
De Novo Large Deletions in the PHEX Gene Caused X-Linked Hypophosphataemic Rickets in Two Italian Female Infants Successfully Treated with Burosumab

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Probands’ Clinical Features
3.2. Molecular Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burton, A.; Castaño, A.; Bruno, M.; Riley, S.; Schumacher, J.; Sultan, M.B.; See Tai, S.; Judge, D.P.; Patel, J.K.; Kelly, J.W. Drug Discovery and Development in Rare Diseases: Taking a Closer Look at the Tafamidis Story. Drug Des. Devel Ther. 2021, 15, 1225–1243. [Google Scholar] [CrossRef]
- Carpenter, T.O.; Whyte, M.P.; Imel, E.A.; Boot, A.M.; Högler, W.; Linglart, A.; Padidela, R.; Van’t Hoff, W.; Mao, M.; Chen, C.Y.; et al. Burosumab Therapy in Children with X-Linked Hypophosphatemia. N. Engl. J. Med. 2018, 378, 1987–1998. [Google Scholar] [CrossRef]
- Trombetti, A.; Al-Daghri, N.; Brandi, M.L.; Cannata-Andía, J.B.; Cavalier, E.; Chandran, M.; Chaussain, C.; Cipullo, L.; Cooper, C.; Haffner, D.; et al. Interdisciplinary management of FGF23-related phosphate wasting syndromes: A Consensus Statement on the evaluation; diagnosis and care of patients with X-linked hypophosphataemia. Nat. Rev. Endocrinol. 2022, 18, 366–384. [Google Scholar] [CrossRef]
- Marques, J.V.O.; Moreira, C.A.; Borba, V.Z.C. New treatments for rare bone diseases: Hypophosphatemic rickets/osteomalácia. Arch. Endocrinol. Metab. 2022, 66, 658–665. [Google Scholar] [CrossRef]
- Baroncelli, G.I.; Mora, S. X-Linked Hypophosphatemic Rickets: Multisystemic Disorder in Children Requiring Multidisciplinary Management. Front. Endocrinol. 2021, 12, 688309. [Google Scholar] [CrossRef]
- Acar, S.; Demir, K.; Shi, Y. Genetic Causes of Rickets. J. Clin. Res. Pediatr. Endocrinol. 2017, 9, 88–105. [Google Scholar] [CrossRef] [PubMed]
- Beck-Nielsen, S.S.; Mughal, Z.; Haffner, D.; Nilsson, O.; Levtchenko, E.; Ariceta, G.; de Lucas Collantes, C.; Schnabel, D.; Jandhyala, R.; Mäkitie, O. FGF23 and its role in X-linked hypophosphatemia-related morbidity. Orphanet J. Rare Dis. 2019, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Rush, E.T.; Johnson, B.; Aradhya, S.; Beltran, D.; Bristow, S.L.; Eisenbeis, S.; Guerra, N.E.; Krolczyk, S.; Miller, N.; Morales, A.; et al. Molecular Diagnoses of X-Linked and Other Genetic Hypophosphatemias: Results From a Sponsored Genetic Testing Program. J. Bone Min. Res. 2022, 37, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Ruppe, M.D. X-Linked Hypophosphatemia. 2012 Feb 9 [Updated 2017 Apr 13]. In GeneReviews®. Available online: https://www.ncbi.nlm.nih.gov/books/NBK83985/ (accessed on 9 July 2023).
