
An RBD-Based Diagnostic Method Useful for the Surveillance of Protective Immunity against SARS-CoV-2 in the Population

 , , ,
, , ,  , ,
, ,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sera Samples
2.2. ELISA Testing of Sera
2.3. Microneutralization Assays
2.4. Statistical Analysis
2.5. Results
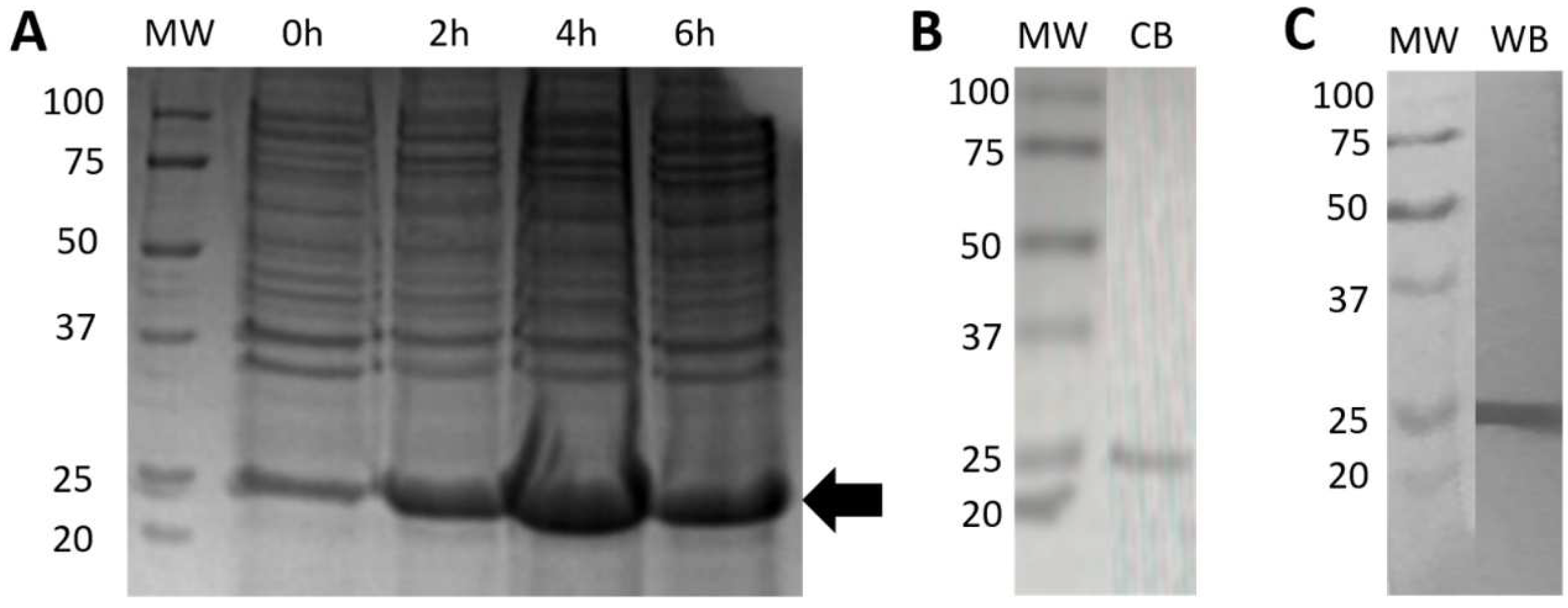
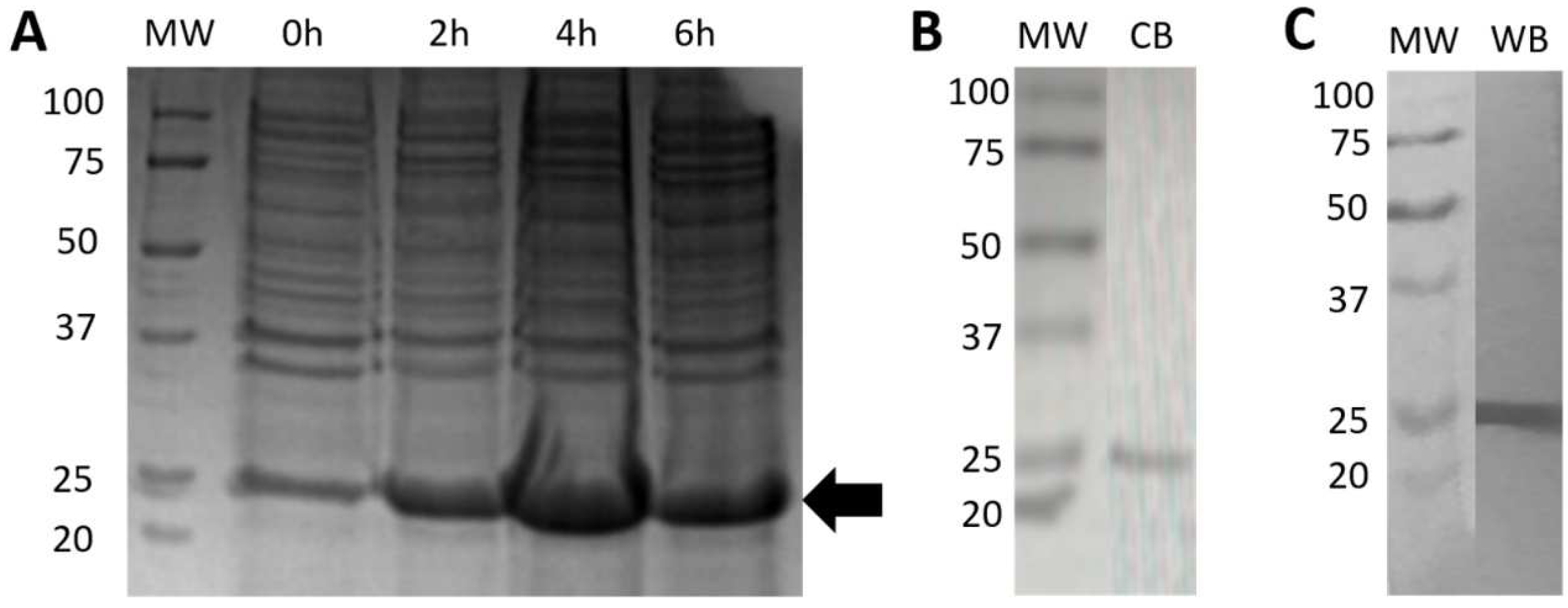
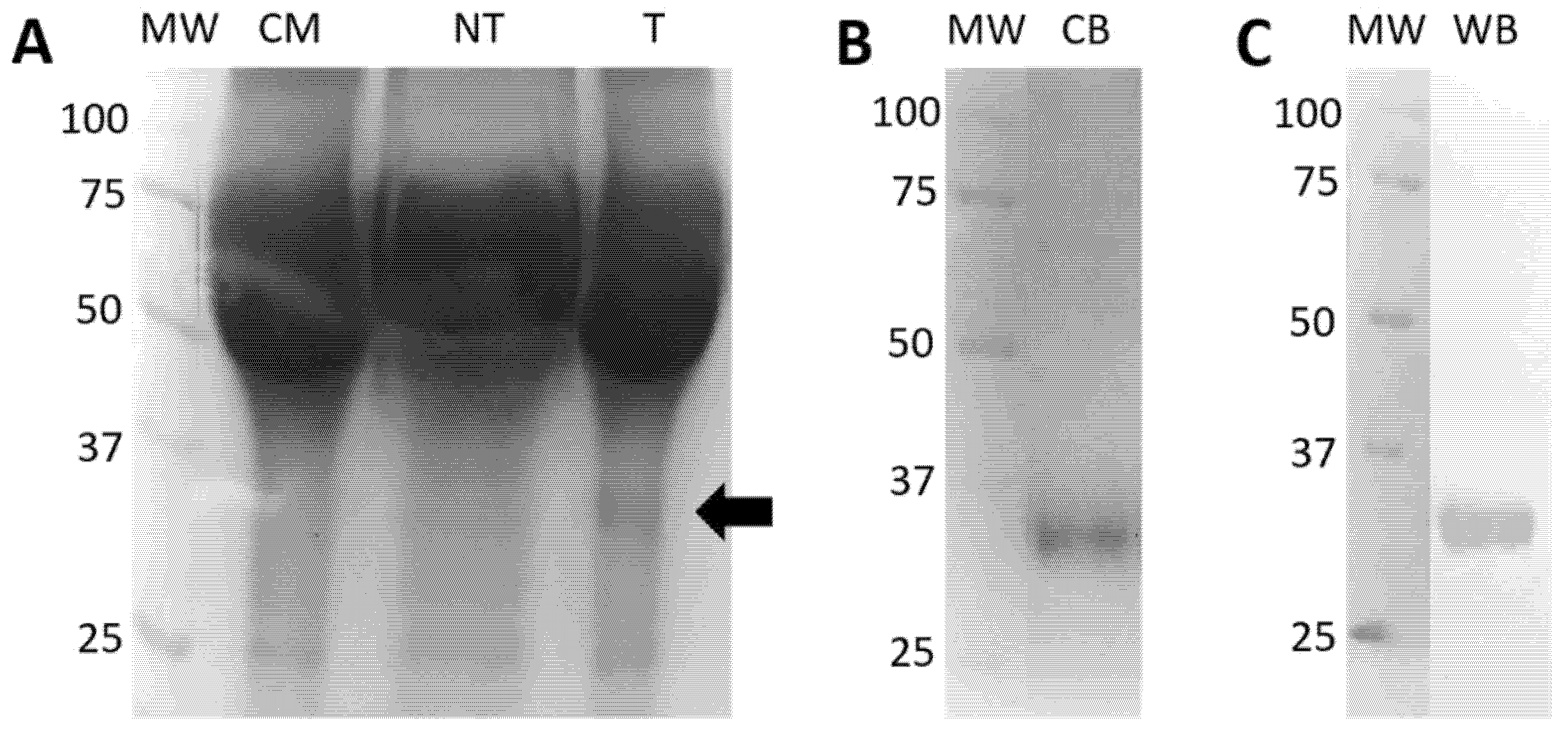
2.5.1. Expression and Purification of Bacterial rRBD
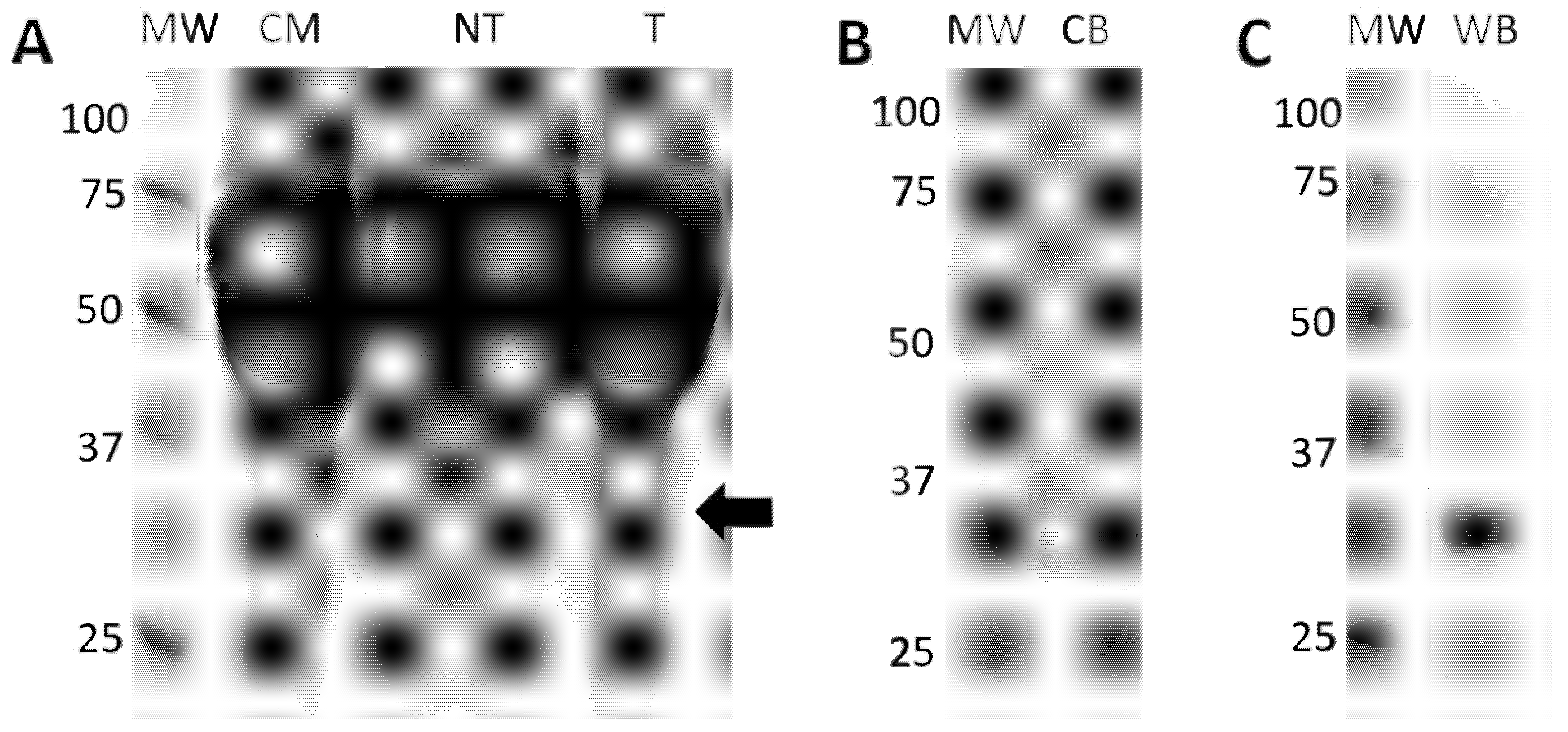
2.5.2. Expression and Purification of Eukaryotic rRBD
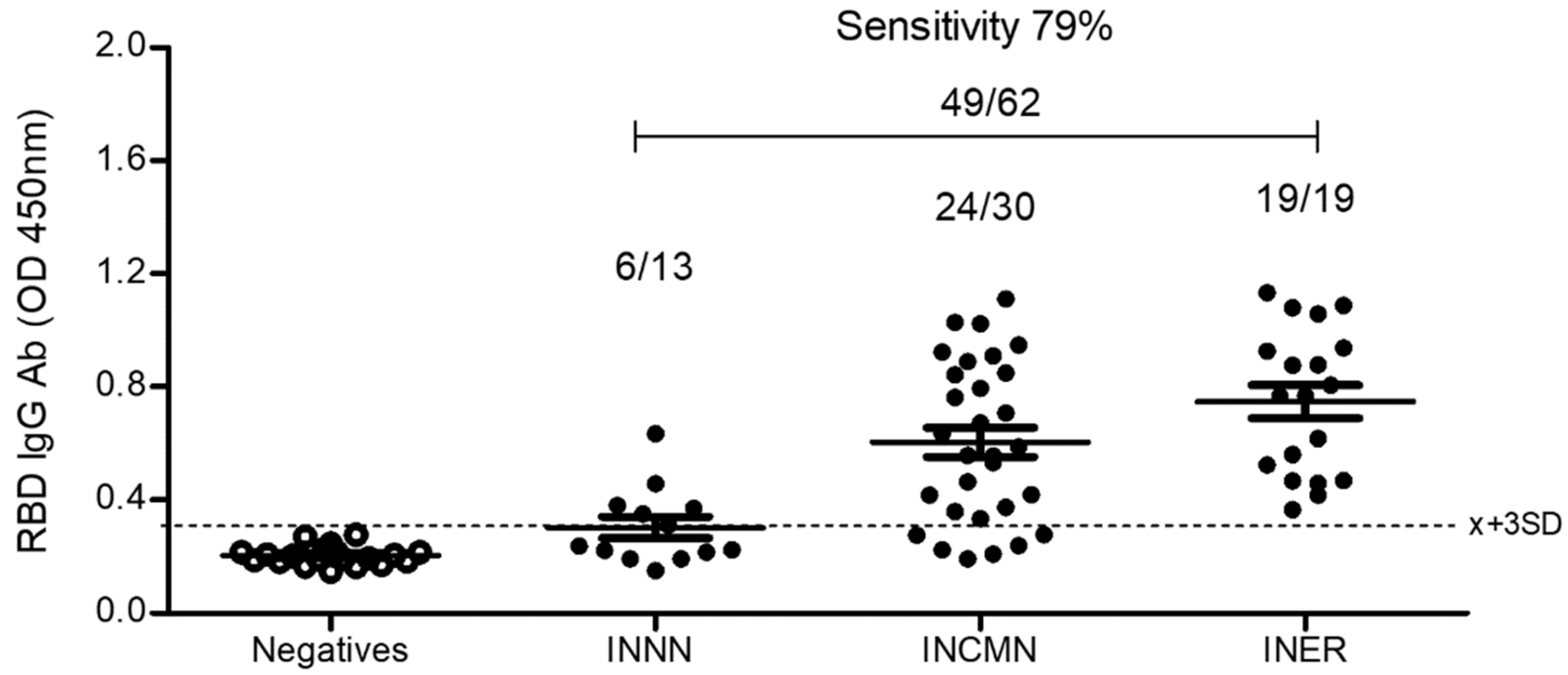
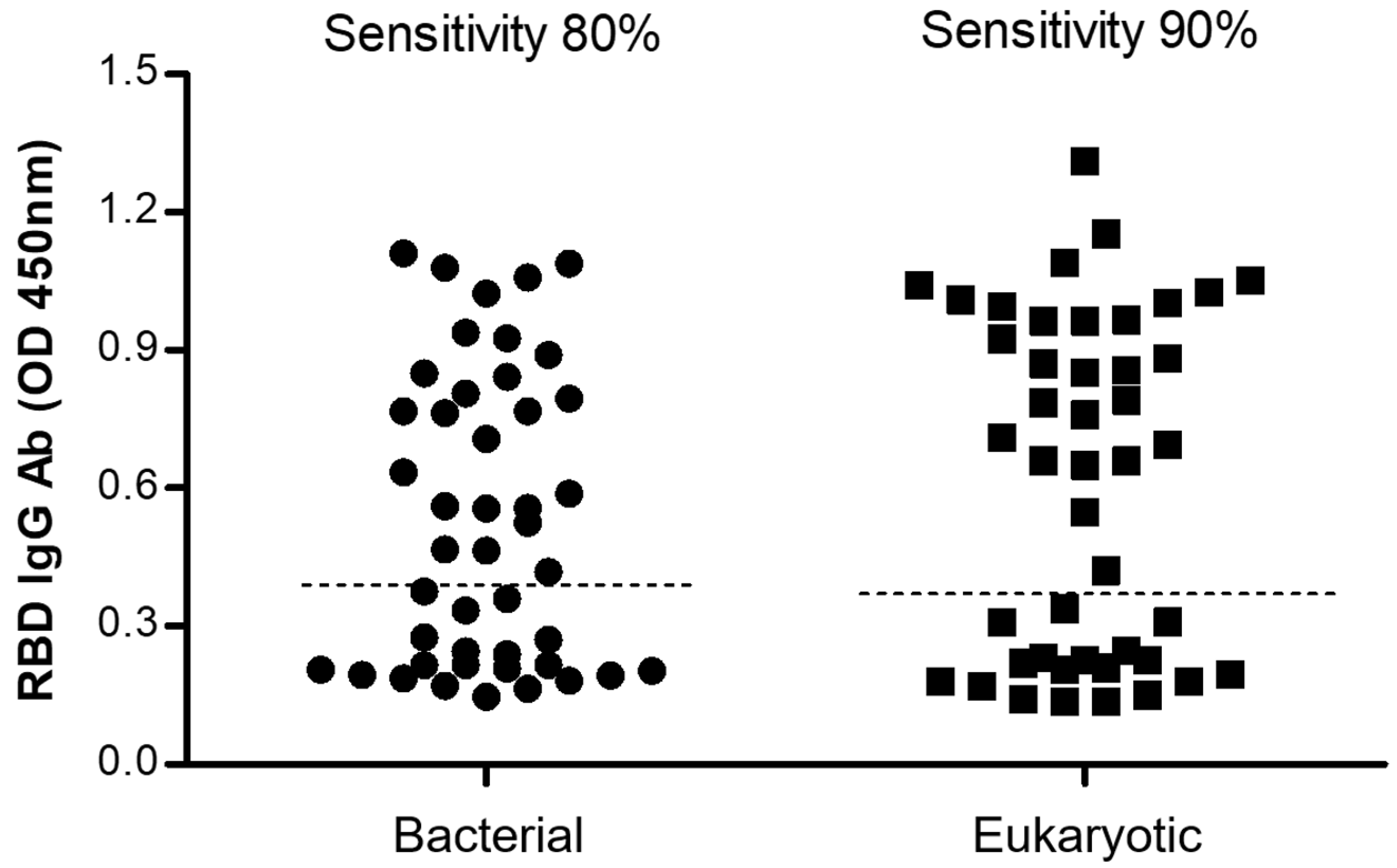
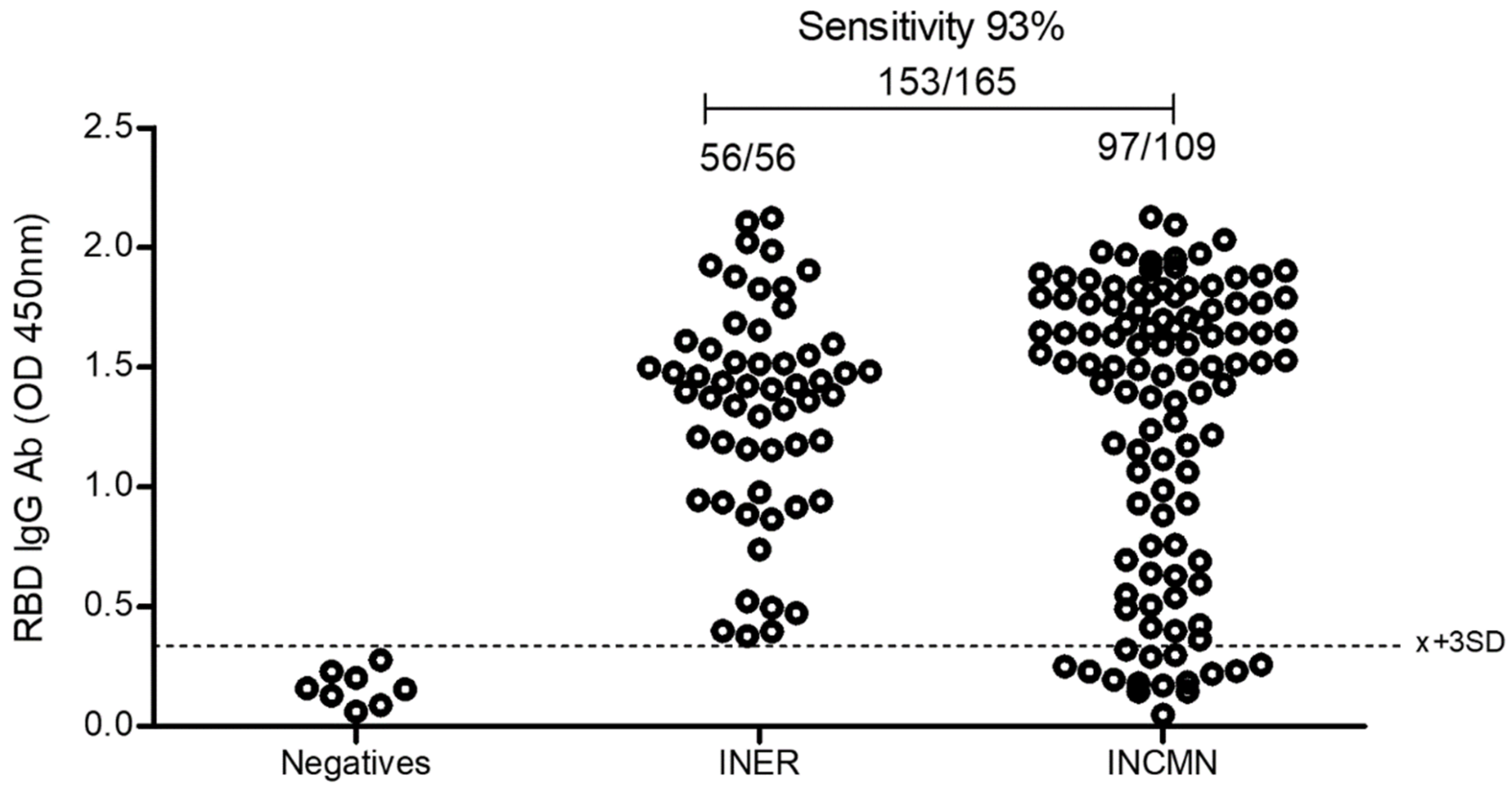
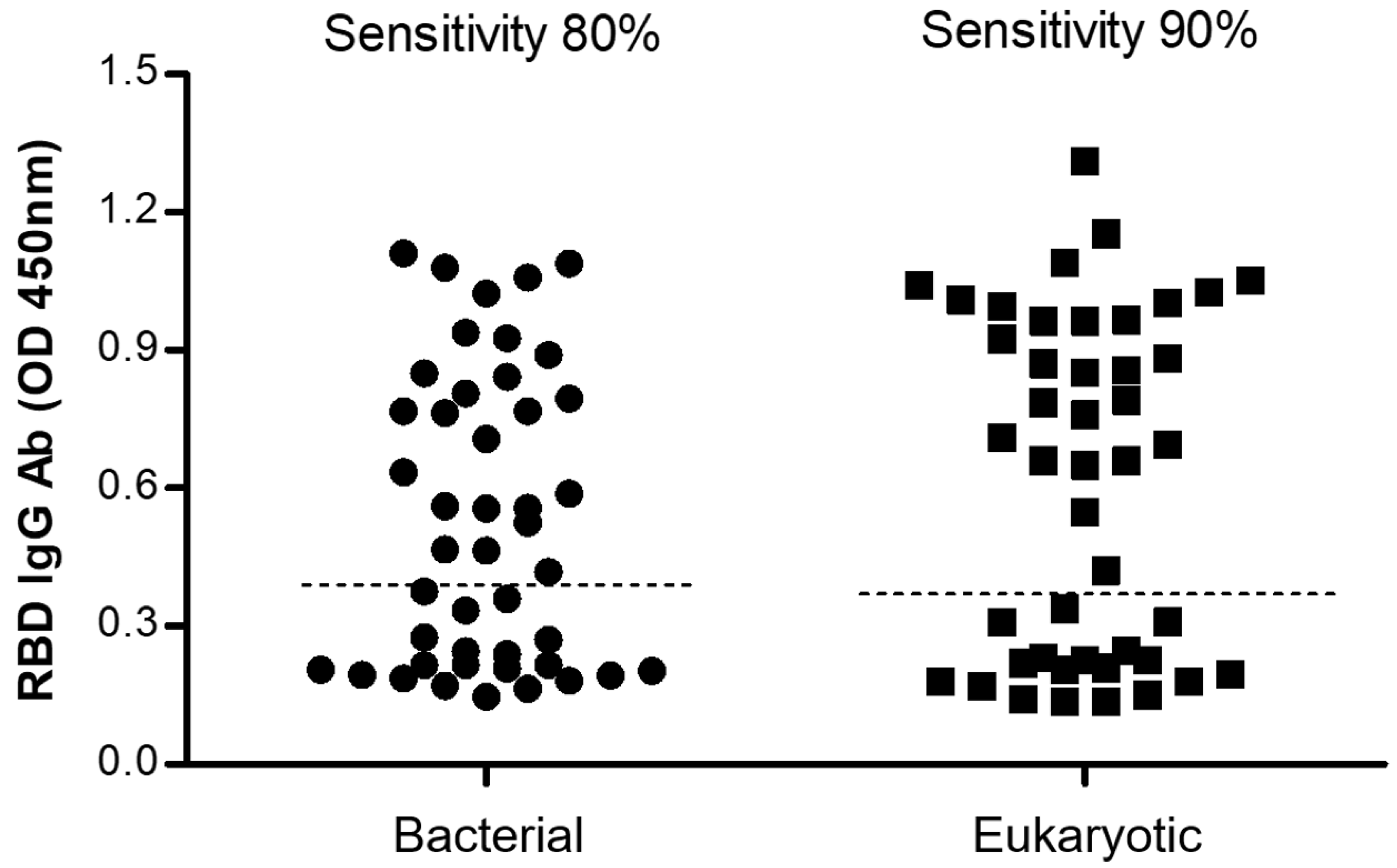
2.5.3. Comparison between the Bacterial and the Eukaryotic rRBD-Based ELISA
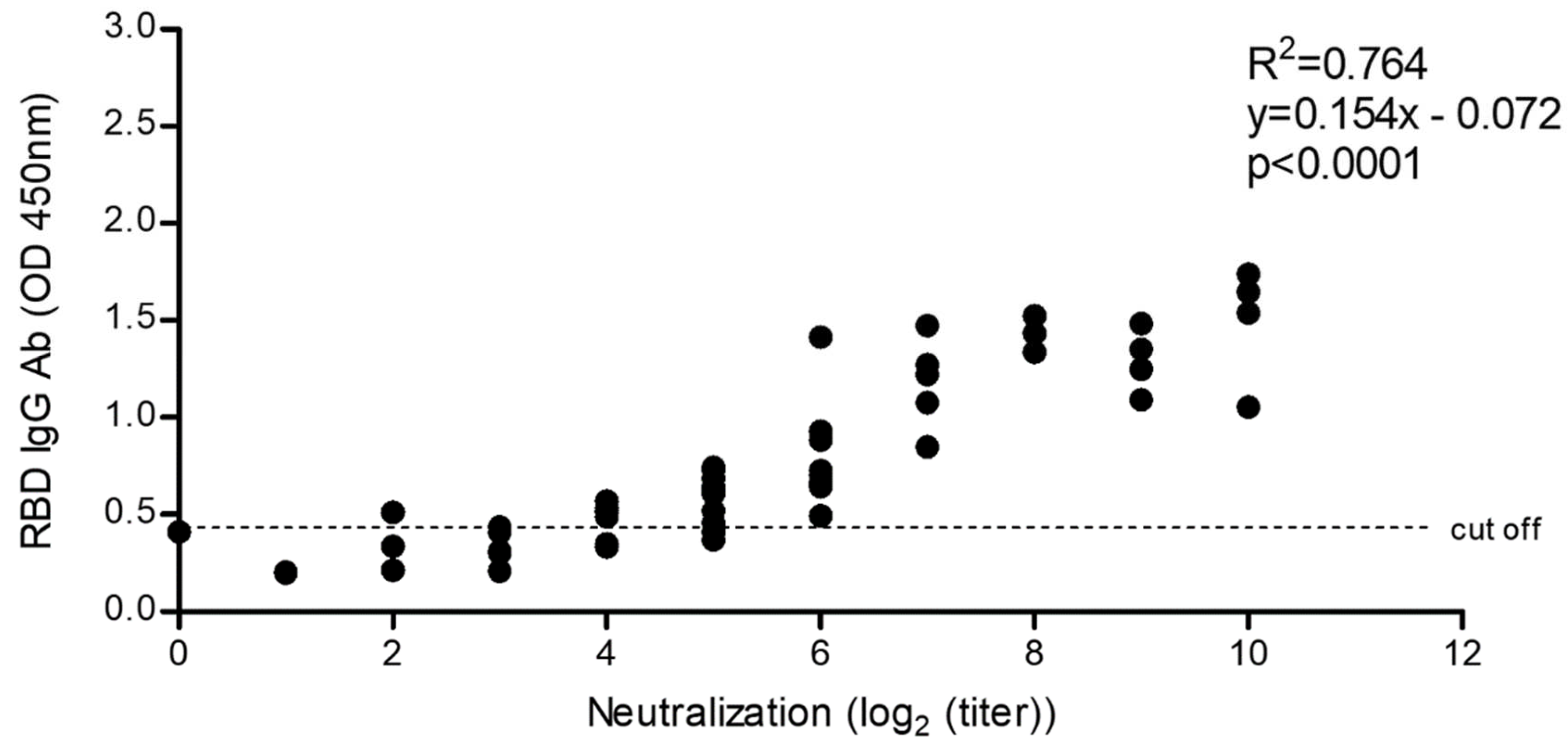
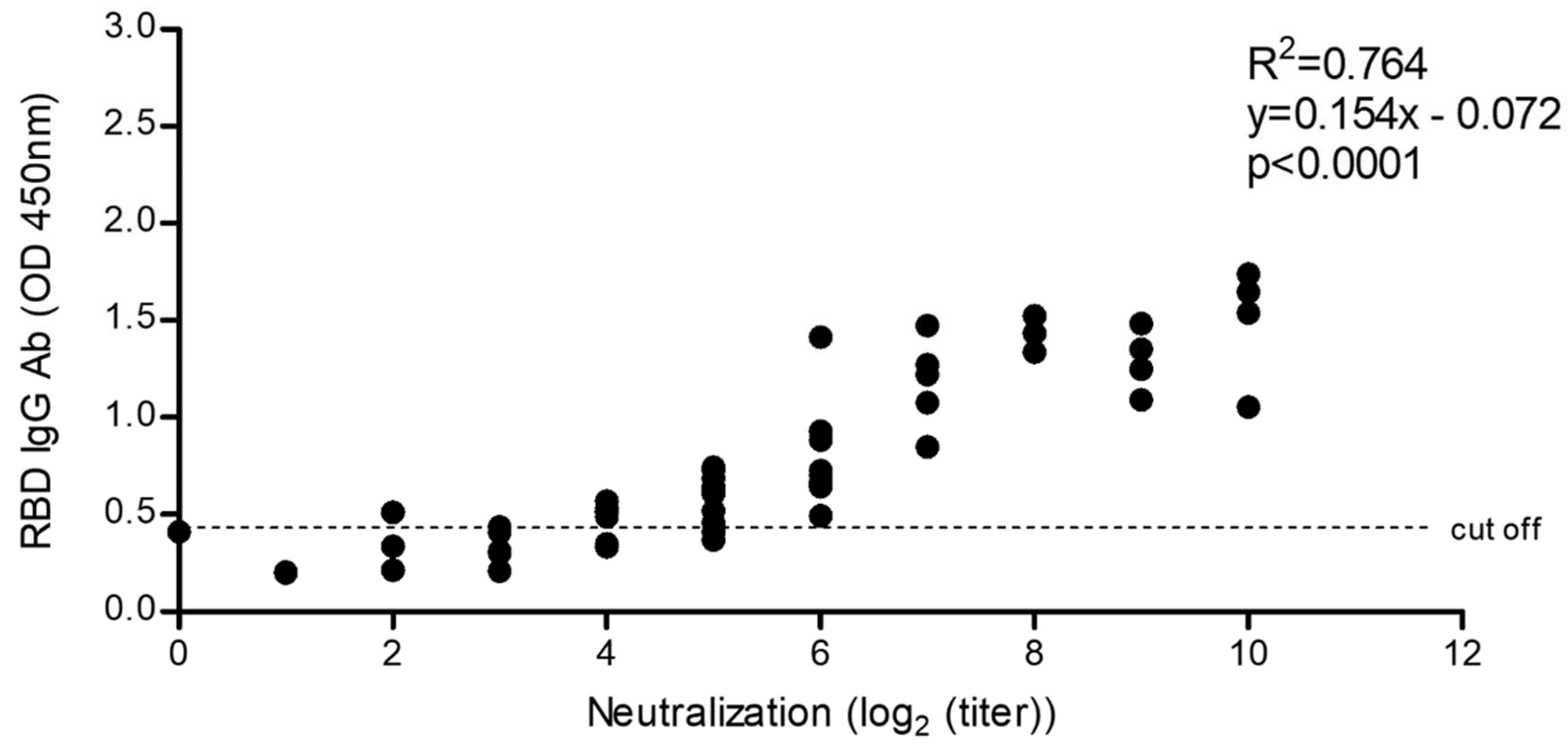
2.5.4. Correlation of SARS-CoV-2 Neutralization Activity and Anti-RBD IgG Antibody Levels
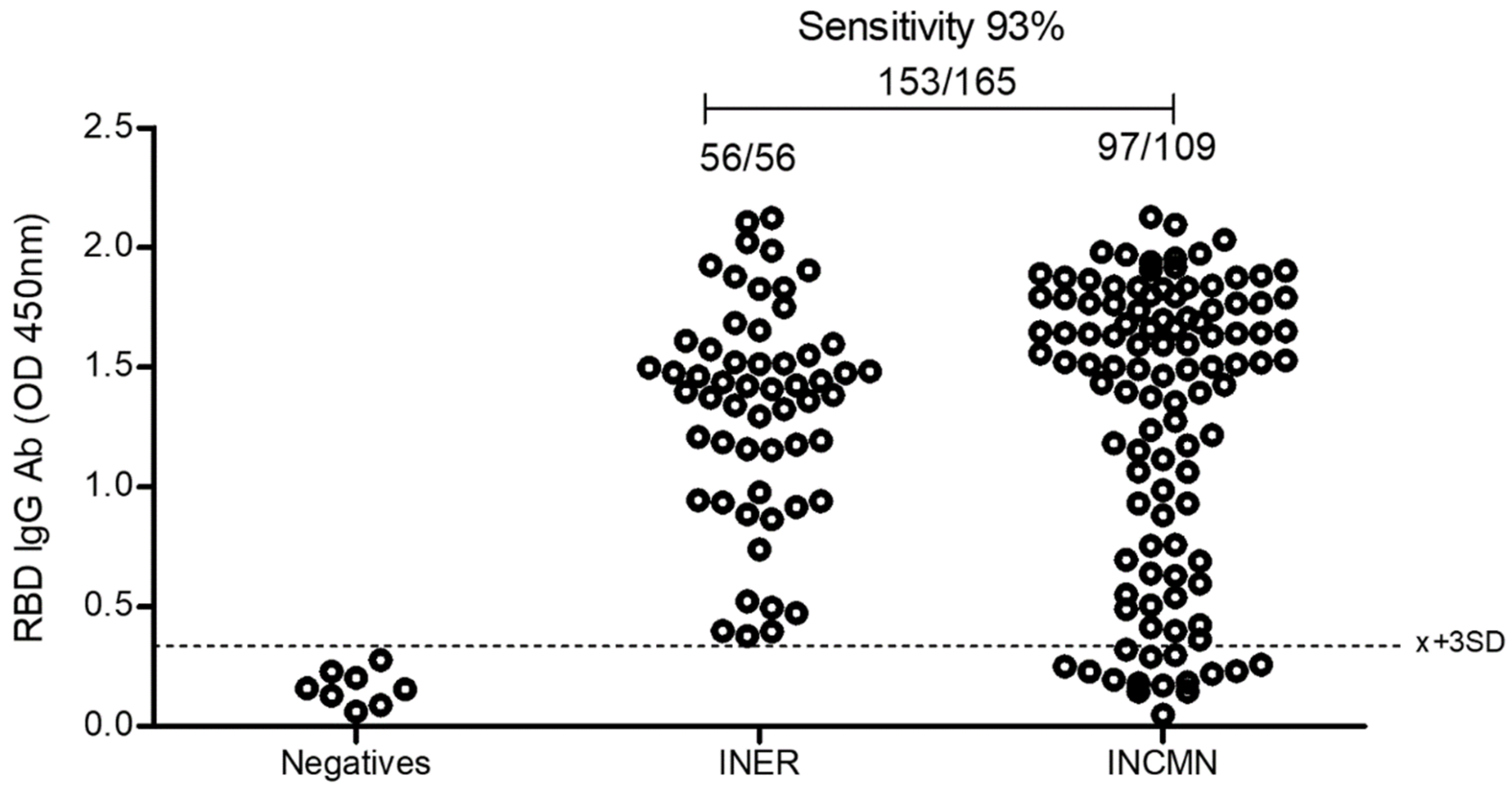
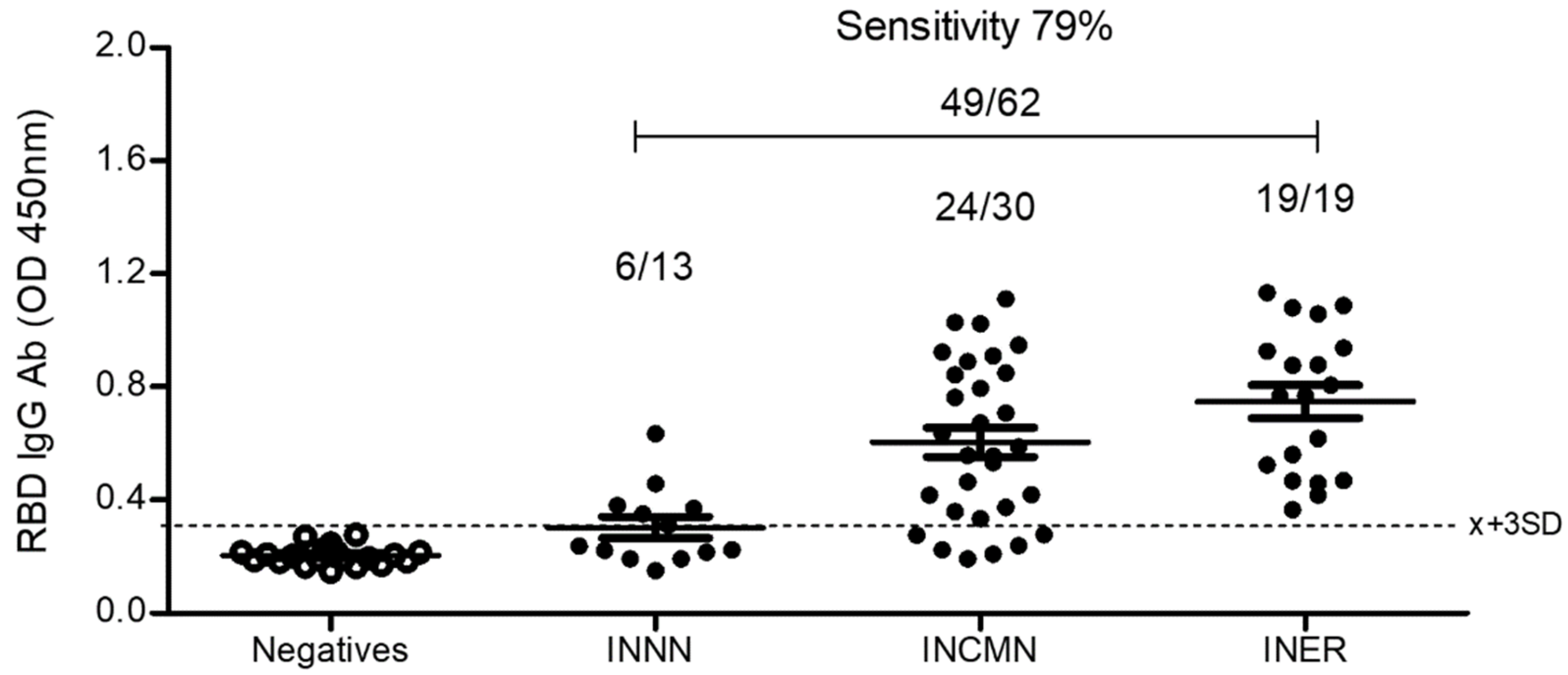
