
Treatment of the Neutropenia Associated with GSD1b and G6PC3 Deficiency with SGLT2 Inhibitors



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Enzymes
2.1. Glucose-6-Phosphatase (G6PC1)
2.2. The Glucose-6-Phosphate Transporter (G6PT)
2.3. The ‘Ubiquitous Glucose-6-Phosphatase’ G6PC3
3. The Diseases
3.1. Glucose-6-Phosphatase (G6PC1) Deficiency or GSD1a
3.2. Glucose-6-Phosphate Transpsorter (G6PT) Deficiency or GSD1b
3.3. G6PC3 Deficiency or Dursum Syndrome
4. Pathophysiological Mechanisms Underlying the Neutropenia
4.1. Diseases of Metabolite Repair
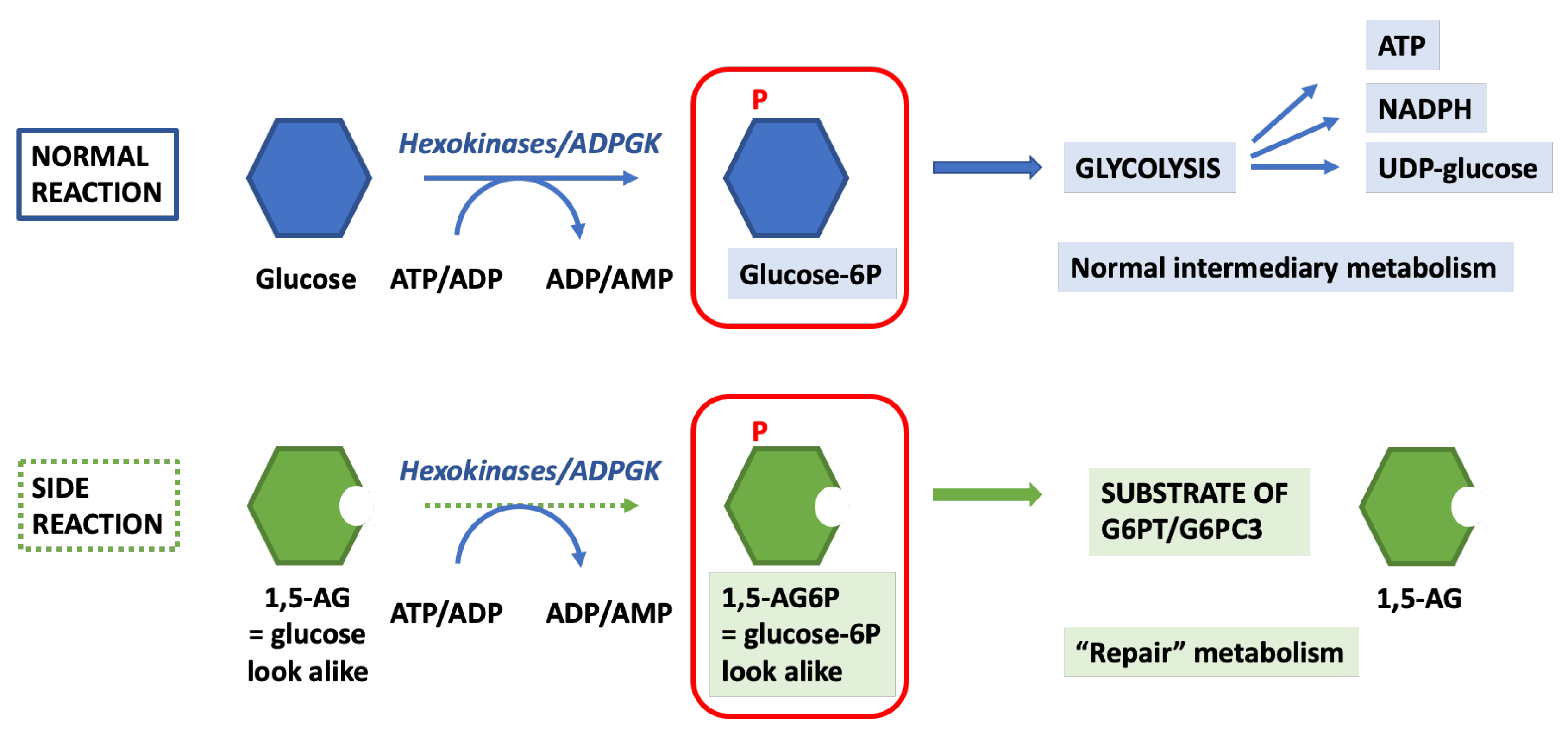
4.2. 1,5-. Anhydroglucitol-6-Phosphate (1,5-AG6P) Accumulation
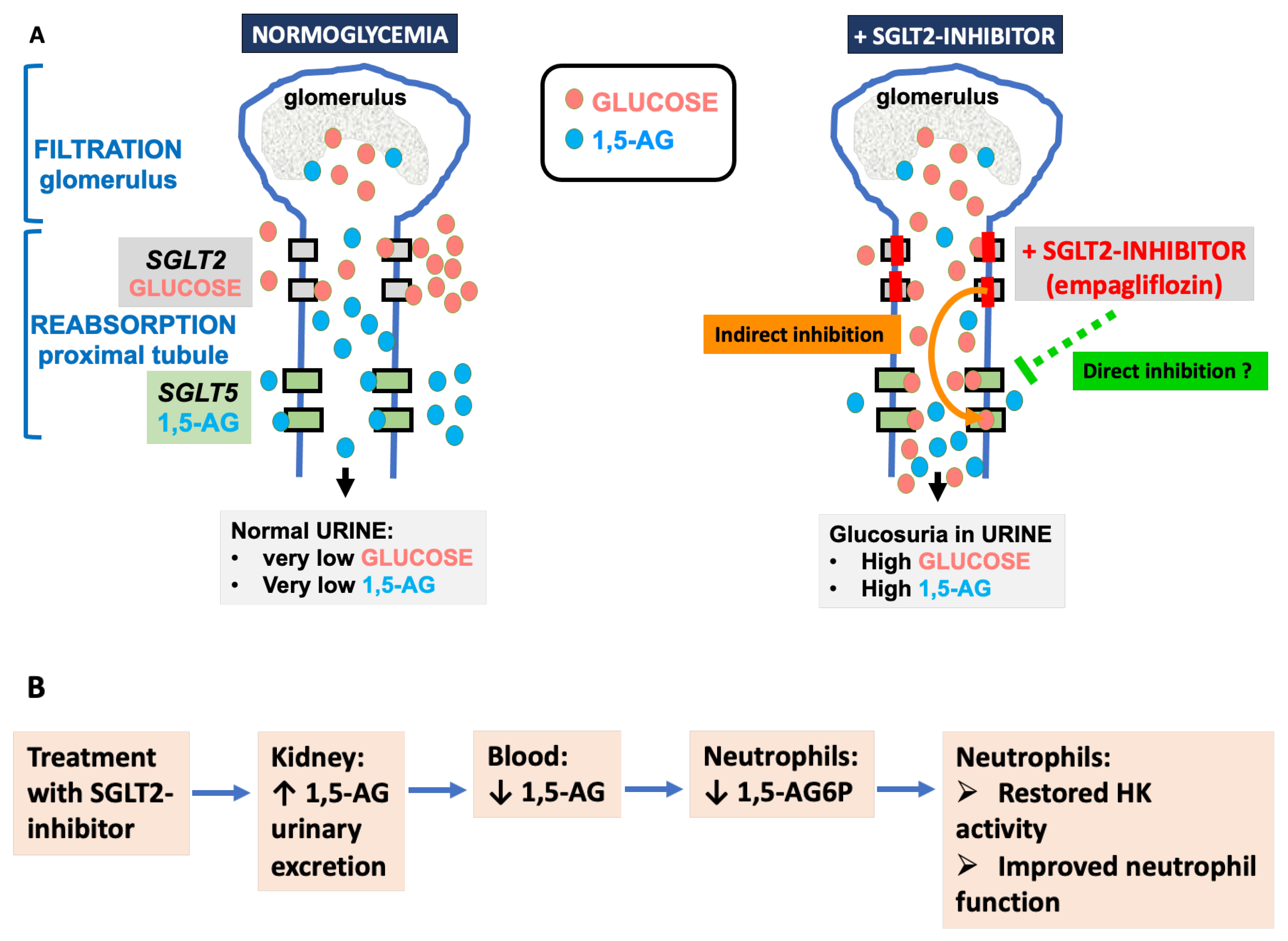
4.3. 1,5-. Anhydroglucitol (1,5-AG)
5. Role of the Neutrophil Dysfunction and Other Aspects of Immune Dysregulation in the Pathophysiology of the Infections
6. Treatment with SGLT2 Inhibitors (Gliflozins)
6.1. Preliminary Note
6.2. Indication
6.3. Dosage, Contraindications and Adverse Effects
6.4. Choice of the Gliflozin
6.5. Effect of Mutations in SGLT5
6.6. Could SGLT2 Inhibitors Help GSD1b Patients beyond Treating Their Neutropenia?
7. Conclusions and Outlooks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cori, G.T.; Cori, C.F. Glucose-6-Phosphatase of the Liver in Glycogen Storage Disease. J. Biol. Chem. 1952, 199, 661–667. [Google Scholar] [CrossRef]
- Korlimarla, A.; Gibson, R.; Kishnani, P.S. Glycogen Storage Diseases. In Nutrition Management of Inherited Metabolic Diseases: Lessons from Metabolic University; Bernstein, L.E., Rohr, F., van Calcar, S., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 349–362. [Google Scholar]
- Narisawa, K.; Igarashi, Y.; Otomo, H.; Tada, K. A new variant of glycogen storage disease Type I probably due to a defect in the glucose-6-phosphate transport system. Biochem. Biophys. Res. Commun. 1978, 83, 1360–1364. [Google Scholar] [CrossRef] [PubMed]
- van Schaftingen, E.; Gerin, I. The glucose-6-phosphatase system. Biochem. J. 2002, 362, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, Y.; Kato, S.; Narisawa, K.; Tada, K.; Amano, Y.; Mori, T.; Takeuchi, S. A direct evidence for defect in glucose-6-phosphate transport system in hepatic microsomal membrane of glycogen storage disease type IB. Biochem. Biophys. Res. Commun. 1984, 119, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Tada, K.; Narisawa, K.; Igarashi, Y.; Kato, S. Glycogen storage disease type IB: A new model of genetic disorders involving the transport system of intracellular membrane. Biochem. Med. 1985, 33, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Beaudet, A.L.; Anderson, D.C.; Michels, V.V.; Arion, W.J.; Lange, A.J. Neutropenia and impaired neutrophil migration in type IB glycogen storage disease. J. Pediatr. 1980, 97, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Gitzelmann, R.; Bosshard, N.U. Defective neutrophil and monocyte functions in glycogen storage disease type Ib: A literature review. Eur. J. Pediatr. 1993, 152 (Suppl. 1), S33–S38. [Google Scholar] [CrossRef] [PubMed]
- Lei, K.J.; Shelly, L.L.; Pan, C.J.; Sidbury, J.B.; Chou, J.Y. Mutations in the glucose-6-phosphatase gene that cause glycogen storage disease type 1a. Science 1993, 262, 580–583. [Google Scholar] [CrossRef]
- Guionie, O.; Clottes, E.; Stafford, K.; Burchell, A. Identification and characterisation of a new human glucose-6-phosphatase isoform. FEBS Lett. 2003, 551, 159–164. [Google Scholar] [CrossRef]
- Shieh, J.J.; Pan, C.J.; Mansfield, B.C.; Chou, J.Y. A glucose-6-phosphate hydrolase, widely expressed outside the liver, can explain age-dependent resolution of hypoglycemia in glycogen storage disease type Ia. J. Biol. Chem. 2003, 278, 47098–47103. [Google Scholar] [CrossRef]
- Ghosh, A.; Shieh, J.J.; Pan, C.J.; Chou, J.Y. Histidine 167 is the phosphate acceptor in glucose-6-phosphatase-beta forming a phosphohistidine enzyme intermediate during catalysis. J. Biol. Chem. 2004, 279, 12479–12483. [Google Scholar] [CrossRef] [PubMed]