- Li, S.S.; Gu, J.M.; Yu, W.J.; He, J.W.; Fu, W.Z.; Zhang, Z.L. Seven novel and six de novo PHEX gene mutations in patients with hypophosphatemic rickets. Int. J. Mol. Med. 2016, 38, 1703–1714. [Google Scholar] [CrossRef]
- Singh, A.K.; Olsen, M.F.; Lavik, L.A.S.; Vold, T.; Drabløs, F.; Sjursen, W. Detecting copy number variation in next generation sequencing data from diagnostic gene panels. BMC Med. Genom. 2021, 14, 214. [Google Scholar] [CrossRef]
- Haffner, D.; Emma, F.; Eastwood, D.M.; Duplan, M.B.; Bacchetta, J.; Schnabel, D.; Wicart, P.; Bockenhauer, D.; Santos, F.; Levtchenko, E.; et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat. Rev. Nephrol. 2019, 15, 435–455. [Google Scholar] [CrossRef] [PubMed]
- Agolini, E.; Chimenz, R.; Fintini, D.; Guarnieri, V.; Guazzarotti, L.; Mora, S.; Salviati, L.; Weber, G. Improving the diagnosis of X-linked hypophosphatemia: Recommendations to optimize diagnostic flow and clinician/geneticist cooperation in the Italian clinical practice. AboutOpen 2021, 8, 29–33. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Fioretti, T.; Auricchio, L.; Piccirillo, A.; Vitiello, G.; Ambrosio, A.; Cattaneo, F.; Ammendola, R.; Esposito, G. Multi-Gene Next-Generation Sequencing for Molecular Diagnosis of Autosomal Recessive Congenital Ichthyosis: A Genotype-Phenotype Study of Four Italian Patients. Diagnostics 2020, 10, 995. [Google Scholar] [CrossRef]
- Padidela, R.; Cheung, M.S.; Saraff, V.; Dharmaraj, P. Clinical guidelines for burosumab in the treatment of XLH in children and adolescents: British paediatric and adolescent bone group recommendations. Endocr. Connect. 2020, 9, 1051–1056. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Fukumoto, S. X-Linked Hypophosphatemia and FGF23-Related Hypophosphatemic Diseases: Prospect for New Treatment. Endocr. Rev. 2018, 39, 274–291. [Google Scholar] [CrossRef] [PubMed]