2.5.5. Comparison of the Eukaryotic rRBD vs. Commercial SARS-CoV-2 ELISA
3. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Shi, Z.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nature reviews Microbiology 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Hong, W.; Pan, X.; Lu, G.; Wei, X. SARS-CoV-2 Omicron variant: Characteristics and prevention. MedComm 2021, 2, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Khandia, R.; Singhal, S.; Alqahtani, T.; Kamal, M.A.; El-Shall, N.A.; Nainu, F.; Desingu, P.A.; Dhama, K. Emergence of SARS-CoV-2 Omicron (B.1.1.529) variant, salient features, high global health concerns and strategies to counter it amid ongoing COVID-19 pandemic. Environ. Res. 2022, 209, 112816. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Huo, J.; Zhou, D.; Zahradník, J.; Supasa, P.; Liu, C.; Duyvesteyn, H.M.; Ginn, H.M.; Mentzer, A.J.; Tuekprakhon, A.; et al. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell 2022, 185, 467–484.e15. [Google Scholar] [CrossRef]
- Gong, W.; Aspatwar, A.; Wang, S.; Parkkila, S.; Wu, X. COVID-19 pandemic: SARS-CoV-2 specific vaccines and challenges, protection via BCG trained immunity, and clinical trials. Expert Rev. Vaccines 2021, 20, 857–880. [Google Scholar] [CrossRef]
- Hoffmann, M.; Krüger, N.; Schulz, S.; Cossmann, A.; Rocha, C.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Moldenhauer, A.S.; Winkler, M.S.; et al. The Omicron variant is highly resistant against antibody-mediated neutralization: Implications for control of the COVID-19 pandemic. Cell 2021, 185, 447–456.e11. [Google Scholar] [CrossRef]
- Motamedi, H.; Ari, M.M.; Dashtbin, S.; Fathollahi, M.; Hossainpour, H.; Alvandi, A.; Moradi, J.; Abiri, R. An update review of globally reported SARS-CoV-2 vaccines in preclinical and clinical stages. Int. Immunopharmacol. 2021, 96, 107763. [Google Scholar] [CrossRef]
- Post, N.; Eddy, D.; Huntley, C.; van Schalkwyk, M.; Shrotri, M.; Leeman, D.; Rigby, S.; Williams, S.V.; Bermingham, W.H.; Kellam, P.; et al. Antibody response to SARS-CoV-2 infection in humans: A systematic review. PLoS ONE 2020, 15, e0244126. [Google Scholar] [CrossRef]
- Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e62020. [Google Scholar] [CrossRef]
- Wu, J.; Deng, W.; Li, S.; Yang, X. Advances in research on ACE2 as a receptor for 2019-nCoV. Cell. Mol. Life Sci. CMLS 2021, 78, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, O.; Martiny, D.; Rochas, O.; van Belkum, A.; Kozlakidis, Z. Considerations for diagnostic COVID-19 tests. Nat. Rev. Microbiol. 2021, 19, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F.; Simon, V. Serology assays to manage COVID-19. Science 2021, 368, 1060–1061. [Google Scholar] [CrossRef]
- Kumavath, R.; Barh, D.; Andrade, B.S.; Imchen, M.; Aburjaile, F.F.; Ch, A.; Rodrigues, D.L.N.; Tiwari, S.; Alzahrani, K.J.; Góes-Neto, A.; et al. The Spike of SARS-CoV-2: Uniqueness and Applications. Front. Immunol. 2021, 12, 663912. [Google Scholar] [CrossRef]
- Amanat, F.; Stadlbauer, D.; Strohmeier, S.; Nguyen, T.H.O.; Chromikova, V.; McMahon, M.; Jiang, K.; Arunkumar, G.A.; Jurczyszak, D.; Polanco, J.; et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med. 2020, 26, 1033–1036. [Google Scholar] [CrossRef]
