
Target Enrichment Approaches for Next-Generation Sequencing Applications in Oncology

Abstract
:1. Introduction
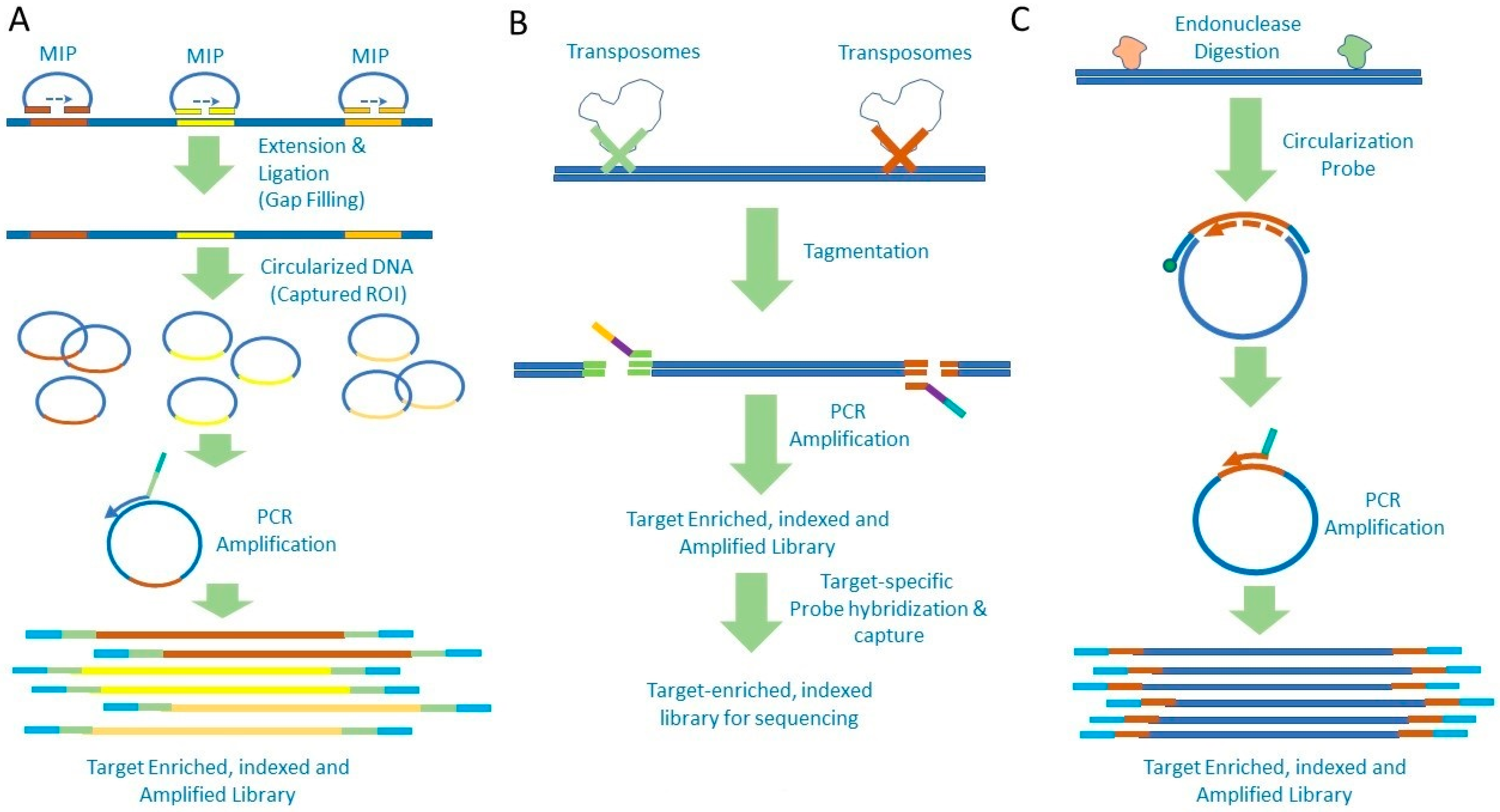
2. Amplicon-Based Target Enrichment
3. Target Enrichment by Hybrid Capture
4. Automation of Target Enrichment
5. Comparison of Target Enrichment Methodologies
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, J.; Chiodini, R.; Badr, A.; Zhang, G. The Impact of Next-Generation Sequencing on Genomics. J. Genet. Genom. 2011, 38, 95–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rennert, H.; Eng, K.; Zhang, T.; Tan, A.; Xiang, J.; Romanel, A.; Kim, R.; Tam, W.; Liu, Y.-C.; Bhinder, B.; et al. Development and Validation of a Whole-Exome Sequencing Test for Simultaneous Detection of Point Mutations, Indels and Copy-Number Alterations for Precision Cancer Care. NPJ Genom. Med. 2016, 1, 16019. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.S.; Ashino, R.; Oota, H.; Ishida, H.; Niimura, Y.; Touhara, K.; Melin, A.D.; Kawamura, S. Genetic Variation of Olfactory Receptor Gene Family in a Japanese Population. Anthropol. Sci. 2022, 211024. [Google Scholar] [CrossRef]
- Jones, M.R.; Good, J.M. Targeted Capture in Evolutionary and Ecological Genomics. Mol. Ecol. 2016, 25, 185–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefterova, M.I.; Suarez, C.J.; Banaei, N.; Pinsky, B.A. Next-Generation Sequencing for Infectious Disease Diagnosis and Management: A Report of the Association for Molecular Pathology. J. Mol. Diagn. 2015, 17, 623–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grover, C.E.; Salmon, A.; Wendel, J.F. Targeted Sequence Capture as a Powerful Tool for Evolutionary Analysis1. Am. J. Bot. 2012, 99, 312–319. [Google Scholar] [CrossRef] [Green Version]
- Kozarewa, I.; Armisen, J.; Gardner, A.F.; Slatko, B.E.; Hendrickson, C.L. Overview of Target Enrichment Strategies. Curr. Protoc. Mol. Biol. 2015, 112, 7–21. [Google Scholar] [CrossRef]
- Hehir-Kwa, J.Y.; Claustres, M.; Hastings, R.J.; van Ravenswaaij-Arts, C.; Christenhusz, G.; Genuardi, M.; Melegh, B.; Cambon-Thomsen, A.; Patsalis, P.; Vermeesch, J. Towards a European Consensus for Reporting Incidental Findings during Clinical NGS Testing. Eur. J. Hum. Genet. 2015, 23, 1601–1606. [Google Scholar] [CrossRef]
- Mertes, F.; ElSharawy, A.; Sauer, S.; van Helvoort, J.M.; van der Zaag, P.J.; Franke, A.; Nilsson, M.; Lehrach, H.; Brookes, A.J. Targeted Enrichment of Genomic DNA Regions for Next-Generation Sequencing. Brief. Funct. Genom. 2011, 10, 374–386. [Google Scholar] [CrossRef]
- Mamanova, L.; Coffey, A.J.; Scott, C.E.; Kozarewa, I.; Turner, E.H.; Kumar, A.; Howard, E.; Shendure, J.; Turner, D.J. Target-Enrichment Strategies for next-Generation Sequencing. Nat. Methods 2010, 7, 111–118. [Google Scholar] [CrossRef]
