81] | 26 [18–39] [2–81] | 24 [8–40] [2–75] | 6 [2–13] [1–49] |
| Distribution by country, n | |||||
| Israel (2) | 55 | 53 | 46 | 7 | 2 |
| Russia (1) | 26 | 25 | 10 | 15 | 1 |
| Pakistan (1) | 24 | 1 | - | 1 | 23 |
| Egypt (1) | 12 | 7 | 1 | 6 | 5 |
| Iran (1) | 12 | 6 | 3 | 3 | 6 |
| Morocco (2) | 9 | 7 | 1 | 6 | 2 |
| Algeria (4) | 8 | 5 | 3 | 2 | 3 |
| India (3) | 4 | - | - | - | 4 |
| Spain (2) | 3 | 3 | - | 3 | - |
| Albania (1) | 3 | 3 | - | 3 | - |
| Greece (1) | 1 | 1 | - | 1 | - |
| Sweden (1) | 1 | 1 | 1 | - | - |
| Colombia (1) | 1 | 1 | 1 | - | - |
| Tunisia (1) | 1 | 1 | - | 1 | - |
| Condition | Present | ||||
|---|---|---|---|---|---|
| Clinical Symptoms | All Patients N = 160 | Type 1 N = 114 | Mild Type 1 N = 66 | Severe Type 1 N = 48 | Not Classified N = 46 |
| (%) N present/N total | (%) N present/N total | (%) N present/N total | (%) N present/N total | (%) N present/N total | |
| Splenomegaly | (75.0) 117/156 | (68.8) 77/112 | (54.5) 36/66 | (89.1) 41/46 | (90.9) 40/44 |
| Thrombocytopenia | (74.4) 116/156 | (74.1) 83/112 | (68.2) 45/66 | (82.6) 38/46 | (75.0) 33/44 |
| Hepatomegaly | (62.4) 98/157 | (53.6) 60/112 | (39.4) 26/66 | (73.9) 34/46 | (84.4) 38/45 |
| Anemia | (56.3) 89/158 | (53.1) 60/113 | (39.4) 26/66 | (72.3) 34/47 | (64.4) 29/45 |
| Gaucher cells in bone marrow | (75.8) 75/99 | (71.4) 45/63 | (59.4) 19/32 | (83.9) 26/31 | (83.3) 30/36 |
| Bone involvement | (43.1) 69/160 | (50.9) 58/114 | (39.4) 26/66 | (66.7) 32/48 | (23.9) 11/46 |
| All Patients N = 160 | Type 1 N = 114 | Mild—Type 1 N = 66 | Severe—Type 1 N = 48 | Not Classified N = 46 | |
|---|---|---|---|---|---|
| GD-DS3 | N = 96 | N = 66 | N = 48 | N = 18 | N = 30 |
| Mean (SD) | 2.34 (2.0) | 1.67 (1.5) | 1.35 (1.3) | 2.52 (1.7) | 3.81 (2.2) |
| Median [IQR] [range] | 1.7 [0.7–3.3] [0–9.7] | 1.4 [0.7–1.9] [0–7.9] | 1.4 [0.7–1.7] [0–7.9] | 2.1 [1.3–3.7] [0–5.6] | 3.6 [2.4–5.2] [0–9.7] |
| Lyso-Gb1 in ng/mL | N = 158 | N = 114 | N = 66 | N = 48 | N = 44 |
| Mean (SD) | 337.5 (270.2) | 283.2 (213.8) | 208.1 (162.1) | 386.5 (233.9) | 478.2 (344.1) |
| Median [IQR] [range] | 253 [129–499] [13–1520] | 230 [119–400] [16–911] | 167 [74–293] [16–677] | 320 [213–528] [76–911] | 442 [178–732] [13–1520] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curado, F.; Rösner, S.; Zielke, S.; Westphal, G.; Grittner, U.; Skrahina, V.; Alasel, M.; Malik, A.M.; Beetz, C.; Böttcher, T.; et al. Insights into the Value of Lyso-Gb1 as a Predictive Biomarker in Treatment-Naïve Patients with Gaucher Disease Type 1 in the LYSO-PROOF Study. Diagnostics 2023, 13, 2812. https://doi.org/10.3390/diagnostics13172812
Curado F, Rösner S, Zielke S, Westphal G, Grittner U, Skrahina V, Alasel M, Malik AM, Beetz C, Böttcher T, et al. Insights into the Value of Lyso-Gb1 as a Predictive Biomarker in Treatment-Naïve Patients with Gaucher Disease Type 1 in the LYSO-PROOF Study. Diagnostics. 2023; 13(17):2812. https://doi.org/10.3390/diagnostics13172812
Chicago/Turabian StyleCurado, Filipa, Sabine Rösner, Susanne Zielke, Gina Westphal, Ulrike Grittner, Volha Skrahina, Mohammed Alasel, Ahmad Mehmood Malik, Christian Beetz, Tobias Böttcher, and et al. 2023. "Insights into the Value of Lyso-Gb1 as a Predictive Biomarker in Treatment-Naïve Patients with Gaucher Disease Type 1 in the LYSO-PROOF Study" Diagnostics 13, no. 17: 2812. https://doi.org/10.3390/diagnostics13172812
APA StyleCurado, F., Rösner, S., Zielke, S., Westphal, G., Grittner, U., Skrahina, V., Alasel, M., Malik, A. M., Beetz, C., Böttcher, T., Barel, G., Sah, A. P., Dinur, T., Anjum, N., Ichraf, Q., Kriouile, Y., Hadipour, Z., Hadipour, F., Revel-Vilk, S., ... Rolfs, A., on behalf of the LYSO-PROOF Study Group. (2023). Insights into the Value of Lyso-Gb1 as a Predictive Biomarker in Treatment-Naïve Patients with Gaucher Disease Type 1 in the LYSO-PROOF Study. Diagnostics, 13(17), 2812. https://doi.org/10.3390/diagnostics13172812