- Law, J.; Logan, M.; Joyce, M.A.; Landi, A.; Hockman, D.; Crawford, K.; Johnson, J.; LaChance, G.; Saffran, H.A.; Shields, J.; et al. SARS-CoV-2 recombinant Receptor-Binding-Domain (RBD) induces neutralizing antibodies against variant strains of SARS-CoV-2 and SARS-CoV-1. Vaccine 2021, 39, 5769–5779. [Google Scholar] [CrossRef]
- Gaebler, C.; Wang, Z.; Lorenzi, J.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y.; et al. Evolution of antibody immunity to SARS-CoV-2. Nature 2021, 591, 639–644. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Conceicao, C.; Thakur, N.; Human, S.; Kelly, J.T.; Logan, L.; Bialy, D.; Bhat, S.; Stevenson-Leggett, P.; Zagrajek, A.K.; Hollinghurst, P.; et al. The SARS-CoV-2 Spike protein has a broad tropism for mammalian ACE2 proteins. PLoS Biol. 2020, 18, e3001016. [Google Scholar] [CrossRef]
- Medina-Enríquez, M.M.; Lopez-León, S.; Carlos-Escalante, J.A.; Aponte-Torres, Z.; Cuapio, A.; Wegman-Ostrosky, T. ACE2: The molecular doorway to SARS-CoV-2. Cell Biosci. 2020, 10, 148. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, L.; Park, Y.J.; Tortorici, M.A.; Czudnochowski, N.; Walls, A.C.; Beltramello, M.; Silacci-Fregni, C.; Pinto, D.; Rosen, L.E.; Bowen, J.E.; et al. Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell 2020, 183, 1024–1042.e21. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual-Iglesias, A.; Canton, J.; Ortega-Prieto, A.M.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A. An Overview of Vaccines against SARS-CoV-2 in the COVID-19 Pandemic Era. Pathogens 2021, 10, 1030. [Google Scholar] [CrossRef]
- Eyre, D.W.; Lumley, S.F.; Wei, J.; Cox, S.; James, T.; Justice, A.; Jesuthasan, G.; O’Donnell, D.; Howarth, A.; Hatch, S.B.; et al. Quantitative SARS-CoV-2 anti-spike responses to Pfizer-BioNTech and Oxford-AstraZeneca vaccines by previous infection status. Clin. Microbiol. Infect. 2021, 27, 1516.e7–1516.e14. [Google Scholar] [CrossRef]
- Vályi-Nagy, I.; Matula, Z.; Gönczi, M.; Tasnády, S.; Bekő, G.; Réti, M.; Ajzner, É.; Uher, F. Comparison of antibody and T cell responses elicited by BBIBP-CorV (Sinopharm) and BNT162b2 (Pfizer-BioNTech) vaccines against SARS-CoV-2 in healthy adult humans. Geroscience 2021, 43, 2321–2331. [Google Scholar] [CrossRef]