- Singh, R.R.; Patel, K.P.; Routbort, M.J.; Reddy, N.G.; Barkoh, B.A.; Handal, B.; Kanagal-Shamanna, R.; Greaves, W.O.; Medeiros, L.J.; Aldape, K.D. Clinical Validation of a Next-Generation Sequencing Screen for Mutational Hotspots in 46 Cancer-Related Genes. J. Mol. Diagn. 2013, 15, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Patel, K.P.; Routbort, M.J.; Aldape, K.; Lu, X.; Manekia, J.; Abraham, R.; Reddy, N.G.; Barkoh, B.A.; Veliyathu, J. Clinical Massively Parallel Next-Generation Sequencing Analysis of 409 Cancer-Related Genes for Mutations and Copy Number Variations in Solid Tumours. Br. J. Cancer 2014, 111, 2014–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingo, T.S.; Kotlar, A.; Cutler, D.J. MPD: Multiplex Primer Design for next-Generation Targeted Sequencing. BMC Bioinformat. 2017, 18, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kechin, A.; Borobova, V.; Boyarskikh, U.; Khrapov, E.; Subbotin, S.; Filipenko, M. NGS-PrimerPlex: High-Throughput Primer Design for Multiplex Polymerase Chain Reactions. PLoS Comput. Biol. 2020, 16, e1008468. [Google Scholar] [CrossRef] [PubMed]
- Barnes, W.M. The Fidelity of Taq Polymerase Catalyzing PCR Is Improved by an N-Terminal Deletion. Gene 1992, 112, 29–35. [Google Scholar] [CrossRef]
- Jia, H.; Guo, Y.; Zhao, W.; Wang, K. Long-Range PCR in next-Generation Sequencing: Comparison of Six Enzymes and Evaluation on the MiSeq Sequencer. Sci. Rep. 2014, 4, 5737. [Google Scholar] [CrossRef] [Green Version]
- Ozcelik, H.; Shi, X.; Chang, M.C.; Tram, E.; Vlasschaert, M.; Di Nicola, N.; Kiselova, A.; Yee, D.; Goldman, A.; Dowar, M. Long-Range PCR and next-Generation Sequencing of BRCA1 and BRCA2 in Breast Cancer. J. Mol. Diagn. 2012, 14, 467–475. [Google Scholar] [CrossRef]
- Tewhey, R.; Warner, J.B.; Nakano, M.; Libby, B.; Medkova, M.; David, P.H.; Kotsopoulos, S.K.; Samuels, M.L.; Hutchison, J.B.; Larson, J.W. Microdroplet-Based PCR Enrichment for Large-Scale Targeted Sequencing. Nat. Biotechnol. 2009, 27, 1025–1031. [Google Scholar] [CrossRef] [Green Version]
- Judkins, T.; Leclair, B.; Bowles, K.; Gutin, N.; Trost, J.; McCulloch, J.; Bhatnagar, S.; Murray, A.; Craft, J.; Wardell, B. Development and Analytical Validation of a 25-Gene next Generation Sequencing Panel That Includes the BRCA1 and BRCA2 Genes to Assess Hereditary Cancer Risk. BMC Cancer 2015, 15, 215. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Lindsay, D.; Chen, Q.B.; Garrett, A.L.; Tan, X.M.; Anders, C.K.; Carey, L.A.; Gupta, G.P. Tracking Plasma DNA Mutation Dynamics in Estrogen Receptor Positive Metastatic Breast Cancer with DPCR-SEQ. NPJ Breast Cancer 2018, 4, 39. [Google Scholar] [CrossRef]