- Gerin, I.; Veiga-da-Cunha, M.; Achouri, Y.; Collet, J.F.; Van Schaftingen, E. Sequence of a putative glucose 6-phosphate translocase, mutated in glycogen storage disease type Ib. FEBS Lett. 1997, 419, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Veiga-da-Cunha, M.; Gerin, I.; Chen, Y.T.; de Barsy, T.; de Lonlay, P.; Dionisi-Vici, C.; Fenske, C.D.; Lee, P.J.; Leonard, J.V.; Maire, I.; et al. A gene on chromosome 11q23 coding for a putative glucose- 6-phosphate translocase is mutated in glycogen-storage disease types Ib and Ic. Am. J. Hum. Genet. 1998, 63, 976–983. [Google Scholar] [CrossRef]
- Cheung, Y.Y.; Kim, S.Y.; Yiu, W.H.; Pan, C.J.; Jun, H.S.; Ruef, R.A.; Lee, E.J.; Westphal, H.; Mansfield, B.C.; Chou, J.Y. Impaired neutrophil activity and increased susceptibility to bacterial infection in mice lacking glucose-6-phosphatase-beta. J. Clin. Investig. 2007, 117, 784–793. [Google Scholar] [CrossRef]
- Boztug, K.; Appaswamy, G.; Ashikov, A.; Schaffer, A.A.; Salzer, U.; Diestelhorst, J.; Germeshausen, M.; Brandes, G.; Lee-Gossler, J.; Noyan, F.; et al. A syndrome with congenital neutropenia and mutations in G6PC3. N. Engl. J. Med. 2009, 360, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Veiga-da-Cunha, M.; Chevalier, N.; Stephenne, X.; Defour, J.P.; Paczia, N.; Ferster, A.; Achouri, Y.; Dewulf, J.P.; Linster, C.L.; Bommer, G.T.; et al. Failure to eliminate a phosphorylated glucose analog leads to neutropenia in patients with G6PT and G6PC3 deficiency. Proc. Natl. Acad. Sci. USA 2019, 116, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Bommer, G.T.; Van Schaftingen, E.; Veiga-da-Cunha, M. Metabolite Repair Enzymes Control Metabolic Damage in Glycolysis. Trends Biochem. Sci. 2019, 45, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Wortmann, S.B.; Van Hove, J.L.K.; Derks, T.G.J.; Chevalier, N.; Knight, V.; Koller, A.; Oussoren, E.; Mayr, J.A.; van Spronsen, F.J.; Lagler, F.B.; et al. Treating neutropenia and neutrophil dysfunction in glycogen storage disease type Ib with an SGLT2 inhibitor. Blood 2020, 136, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, C.; Stephenne, X.; Diederich, J.; Mounkoro, P.; Chevalier, N.; Ferster, A.; Van Schaftingen, E.; Veiga-da-Cunha, M. Successful use of empagliflozin to treat neutropenia in two G6PC3-deficient children: Impact of a mutation in SGLT5. J. Inherit. Metab. Dis. 2022, 45, 759–768. [Google Scholar] [CrossRef]
- Diederich, J.; Mounkoro, P.; Tirado, H.A.; Chevalier, N.; Van Schaftingen, E.; Veiga-da-Cunha, M. SGLT5 is the renal transporter for 1,5-anhydroglucitol, a major driver in two rare forms of neutropenia. Cell. Mol. Life Sci. 2023, submitted.
- Ockerman, P.A.; Lundborg, H. Conversion of fructose to glucose by human jejunum absence of galactose-to-glucose conversion. Biochim. Biophys. Acta 1965, 105, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Bismut, H.; Hers, H.G.; Van Schaftingen, E. Conversion of fructose to glucose in the rabbit small intestine. A reappraisal of the direct pathway. Eur. J. Biochem. 1993, 213, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Arion, W.J.; Wallin, B.K.; Carlson, P.W.; Lange, A.J. The specificity of glucose 6-phosphatase of intact liver microsomes. J. Biol. Chem. 1972, 247, 2558–2565. [Google Scholar] [CrossRef] [PubMed]
- Pao, S.S.; Paulsen, I.T.; Saier, M.H., Jr. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 1998, 62, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Bartoloni, L.; Antonarakis, S.E. The human sugar-phosphate/phosphate exchanger family SLC37. Pflugers Arch 2004, 447, 780–783. [Google Scholar] [CrossRef]
- Chou, J.Y.; Mansfield, B.C. The SLC37 family of sugar-phosphate/phosphate exchangers. Curr. Top. Membr. 2014, 73, 357–382. [Google Scholar]
- Cappello, A.R.; Curcio, R.; Lappano, R.; Maggiolini, M.; Dolce, V. The Physiopathological Role of the Exchangers Belonging to the SLC37 Family. Front. Chem. 2018, 6, 122. [Google Scholar] [CrossRef]