- Guzmán-Martínez, O.; Guardado, K.; de Guevara, E.L.; Navarro, S.; Hernández, C.; Zenteno-Cuevas, R.; Montero, H. IgG Antibodies Generation and Side Effects Caused by Ad5-nCoV Vaccine (CanSino Biologics) and BNT162b2 Vaccine (Pfizer/BioNTech) among Mexican Population. Vaccines 2021, 9, 999. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayón-Núñez, D.A.; Cervantes-Torres, J.; Cabello-Gutiérrez, C.; Rosales-Mendoza, S.; Rios-Valencia, D.; Huerta, L.; Bobes, R.J.; Carrero, J.C.; Segura-Velázquez, R.; Fierro, N.A.; et al. An RBD-Based Diagnostic Method Useful for the Surveillance of Protective Immunity against SARS-CoV-2 in the Population. Diagnostics 2022, 12, 1629. https://doi.org/10.3390/diagnostics12071629
Ayón-Núñez DA, Cervantes-Torres J, Cabello-Gutiérrez C, Rosales-Mendoza S, Rios-Valencia D, Huerta L, Bobes RJ, Carrero JC, Segura-Velázquez R, Fierro NA, et al. An RBD-Based Diagnostic Method Useful for the Surveillance of Protective Immunity against SARS-CoV-2 in the Population. Diagnostics. 2022; 12(7):1629. https://doi.org/10.3390/diagnostics12071629