- Murphy, T.W.; Hsieh, Y.-P.; Zhu, B.; Naler, L.B.; Lu, C. Microfluidic Platform for Next-Generation Sequencing Library Preparation with Low-Input Samples. Anal. Chem. 2020, 92, 2519–2526. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.F.; Kotrová, M.; Calabrese, S.; Darzentas, N.; Hutzenlaub, T.; Zengerle, R.; Brüggemann, M.; Paust, N. Automation of Amplicon-Based Library Preparation for next-Generation Sequencing by Centrifugal Microfluidics. Anal. Chem. 2020, 92, 12833–12841. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Liebers, M.; Zhelyazkova, B.; Cao, Y.; Panditi, D.; Lynch, K.D.; Chen, J.; Robinson, H.E.; Shim, H.S.; Chmielecki, J. Anchored Multiplex PCR for Targeted Next-Generation Sequencing. Nat. Med. 2014, 20, 1479–1484. [Google Scholar] [CrossRef]
- Beg, S.; Bareja, R.; Ohara, K.; Eng, K.W.; Wilkes, D.C.; Pisapia, D.J.; Al Zoughbi, W.; Kudman, S.; Zhang, W.; Rao, R. Integration of Whole-Exome and Anchored PCR-Based next Generation Sequencing Significantly Increases Detection of Actionable Alterations in Precision Oncology. Transl. Oncol. 2021, 14, 100944. [Google Scholar] [CrossRef]
- Racanelli, D.; Brenca, M.; Baldazzi, D.; Goeman, F.; Casini, B.; de Angelis, B.; Guercio, M.; Milano, G.M.; Tamborini, E.; Busico, A. Next-Generation Sequencing Approaches for the Identification of Pathognomonic Fusion Transcripts in Sarcomas: The Experience of the Italian ACC Sarcoma Working Group. Front. Oncol. 2020, 10, 489. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, L.; Mamon, H.; Kulke, M.H.; Berbeco, R.; Makrigiorgos, G.M. Replacing PCR with COLD-PCR Enriches Variant DNA Sequences and Redefines the Sensitivity of Genetic Testing. Nat. Med. 2008, 14, 579–584. [Google Scholar] [CrossRef]
- Milbury, C.A.; Li, J.; Makrigiorgos, G.M. Ice-COLD-PCR Enables Rapid Amplification and Robust Enrichment for Low-Abundance Unknown DNA Mutations. Nucleic Acids Res. 2011, 39, e2. [Google Scholar] [CrossRef]
- Milbury, C.A.; Correll, M.; Quackenbush, J.; Rubio, R.; Makrigiorgos, G.M. COLD-PCR Enrichment of Rare Cancer Mutations Prior to Targeted Amplicon Resequencing. Clin. Chem. 2012, 58, 580–589. [Google Scholar] [CrossRef] [Green Version]
- Luthra, R.; Patel, K.P.; Reddy, N.G.; Haghshenas, V.; Routbort, M.J.; Harmon, M.A.; Barkoh, B.A.; Kanagal-Shamanna, R.; Ravandi, F.; Cortes, J.E. Next-Generation Sequencing-Based Multigene Mutational Screening for Acute Myeloid Leukemia Using MiSeq: Applicability for Diagnostics and Disease Monitoring. Haematologica 2014, 99, 465. [Google Scholar] [CrossRef]
- Lesnik, E.A.; Freier, S.M. Relative Thermodynamic Stability of DNA, RNA, and DNA: RNA Hybrid Duplexes: Relationship with Base Composition and Structure. Biochemistry 1995, 34, 10807–10815. [Google Scholar] [CrossRef]