- Gerin, I.; Veiga-da-Cunha, M.; Noel, G.; Van Schaftingen, E. Structure of the gene mutated in glycogen storage disease type Ib. Gene 1999, 227, 189–195. [Google Scholar] [CrossRef]
- Ihara, K.; Nomura, A.; Hikino, S.; Takada, H.; Hara, T. Quantitative analysis of glucose-6-phosphate translocase gene expression in various human tissues and haematopoietic progenitor cells. J. Inherit. Metab. Dis. 2000, 23, 583–592. [Google Scholar] [CrossRef]
- Mandula, B.; Srivastava, S.K.; Beutler, E. Hexose-6-phosphate dehydrogenase: Distribution in rat tissues and effect of diet, age and steroids. Arch. Biochem. Biophys. 1970, 141, 155–161. [Google Scholar] [CrossRef]
- Takahashi, T.; Hori, S.H. Intramembraneous localization of rat liver microsomal hexose-6-phosphate dehydrogenase and membrane permeability to its substrates. Biochim. Biophys. Acta 1978, 524, 262–276. [Google Scholar] [CrossRef] [PubMed]
- Zielinska, A.E.; Walker, E.A.; Stewart, P.M.; Lavery, G.G. Biochemistry and physiology of hexose-6-phosphate knockout mice. Mol. Cell Endocrinol. 2011, 336, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Odermatt, A.; Klusonova, P. 11beta-Hydroxysteroid dehydrogenase 1: Regeneration of active glucocorticoids is only part of the story. J. Steroid Biochem. Mol. Biol. 2015, 151, 85–92. [Google Scholar] [CrossRef]
- Arden, S.D.; Zahn, T.; Steegers, S.; Webb, S.; Bergman, B.; O′Brien, R.M.; Hutton, J.C. Molecular cloning of a pancreatic islet-specific glucose-6-phosphatase catalytic subunit-related protein. Diabetes 1999, 48, 531–542. [Google Scholar] [CrossRef]
- Martin, C.C.; Oeser, J.K.; Svitek, C.A.; Hunter, S.I.; Hutton, J.C.; O’Brien, R.M. Identification and characterization of a human cDNA and gene encoding a ubiquitously expressed glucose-6-phosphatase catalytic subunit-related protein. J. Mol. Endocrinol. 2002, 29, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Hutton, J.C.; O’Brien, R.M. Glucose-6-phosphatase catalytic subunit gene family. J. Biol. Chem. 2009, 284, 29241–29245. [Google Scholar] [CrossRef] [PubMed]
- Bouatia-Naji, N.; Rocheleau, G.; Van Lommel, L.; Lemaire, K.; Schuit, F.; Cavalcanti-Proenca, C.; Marchand, M.; Hartikainen, A.L.; Sovio, U.; De Graeve, F.; et al. A polymorphism within the G6PC2 gene is associated with fasting plasma glucose levels. Science 2008, 320, 1085–1088. [Google Scholar] [CrossRef]
- Weston, B.W.; Lin, J.L.; Muenzer, J.; Cameron, H.S.; Arnold, R.R.; Seydewitz, H.H.; Mayatepek, E.; Van Schaftingen, E.; Veiga-da-Cunha, M.; Matern, D.; et al. Glucose-6-phosphatase mutation G188R confers an atypical glycogen storage disease type 1b phenotype. Pediatr. Res. 2000, 48, 329–334. [Google Scholar] [CrossRef]
- Lawrence, N.T.; Chengsupanimit, T.; Brown, L.M.; Weinstein, D.A. High Incidence of Serologic Markers of Inflammatory Bowel Disease in Asymptomatic Patients with Glycogen Storage Disease Type Ia. JIMD Rep. 2015, 24, 123–128. [Google Scholar]
- Kilpatrick, L.; Garty, B.Z.; Lundquist, K.F.; Hunter, K.; Stanley, C.A.; Baker, L.; Douglas, S.D.; Korchak, H.M. Impaired metabolic function and signaling defects in phagocytic cells in glycogen storage disease type 1b. J. Clin. Investig. 1990, 86, 196–202. [Google Scholar] [CrossRef]
- Jun, H.S.; Weinstein, D.A.; Lee, Y.M.; Mansfield, B.C.; Chou, J.Y. Molecular mechanisms of neutrophil dysfunction in glycogen storage disease type Ib. Blood 2014, 123, 2843–2853. [Google Scholar] [CrossRef] [PubMed]
- Wicker, C.; Roda, C.; Perry, A.; Arnoux, J.B.; Brassier, A.; Castelle, M.; Servais, A.; Donadieu, J.; Bouchereau, J.; Pigneur, B.; et al. Infectious and digestive complications in glycogen storage disease type Ib: Study of a French cohort. Mol. Genet. Metab. Rep. 2020, 23, 100581. [Google Scholar] [CrossRef] [PubMed]
- Dieckgraefe, B.K.; Korzenik, J.R.; Husain, A.; Dieruf, L. Association of glycogen storage disease 1b and Crohn disease: Results of a North American survey. Eur. J. Pediatr. 2002, 161 (Suppl. S1), S88–S92. [Google Scholar] [CrossRef] [PubMed]