Chicago/Turabian StyleAyón-Núñez, Dolores Adriana, Jacquelynne Cervantes-Torres, Carlos Cabello-Gutiérrez, Sergio Rosales-Mendoza, Diana Rios-Valencia, Leonor Huerta, Raúl J. Bobes, Julio César Carrero, René Segura-Velázquez, Nora Alma Fierro, and et al. 2022. "An RBD-Based Diagnostic Method Useful for the Surveillance of Protective Immunity against SARS-CoV-2 in the Population" Diagnostics 12, no. 7: 1629. https://doi.org/10.3390/diagnostics12071629
APA StyleAyón-Núñez, D. A., Cervantes-Torres, J., Cabello-Gutiérrez, C., Rosales-Mendoza, S., Rios-Valencia, D., Huerta, L., Bobes, R. J., Carrero, J. C., Segura-Velázquez, R., Fierro, N. A., Hernández, M., Zúñiga-Ramos, J., Gamba, G., Cárdenas, G., Frías-Jiménez, E., Herrera, L. A., Fragoso, G., Sciutto, E., Suárez-Güemes, F., & Laclette, J. P. (2022). An RBD-Based Diagnostic Method Useful for the Surveillance of Protective Immunity against SARS-CoV-2 in the Population. Diagnostics, 12(7), 1629. https://doi.org/10.3390/diagnostics12071629