- Brandi, M.L.; Ariceta, G.; Beck-Nielsen, S.S.; Boot, A.M.; Briot, K.; de Lucas Collantes, C.; Emma, F.; Giannini, S.; Haffner, D.; Keen, R.; et al. Post-authorisation safety study of burosumab use in paediatric; adolescent and adult patients with X-linked hypophosphataemia: Rationale and description. Ther. Adv. Chronic Dis. 2022, 13, 20406223221117471. [Google Scholar] [CrossRef]
- Imel, E.A.; Glorieux, F.H.; Whyte, M.P.; Munns, C.F.; Ward, L.M.; Nilsson, O.; Simmons, J.H.; Padidela, R.; Namba, N.; Cheong, H.I.; et al. Burosumab versus conventional therapy in children with X-linked hypophosphataemia: A randomised; active-controlled; open-label; phase 3 trial. Lancet 2019, 393, 2416–2427. [Google Scholar] [CrossRef]
- Ward, L.M.; Glorieux, F.H.; Whyte, M.P.; Munns, C.F.; Portale, A.A.; Högler, W.; Simmons, J.H.; Gottesman, G.S.; Padidela, R.; Namba, N.; et al. Effect of Burosumab Compared with Conventional Therapy on Younger vs Older Children With X-linked Hypophosphatemia. J. Clin. Endocrinol. Metab. 2022, 107, e3241–e3253. [Google Scholar] [CrossRef]
- Beecroft, S.J.; Lamont, P.J.; Edwards, S.; Goullée, H.; Davis, M.R.; Laing, N.G.; Ravenscroft, G. The Impact of Next-Generation Sequencing on the Diagnosis; Treatment; and Prevention of Hereditary Neuromuscular Disorders. Mol. Diagn. Ther. 2020, 24, 641–652. [Google Scholar] [CrossRef]
- De Palma, F.D.E.; Nunziato, M.; D’Argenio, V.; Savarese, M.; Esposito, G.; Salvatore, F. Comprehensive Molecular Analysis of DMD Gene Increases the Diagnostic Value of Dystrophinopathies: A Pilot Study in a Southern Italy Cohort of Patients. Diagnostics 2021, 11, 1910. [Google Scholar] [CrossRef]
- Carsana, A.; Frisso, G.; Intrieri, M.; Tremolaterra, M.R.; Savarese, G.; Scapagnini, G.; Esposito, G.; Santoro, L.; Salvatore, F. A 15-year molecular analysis of DMD/BMD: Genetic features in a large cohort. Front. Biosci. 2010, 2, 547–558. [Google Scholar] [CrossRef]