- Dale, D.C.; Bolyard, A.A.; Marrero, T.; Kelley, M.L.; Makaryan, V.; Tran, E.; Leung, J.; Boxer, L.A.; Kishnani, P.S.; Austin, S.; et al. Neutropenia in glycogen storage disease Ib: Outcomes for patients treated with granulocyte colony-stimulating factor. Curr. Opin. Hematol. 2019, 26, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, P.S.; Alter, B.P.; Bolyard, A.A.; Bonilla, M.A.; Boxer, L.A.; Cham, B.; Fier, C.; Freedman, M.; Kannourakis, G.; Kinsey, S.; et al. The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood 2006, 107, 4628–4635. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Ihara, K.; Matsumoto, T.; Tsutsumi, Y.; Nomura, A.; Ohga, S.; Hara, T. Inflammatory bowel disease-like colitis in glycogen storage disease type 1b. Inflamm. Bowel Dis. 2001, 7, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Bolton, C.; Burch, N.; Morgan, J.; Harrison, B.; Pandey, S.; Pagnamenta, A.T.; Taylor, J.C.; Taylor, J.M.; Marsh, J.C.W.; Potter, V.; et al. Remission of inflammatory bowel disease in Glucose-6-Phosphatase 3 deficiency by allogeneic haematopoietic stem cell transplantation. J. Crohns Colitis 2019, 14, 142–147. [Google Scholar] [CrossRef]
- Desplantes, C.; Fremond, M.L.; Beaupain, B.; Harousseau, J.L.; Buzyn, A.; Pellier, I.; Roques, G.; Morville, P.; Paillard, C.; Bruneau, J.; et al. Clinical spectrum and long-term follow-up of 14 cases with G6PC3 mutations from the French Severe Congenital Neutropenia Registry. Orphanet J. Rare Dis. 2014, 9, 183. [Google Scholar] [CrossRef]
- Visser, G.; Rake, J.P.; Labrune, P.; Leonard, J.V.; Moses, S.; Ullrich, K.; Wendel, U.; Groenier, K.H.; Smit, G.P. Granulocyte colony-stimulating factor in glycogen storage disease type 1b. Results of the European Study on Glycogen Storage Disease Type 1. Eur. J. Pediatr. 2002, 161 (Suppl. S1), S83–S87. [Google Scholar] [CrossRef]
- Rossi, A.; Miele, E.; Fecarotta, S.; Veiga-da-Cunha, M.; Martinelli, M.; Mollica, C.; D’Armiento, M.; Mozzillo, E.; Strisciuglio, P.; Derks, T.G.J.; et al. Crohn disease-like enterocolitis remission after empagliflozin treatment in a child with glycogen storage disease type Ib: A case report. Ital. J. Pediatr. 2021, 47, 149. [Google Scholar] [CrossRef]
- Mikami, M.; Arai, A.; Mizumoto, H. Empagliflozin ameliorated neutropenia in a girl with glycogen storage disease Ib. Pediatr. Int. 2021, 63, 1394–1396. [Google Scholar] [CrossRef]
- Makrilakis, K.; Barmpagianni, A.; Veiga-da-Cunha, M. Repurposing of Empagliflozin as a Possible Treatment for Neutropenia and Inflammatory Bowel Disease in Glycogen Storage Disease Type Ib: A Case Report. Cureus 2022, 14, e27264. [Google Scholar] [CrossRef] [PubMed]
- Halligan, R.K.; Dalton, R.N.; Turner, C.; Lewis, K.A.; Mundy, H.R. Understanding the role of SGLT2 inhibitors in glycogen storage disease type Ib: The experience of one UK centre. Orphanet J. Rare Dis. 2022, 17, 195. [Google Scholar] [CrossRef] [PubMed]
- Grunert, S.C.; Rosenbaum-Fabian, S.; Schumann, A.; Selbitz, A.C.; Merz, W.; Gieselmann, A.; Spiekerkoetter, U. Two successful pregnancies and first use of empagliflozin during pregnancy in glycogen storage disease type Ib. JIMD Rep. 2022, 63, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Bidiuk, J.; Gaciong, Z.A.; Sobieraj, P. The overall benefits of empagliflozin treatment in adult siblings with glycogen storage disease type Ib: One year experience. Arch. Med. Sci. 2022, 18, 1095–1099. [Google Scholar] [CrossRef]
- Grunert, S.C.; Elling, R.; Maag, B.; Wortmann, S.B.; Derks, T.G.J.; Hannibal, L.; Schumann, A.; Rosenbaum-Fabian, S.; Spiekerkoetter, U. Improved inflammatory bowel disease, wound healing and normal oxidative burst under treatment with empagliflozin in glycogen storage disease type Ib. Orphanet J. Rare Dis. 2020, 15, 218. [Google Scholar] [CrossRef]
- Banka, S.; Newman, W.G. A clinical and molecular review of ubiquitous glucose-6-phosphatase deficiency caused by G6PC3 mutations. Orphanet J. Rare Dis. 2013, 8, 84. [Google Scholar] [CrossRef]
- Velez-Tirado, N.; Yamazaki-Nakashimada, M.A.; Lopez Valentin, E.; Partida-Gaytan, A.; Scheffler-Mendoza, S.C.; Chaia Semerena, G.M.; Alvarez-Cardona, A.; Suarez Gutierrez, M.A.; Medina Torres, E.A.; Baeza Capetillo, P.; et al. Severe congenital neutropenia due to G6PC3 deficiency: Case series of five patients and literature review. Scand. J. Immunol. 2022, 95, e13136. [Google Scholar] [CrossRef]
- McKinney, C.; Ellison, M.; Briones, N.J.; Baroffio, A.; Murphy, J.; Tran, A.D.; Reisz, J.A.; D’Alessandro, A.; Ambruso, D.R. Metabolic abnormalities in G6PC3-deficient human neutrophils result in severe functional defects. Blood Adv. 2020, 4, 5888–5901. [Google Scholar] [CrossRef]
- Van Schaftingen, E.; Rzem, R.; Veiga-da-Cunha, M. L: -2-Hydroxyglutaric aciduria, a disorder of metabolite repair. J. Inherit. Metab. Dis. 2009, 32, 135–142. [Google Scholar] [CrossRef]
- Rzem, R.; Veiga-da-Cunha, M.; Noel, G.; Goffette, S.; Nassogne, M.C.; Tabarki, B.; Scholler, C.; Marquardt, T.; Vikkula, M.; Van Schaftingen, E. A gene encoding a putative FAD-dependent L-2-hydroxyglutarate dehydrogenase is mutated in L-2-hydroxyglutaric aciduria. Proc. Natl. Acad. Sci. USA 2004, 101, 16849–16854. [Google Scholar] [CrossRef] [PubMed]
- Rzem, R.; Achouri, Y.; Marbaix, E.; Schakman, O.; Wiame, E.; Marie, S.; Gailly, P.; Vincent, M.F.; Veiga-da-Cunha, M.; Van Schaftingen, E. A mouse model of L-2-hydroxyglutaric aciduria, a disorder of metabolite repair. PLoS ONE 2015, 10, e0119540. [Google Scholar] [CrossRef] [PubMed]
- Veiga-da-Cunha, M.; Van Schaftingen, E.; Bommer, G.T. Inborn errors of metabolite repair. J. Inherit. Metab. Dis. 2019, 43, 14–24. [Google Scholar] [CrossRef]
- Crane, R.K.; Sols, A. The non-competitive inhibition of brain hexokinase by glucose-6-phosphate and related compounds. J. Biol. Chem. 1954, 210, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.Y.; Alechina, O.; Aleshin, A.E.; Fromm, H.J.; Honzatko, R.B. Identification of a phosphate regulatory site and a low affinity binding site for glucose 6-phosphate in the N-terminal half of human brain hexokinase. J. Biol. Chem. 1998, 273, 19548–19553. [Google Scholar] [CrossRef]
- Maianski, N.A.; Geissler, J.; Srinivasula, S.M.; Alnemri, E.S.; Roos, D.; Kuijpers, T.W. Functional characterization of mitochondria in neutrophils: A role restricted to apoptosis. Cell Death Differ. 2004, 11, 143–153. [Google Scholar] [CrossRef]
- Borregaard, N.; Herlin, T. Energy metabolism of human neutrophils during phagocytosis. J. Clin. Investig. 1982, 70, 550–557. [Google Scholar] [CrossRef]
- van Raam, B.J.; Sluiter, W.; de Wit, E.; Roos, D.; Verhoeven, A.J.; Kuijpers, T.W. Mitochondrial membrane potential in human neutrophils is maintained by complex III activity in the absence of supercomplex organisation. PLoS ONE 2008, 3, e2013. [Google Scholar] [CrossRef]
- Toller-Kawahisa, J.E.; O’Neill, L.A.J. How neutrophil metabolism affects bacterial killing. Open Biol. 2022, 12, 220248. [Google Scholar] [CrossRef]
- Hayee, B.; Antonopoulos, A.; Murphy, E.J.; Rahman, F.Z.; Sewell, G.; Smith, B.N.; McCartney, S.; Furman, M.; Hall, G.; Bloom, S.L.; et al. G6PC3 mutations are associated with a major defect of glycosylation: A novel mechanism for neutrophil dysfunction. Glycobiology 2011, 21, 914–924. [Google Scholar] [CrossRef]
- Chou, J.Y.; Jun, H.S.; Mansfield, B.C. Neutropenia in type Ib glycogen storage disease. Curr. Opin. Hematol. 2010, 17, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.S.; Lee, Y.M.; Cheung, Y.Y.; McDermott, D.H.; Murphy, P.M.; De Ravin, S.S.; Mansfield, B.C.; Chou, J.Y. Lack of glucose recycling between endoplasmic reticulum and cytoplasm underlies cellular dysfunction in glucose-6-phosphatase-beta-deficient neutrophils in a congenital neutropenia syndrome. Blood 2010, 116, 2783–2792. [Google Scholar] [CrossRef] [PubMed]
- Pitkanen, E. 1,5-Anhydro-D-glucitol--a novel type of sugar in the human organism. Scand. J. Clin. Lab. Invest. Suppl. 1990, 201, 55–62. [Google Scholar]
- Yamanouchi, T.; Tachibana, Y.; Akanuma, H.; Minoda, S.; Shinohara, T.; Moromizato, H.; Miyashita, H.; Akaoka, I. Origin and disposal of 1,5-anhydroglucitol, a major polyol in the human body. Am. J. Physiol. 1992, 263, E268–E273. [Google Scholar] [CrossRef]