- Esposito, G.; Carsana, A. Metabolic Alterations in Cardiomyocytes of Patients with Duchenne and Becker Muscular Dystrophies. J. Clin. Med. 2019, 8, 2151. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Li, X.; Sheng, H.; Su, L.; Wu, M.; Cheng, J.; Huang, Y.; Mao, X.; Zhou, Z.; et al. Novel variants and uncommon cases among southern Chinese children with X-linked hypophosphatemia. J. Endocrinol. Investig. 2020, 43, 1577–1590. [Google Scholar] [CrossRef]
- El Hakam, C.; Parenté, A.; Baraige, F.; Magnol, L.; Forestier, L.; Di Meo, F.; Blanquet, V. PHEX L222P Mutation Increases Phex Expression in a New ENU Mouse Model for XLH Disease. Genes 2022, 13, 1356. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Zhu, H.M.; Liu, H.Q.; Li, L.P.; Liu, S.L.; Wang, H. Two novel variants of the PHEX gene in patients with X-linked dominant hypophosphatemic rickets and prenatal diagnosis for fetuses in these families. Int. J. Mol. Med. 2018, 41, 2012–2020. [Google Scholar] [CrossRef] [PubMed]
- Gan, Y.M.; Zhang, Y.P.; Ruan, D.D.; Huang, J.B.; Zhu, Y.B.; Lin, X.F.; Xiao, X.P.; Cheng, Q.; Geng, Z.B.; Liao, L.S.; et al. Function of PHEX mutations p.Glu145* and p.Trp749Arg in families with X-linked hypophosphatemic rickets by the negative regulation mechanism on FGF23 promoter transcription. Cell Death Dis. 2022, 13, 518. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Tremolaterra, M.R.; Marsocci, E.; Tandurella, I.C.; Fioretti, T.; Savarese, M.; Carsana, A. Precise mapping of 17 deletion breakpoints within the central hotspot deletion region (introns 50 and 51) of the DMD gene. J. Hum. Genet. 2017, 62, 1057–1063. [Google Scholar] [CrossRef]
- Fioretti, T.; Di Iorio, V.; Lombardo, B.; De Falco, F.; Cevenini, A.; Cattaneo, F.; Testa, F.; Pastore, L.; Simonelli, F.; Esposito, G. Molecular Characterization of Choroideremia-Associated Deletions Reveals an Unexpected Regulation of CHM Gene Transcription. Genes 2021, 12, 1111. [Google Scholar] [CrossRef]