- Pitkanen, E. Serum 1,5-anhydroglucitol in normal subjects and in patients with insulin-dependent diabetes mellitus. Scand. J. Clin. Lab. Invest. 1982, 42, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Stickle, D.; Turk, J. A kinetic mass balance model for 1,5-anhydroglucitol: Applications to monitoring of glycemic control. Am. J. Physiol. 1997, 273, E821–E830. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Bojsen, K.; Svensson, B.; Marcussen, J. Alpha-1,4-glucan lyases producing 1,5-anhydro-D-fructose from starch and glycogen have sequence similarity to alpha-glucosidases. Biochim. Biophys. Acta 1999, 1433, 1–15. [Google Scholar] [CrossRef]
- Yu, S. The anhydrofructose pathway of glycogen catabolism. IUBMB Life 2008, 60, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, M.; Kubota, S. Mouse AKR1E1 is an ortholog of pig liver NADPH dependent 1,5-anhydro-D-fructose reductase. Biosci. Biotechnol. Biochem. 2008, 72, 872–876. [Google Scholar] [CrossRef]
- Ying, L.; Ma, X.; Yin, J.; Wang, Y.; He, X.; Peng, J.; Bao, Y.; Zhou, J.; Jia, W. The metabolism and transport of 1,5-anhydroglucitol in cells. Acta Diabetol. 2018, 55, 279–286. [Google Scholar] [CrossRef]
- Kametani, S.; Hashimoto, Y.; Yamanouchi, T.; Akanuma, Y.; Akanuma, H. Reduced renal reabsorption of 1,5-anhydro-D-glucitol in diabetic rats and mice. J. Biochem. 1987, 102, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Pitkanen, E.; Pitkanen, O.M. Renal tubular reabsorption of 1,5-anhydro-D-glucitol and D-mannose in vivo in the rat. Pflug. Arch 1992, 420, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M.; Loo, D.D.; Hirayama, B.A. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef] [PubMed]
- Grempler, R.; Augustin, R.; Froehner, S.; Hildebrandt, T.; Simon, E.; Mark, M.; Eickelmann, P. Functional characterisation of human SGLT-5 as a novel kidney-specific sodium-dependent sugar transporter. FEBS Lett. 2012, 586, 248–253. [Google Scholar] [CrossRef]
- Fukuzawa, T.; Fukazawa, M.; Ueda, O.; Shimada, H.; Kito, A.; Kakefuda, M.; Kawase, Y.; Wada, N.A.; Goto, C.; Fukushima, N.; et al. SGLT5 reabsorbs fructose in the kidney but its deficiency paradoxically exacerbates hepatic steatosis induced by fructose. PLoS ONE 2013, 8, e56681. [Google Scholar] [CrossRef]
- Ying, L.; He, X.; Ma, X.; Shen, Y.; Su, H.; Peng, J.; Wang, Y.; Bao, Y.; Zhou, J.; Jia, W. Serum 1,5-anhydroglucitol when used with fasting plasma glucose improves the efficiency of diabetes screening in a Chinese population. Sci. Rep. 2017, 7, 11968. [Google Scholar] [CrossRef]
- Kappel, B.A.; Moellmann, J.; Thiele, K.; Rau, M.; Artati, A.; Adamski, J.; Ghesquiere, B.; Schuett, K.; Romeo, F.; Stoehr, R.; et al. Human and mouse non-targeted metabolomics identify 1,5-anhydroglucitol as SGLT2-dependent glycemic marker. Clin. Transl. Med. 2021, 11, e470. [Google Scholar] [CrossRef]
- Ghezzi, C.; Loo, D.D.F.; Wright, E.M. Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 2018, 61, 2087–2097. [Google Scholar] [CrossRef]
- Grempler, R.; Thomas, L.; Eckhardt, M.; Himmelsbach, F.; Sauer, A.; Sharp, D.E.; Bakker, R.A.; Mark, M.; Klein, T.; Eickelmann, P. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: Characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes. Metab. 2012, 14, 83–90. [Google Scholar] [CrossRef]
- Skokowa, J.; Dale, D.C.; Touw, I.P.; Zeidler, C.; Welte, K. Severe congenital neutropenias. Nat. Rev. Dis. Primers 2017, 3, 17032. [Google Scholar] [CrossRef]
- Heyworth, P.G.; Cross, A.R.; Curnutte, J.T. Chronic granulomatous disease. Curr. Opin. Immunol. 2003, 15, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.S.; Cheung, Y.Y.; Lee, Y.M.; Mansfield, B.C.; Chou, J.Y. Glucose-6-phosphatase-beta, implicated in a congenital neutropenia syndrome, is essential for macrophage energy homeostasis and functionality. Blood 2012, 119, 4047–4055. [Google Scholar] [CrossRef] [PubMed]
- Volz, M.S.; Nassir, M.; Treese, C.; von Winterfeld, M.; Plockinger, U.; Epple, H.J.; Siegmund, B. Inflammatory bowel disease (IBD)-like disease in a case of a 33-year old man with glycogenosis 1b. BMC Gastroenterol. 2015, 15, 45. [Google Scholar] [CrossRef]
- Goenka, A.; Doherty, J.A.; Al-Farsi, T.; Jagger, C.; Banka, S.; Cheesman, E.; Fagbemi, A.; Hughes, S.M.; Wynn, R.F.; Hussell, T.; et al. Neutrophil dysfunction triggers inflammatory bowel disease in G6PC3 deficiency. J. Leukoc. Biol. 2021, 109, 1147–1154. [Google Scholar] [CrossRef]
- Melis, D.; Carbone, F.; Minopoli, G.; La Rocca, C.; Perna, F.; De Rosa, V.; Galgani, M.; Andria, G.; Parenti, G.; Matarese, G. Cutting Edge: Increased Autoimmunity Risk in Glycogen Storage Disease Type 1b Is Associated with a Reduced Engagement of Glycolysis in T Cells and an Impaired Regulatory T Cell Function. J. Immunol. 2017, 198, 3803–3808. [Google Scholar] [CrossRef]
- Tallis, E.; Karsenty, C.L.; Grimes, A.B.; Karam, L.B.; Elsea, S.H.; Sutton, V.R.; Rawls-Castillo, B.L.; Liu, N.; Soler-Alfonso, C. Untargeted metabolomic profiling in a patient with glycogen storage disease Ib receiving empagliflozin treatment. JIMD Rep. 2022, 63, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Hexner-Erlichman, Z.; Veiga-da-Cunha, M.; Zehavi, Y.; Vadasz, Z.; Sabag, A.D.; Tatour, S.; Spiegel, R. Favorable outcome of empagliflozin treatment in two pediatric glycogen storage disease type 1b patients. Front. Pediatr. 2022, 10, 1071464. [Google Scholar] [CrossRef]
- Kaczor, M.; Greczan, M.; Kierus, K.; Ehmke Vel Emczynska-Seliga, E.; Ciara, E.; Piatosa, B.; Rokicki, D.; Ksiazyk, J.; Wesol-Kucharska, D. Sodium-glucose cotransporter type 2 channel inhibitor: Breakthrough in the treatment of neutropenia in patients with glycogen storage disease type 1b? JIMD Rep. 2022, 63, 199–206. [Google Scholar] [CrossRef]
- Guerra, F.; Gasperini, S.; Bonanomi, S.; Crescitelli, V.; Pretese, R.; Da Dalt, L.; Norata, G.D.; Balzarini, M.; Biondi, A.; Baragetti, A.; et al. Finding balance between mature and immature neutrophils: The effects of empagliflozin in GSD-Ib. eJHaem 2023, 4, 551–554. [Google Scholar] [CrossRef]
- Hiwarkar, P.; Bargir, U.; Pandrowala, A.; Bodhanwala, M.; Thakker, N.; Taur, P.; Madkaikar, M.; Desai, M. SLGT2 Inhibitor Rescues Myelopoiesis in G6PC3 Deficiency. J. Clin. Immunol. 2022, 42, 1653–1659. [Google Scholar] [CrossRef]
- Ledeczi, Z.; Pittner, R.; Krivan, G.; Kardon, T.; Legeza, B. Empagliflozin restores neutropenia and neutrophil dysfunction in a young patient with severe congenital neutropenia type 4. J. Allergy Clin. Immunol. Pract. 2023, 11, 344–346.e341. [Google Scholar] [CrossRef] [PubMed]
- Grunert, S.C.; Derks, T.G.J.; Adrian, K.; Al-Thihli, K.; Ballhausen, D.; Bidiuk, J.; Bordugo, A.; Boyer, M.; Bratkovic, D.; Brunner-Krainz, M.; et al. Efficacy and safety of empagliflozin in glycogen storage disease type Ib: Data from an international questionnaire. Genet. Med. 2022, 24, 1781–1788. [Google Scholar] [CrossRef] [PubMed]
- Schroten, H.; Wendel, U.; Burdach, S.; Roesler, J.; Breidenbach, T.; Schweitzer, S.; Zeidler, C.; Welte, K. Colony-stimulating factors for neutropenia in glycogen storage disease Ib. Lancet 1991, 337, 736–737. [Google Scholar] [CrossRef]
- Roe, T.F.; Coates, T.D.; Thomas, D.W.; Miller, J.H.; Gilsanz, V. Brief report: Treatment of chronic inflammatory bowel disease in glycogen storage disease type Ib with colony-stimulating factors. N. Engl. J. Med. 1992, 326, 1666–1669. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Austin, S.L.; Abdenur, J.E.; Arn, P.; Bali, D.S.; Boney, A.; Chung, W.K.; Dagli, A.I.; Dale, D.; Koeberl, D.; et al. American College of Medical, G. and Genomics. Diagnosis and management of glycogen storage disease type I: A practice guideline of the American College of Medical Genetics and Genomics. Genet. Med. 2014, 16, e1. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Levin, A.; Nangaku, M.; Kadowaki, T.; Agarwal, R.; Hauske, S.J.; Elsasser, A.; Ritter, I.; Steubl, D.; Wanner, C.; et al. Safety of Empagliflozin in Patients With Type 2 Diabetes and Chronic Kidney Disease: Pooled Analysis of Placebo-Controlled Clinical Trials. Diabetes Care 2022, 45, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Donadieu, J.; Alimi, A.; Brassier, A.; Beaupain, B.; Wicker, C.; Alili, J.-M.; Bellanne-Chantelot, C.; Chaussade, A.; Castelle, M.; Lamarque, M.; et al. Oral SGLT2 Inhibitors in Glycogen Storage Disease Type Ib and G6PC3-Deficiency. Preliminary Results from an Off-Label Study of 21 Patients. Blood 2022, 140 (Suppl. S1), 8320–8322. [Google Scholar] [CrossRef]