- Mindler, G.T.; Stauffer, A.; Kranzl, A.; Penzkofer, S.; Ganger, R.; Radler, C.; Haeusler, G.; Raimann, A. Persistent Lower Limb Deformities Despite Amelioration of Rickets in X-Linked Hypophosphatemia (XLH)—A Prospective Observational Study. Front. Endocrinol. 2022, 13, 866170. [Google Scholar] [CrossRef]
- Paloian, N.J.; Nemeth, B.; Sharafinski, M.; Modaff, P.; Steiner, R.D. Real-world effectiveness of burosumab in children with X-linked hypophosphatemic rickets. Pediatr. Nephrol. 2022, 37, 2667–2677. [Google Scholar] [CrossRef] [PubMed]
- Schindeler, A.; Biggin, A.; Munns, C.F. Clinical Evidence for the Benefits of Burosumab Therapy for X-Linked Hypophosphatemia (XLH) and Other Conditions in Adults and Children. Front. Endocrinol. 2020, 11, 338. [Google Scholar] [CrossRef] [PubMed]
- Mughal, M.Z.; Baroncelli, G.I.; de Lucas-Collantes, C.; Linglart, A.; Magnolato, A.; Raimann, A.; Santos, F.; Schnabel, D.; Shaw, N.; Nilsson, O. Burosumab for X-linked hypophosphatemia in children and adolescents: Opinion based on early experience in seven European countries. Front. Endocrinol. 2023, 13, 1034580. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Ruggiero, R.; Savarese, M.; Savarese, G.; Tremolaterra, M.R.; Salvatore, F.; Carsana, A. Prenatal molecular diagnosis of inherited neuromuscular diseases: Duchenne/Becker muscular dystrophy; myotonic dystrophy type 1 and spinal muscular atrophy. Clin. Chem. Lab. Med. 2013, 51, 2239–2245. [Google Scholar] [CrossRef] [PubMed]



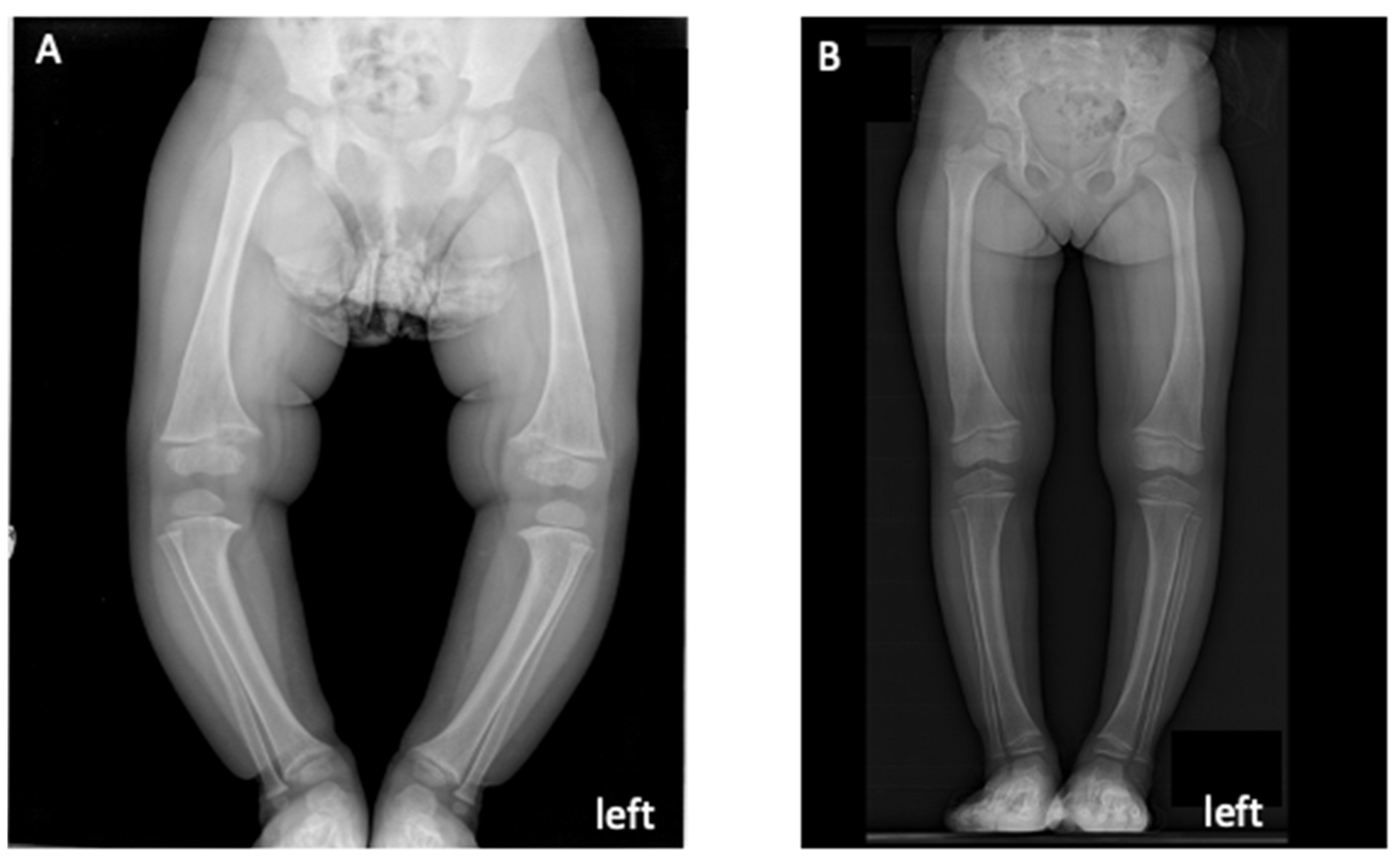{kind=link}
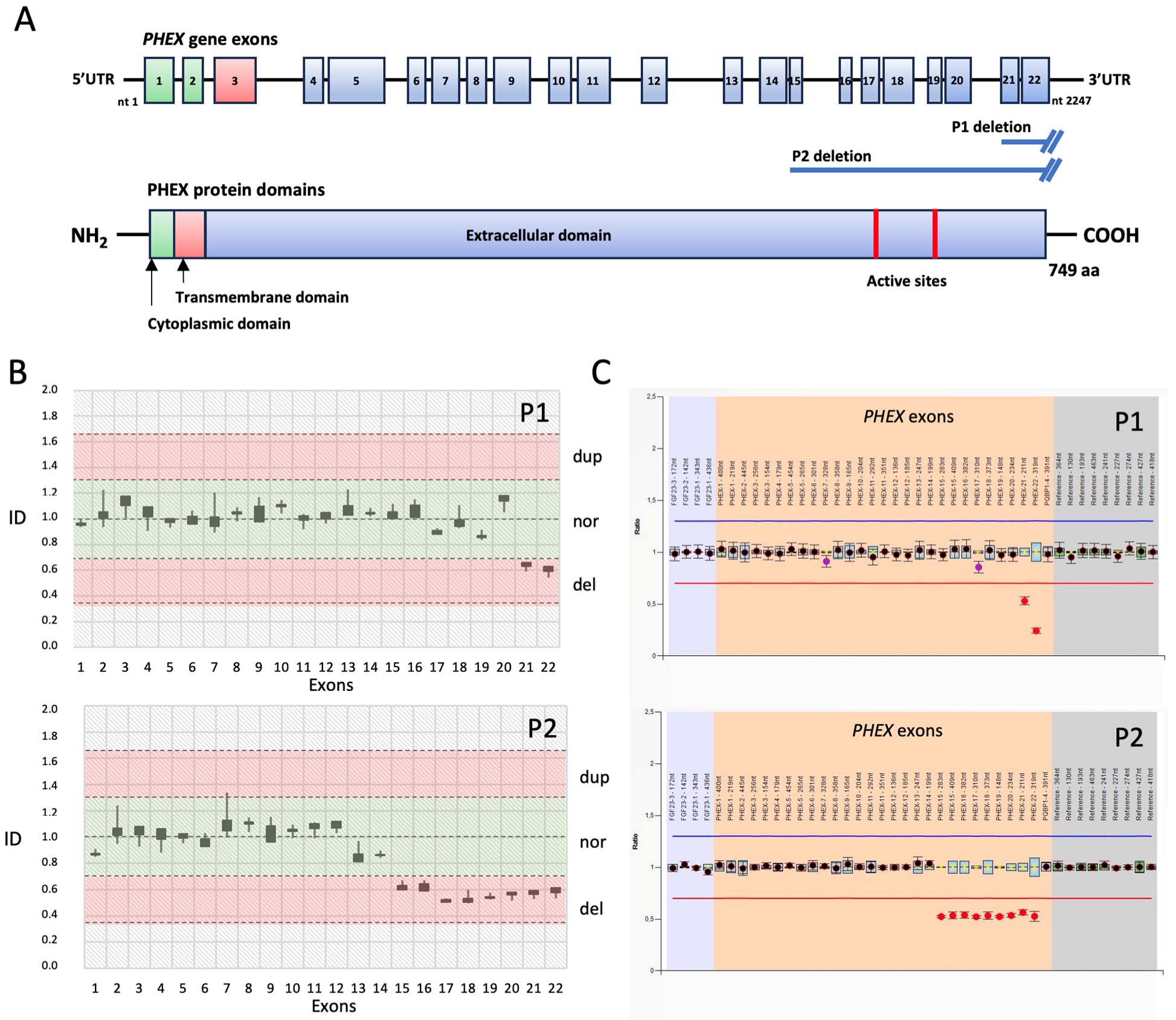{kind=link}
| Height (cm) | Weight (kg) | eGFR (mL/min/1.73 m2) | 25(OH)2D (ng/mL) | ALP (IU/L) | P (mg/dL) | Ca (mg/dL) | PTH (pg/mL) | FGF23 (ng/L) | TRP (%) | TmP/GFR (mg/dL) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | 82 ↓ | 12.7 ↓ | 113 = | 15 ↓ | 642 ↑ | 2.2 ↓ | 8.8 = | 18 = | 79.5 ↑ | 77 ↓ | 1.86 ↓ |
| Patient 2 | 79 ↓ | 13.5 ↓ | 109 = | 30 = | 808 ↑ | 2 ↓ | 9 = | 16 = | 90 ↑ | 75 ↓ | 1.97 ↓ |
| eGFR (mL/min/1.73 m2) | 25(OH)2D (ng/mL) | ALP (IU/L) | P (mg/dL) | Ca (mg/dL) | PTH (pg/mL) | TRP (%) | TmP/GFR (mg/dL) | |
|---|---|---|---|---|---|---|---|---|
| Patient 1 | 111 | 75 | 259 | 3.7 | 9.5 | 22 | 85 | 3.30 |
| Patient 2 | 194 | 65 | 289 | 3.7 | 9.1 | 23 | 87 | 3.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pecoraro, C.; Fioretti, T.; Perruno, A.; Klain, A.; Cioffi, D.; Ambrosio, A.; Passaro, D.; Annicchiarico Petruzzelli, L.; Di Domenico, C.; de Girolamo, D.; et al. De Novo Large Deletions in the PHEX Gene Caused X-Linked Hypophosphataemic Rickets in Two Italian Female Infants Successfully Treated with Burosumab. Diagnostics 2023, 13, 2552. https://doi.org/10.3390/diagnostics13152552
Pecoraro C, Fioretti T, Perruno A, Klain A, Cioffi D, Ambrosio A, Passaro D, Annicchiarico Petruzzelli L, Di Domenico C, de Girolamo D, et al. De Novo Large Deletions in the PHEX Gene Caused X-Linked Hypophosphataemic Rickets in Two Italian Female Infants Successfully Treated with Burosumab. Diagnostics. 2023; 13(15):2552. https://doi.org/10.3390/diagnostics13152552
Chicago/Turabian StylePecoraro, Carmine, Tiziana Fioretti, Assunta Perruno, Antonella Klain, Daniela Cioffi, Adelaide Ambrosio, Diego Passaro, Luigi Annicchiarico Petruzzelli, Carmela Di Domenico, Domenico de Girolamo, and et al. 2023. "De Novo Large Deletions in the PHEX Gene Caused X-Linked Hypophosphataemic Rickets in Two Italian Female Infants Successfully Treated with Burosumab" Diagnostics 13, no. 15: 2552. https://doi.org/10.3390/diagnostics13152552
APA StylePecoraro, C., Fioretti, T., Perruno, A., Klain, A., Cioffi, D., Ambrosio, A., Passaro, D., Annicchiarico Petruzzelli, L., Di Domenico, C., de Girolamo, D., Vallone, S., Cattaneo, F., Ammendola, R., & Esposito, G. (2023). De Novo Large Deletions in the PHEX Gene Caused X-Linked Hypophosphataemic Rickets in Two Italian Female Infants Successfully Treated with Burosumab. Diagnostics, 13(15), 2552. https://doi.org/10.3390/diagnostics13152552