- Atal, S.; Fatima, Z.; Singh, S.; Balakrishnan, S.; Joshi, R. Remogliflozin: The new low cost SGLT-2 inhibitor for type 2 diabetes mellitus. Diabetol. Int. 2020, 12, 247–253. [Google Scholar] [CrossRef]
- Ayoub, B.M.; Mowaka, S.; Elzanfaly, E.S.; Ashoush, N.; Elmazar, M.M.; Mousa, S.A. Pharmacokinetic Evaluation of Empagliflozin in Healthy Egyptian Volunteers Using LC-MS/MS and Comparison with Other Ethnic Populations. Sci. Rep. 2017, 7, 2583. [Google Scholar] [CrossRef]
- Kapur, A.; O’Connor-Semmes, R.; Hussey, E.K.; Dobbins, R.L.; Tao, W.; Hompesch, M.; Smith, G.A.; Polli, J.W.; James, C.D., Jr.; Mikoshiba, I.; et al. First human dose-escalation study with remogliflozin etabonate, a selective inhibitor of the sodium-glucose transporter 2 (SGLT2), in healthy subjects and in subjects with type 2 diabetes mellitus. BMC Pharmacol. Toxicol. 2013, 14, 26. [Google Scholar] [CrossRef]
- Li, M.; Maruthur, N.M.; Loomis, S.J.; Pietzner, M.; North, K.E.; Mei, H.; Morrison, A.C.; Friedrich, N.; Pankow, J.S.; Nauck, M.; et al. Genome-wide association study of 1,5-anhydroglucitol identifies novel genetic loci linked to glucose metabolism. Sci. Rep. 2017, 7, 2812. [Google Scholar] [CrossRef] [PubMed]
- Loomis, S.J.; Kottgen, A.; Li, M.; Tin, A.; Coresh, J.; Boerwinkle, E.; Gibbs, R.; Muzny, D.; Pankow, J.; Selvin, E.; et al. Rare variants in SLC5A10 are associated with serum 1,5-anhydroglucitol (1,5-AG) in the Atherosclerosis Risk in Communities (ARIC) Study. Sci. Rep. 2019, 9, 5941. [Google Scholar] [CrossRef] [PubMed]
- Long, T.; Hicks, M.; Yu, H.C.; Biggs, W.H.; Kirkness, E.F.; Menni, C.; Zierer, J.; Small, K.S.; Mangino, M.; Messier, H.; et al. Whole-genome sequencing identifies common-to-rare variants associated with human blood metabolites. Nat. Genet. 2017, 49, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Aoun, B.; Sanjad, S.; Degheili, J.A.; Barhoumi, A.; Bassyouni, A.; Karam, P.E. Kidney and Metabolic Phenotypes in Glycogen Storage Disease Type-I Patients. Front. Pediatr. 2020, 8, 591. [Google Scholar] [CrossRef]
- D’Acierno, M.; Resaz, R.; Iervolino, A.; Nielsen, R.; Sardella, D.; Siccardi, S.; Costanzo, V.; D’Apolito, L.; Suzumoto, Y.; Segalerba, D.; et al. Dapagliflozin Prevents Kidney Glycogen Accumulation and Improves Renal Proximal Tubule Cell Functions in a Mouse Model of Glycogen Storage Disease Type 1b. J. Am. Soc. Nephrol. 2022, 33, 1864–1875. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veiga-da-Cunha, M.; Wortmann, S.B.; Grünert, S.C.; Van Schaftingen, E. Treatment of the Neutropenia Associated with GSD1b and G6PC3 Deficiency with SGLT2 Inhibitors. Diagnostics 2023, 13, 1803. https://doi.org/10.3390/diagnostics13101803
Veiga-da-Cunha M, Wortmann SB, Grünert SC, Van Schaftingen E. Treatment of the Neutropenia Associated with GSD1b and G6PC3 Deficiency with SGLT2 Inhibitors. Diagnostics. 2023; 13(10):1803. https://doi.org/10.3390/diagnostics13101803
Chicago/Turabian StyleVeiga-da-Cunha, Maria, Saskia B. Wortmann, Sarah C. Grünert, and Emile Van Schaftingen. 2023. "Treatment of the Neutropenia Associated with GSD1b and G6PC3 Deficiency with SGLT2 Inhibitors" Diagnostics 13, no. 10: 1803. https://doi.org/10.3390/diagnostics13101803
APA StyleVeiga-da-Cunha, M., Wortmann, S. B., Grünert, S. C., & Van Schaftingen, E. (2023). Treatment of the Neutropenia Associated with GSD1b and G6PC3 Deficiency with SGLT2 Inhibitors. Diagnostics, 13(10), 1803. https://doi.org/10.3390/diagnostics13101803