- Okou, D.T.; Steinberg, K.M.; Middle, C.; Cutler, D.J.; Albert, T.J.; Zwick, M.E. Microarray-Based Genomic Selection for High-Throughput Resequencing. Nat. Methods 2007, 4, 907–909. [Google Scholar] [CrossRef] [PubMed]
- Gnirke, A.; Melnikov, A.; Maguire, J.; Rogov, P.; LeProust, E.M.; Brockman, W.; Fennell, T.; Giannoukos, G.; Fisher, S.; Russ, C. Solution Hybrid Selection with Ultra-Long Oligonucleotides for Massively Parallel Targeted Sequencing. Nat. Biotechnol. 2009, 27, 182–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodges, E.; Rooks, M.; Xuan, Z.; Bhattacharjee, A.; Gordon, D.B.; Brizuela, L.; McCombie, W.R.; Hannon, G.J. Hybrid Selection of Discrete Genomic Intervals on Custom-Designed Microarrays for Massively Parallel Sequencing. Nat. Protoc. 2009, 4, 960–974. [Google Scholar] [CrossRef] [Green Version]
- Duncavage, E.J.; Magrini, V.; Becker, N.; Armstrong, J.R.; Demeter, R.T.; Wylie, T.; Abel, H.J.; Pfeifer, J.D. Hybrid Capture and Next-Generation Sequencing Identify Viral Integration Sites from Formalin-Fixed, Paraffin-Embedded Tissue. J. Mol. Diagn. 2011, 13, 325–333. [Google Scholar] [CrossRef]
- Clark, T.A.; Chung, J.H.; Kennedy, M.; Hughes, J.D.; Chennagiri, N.; Lieber, D.S.; Fendler, B.; Young, L.; Zhao, M.; Coyne, M. Analytical Validation of a Hybrid Capture–Based Next-Generation Sequencing Clinical Assay for Genomic Profiling of Cell-Free Circulating Tumor DNA. J. Mol. Diagn. 2018, 20, 686–702. [Google Scholar] [CrossRef]
- Dahl, F.; Stenberg, J.; Fredriksson, S.; Welch, K.; Zhang, M.; Nilsson, M.; Bicknell, D.; Bodmer, W.F.; Davis, R.W.; Ji, H. Multigene Amplification and Massively Parallel Sequencing for Cancer Mutation Discovery. Proc. Natl. Acad. Sci. USA 2007, 104, 9387–9392. [Google Scholar] [CrossRef] [Green Version]
- Hiatt, J.B.; Pritchard, C.C.; Salipante, S.J.; O’Roak, B.J.; Shendure, J. Single Molecule Molecular Inversion Probes for Targeted, High-Accuracy Detection of Low-Frequency Variation. Genome Res. 2013, 23, 843–854. [Google Scholar] [CrossRef] [Green Version]
- Caruccio, N. Preparation of Next-Generation Sequencing Libraries Using NexteraTM Technology: Simultaneous DNA Fragmentation and Adaptor Tagging by in Vitro Transposition. Methods Mol. Biol. 2011, 733, 241–255. [Google Scholar]
- Chen, H.; Yao, J.; Fu, Y.; Pang, Y.; Wang, J.; Huang, Y. Tagmentation on Microbeads: Restore Long-Range DNA Sequence Information Using Next Generation Sequencing with Library Prepared by Surface-Immobilized Transposomes. ACS Appl. Mater. Interfaces 2018, 10, 11539–11545. [Google Scholar] [CrossRef]
- Ren, H.; Xi, Y.; Li, Z.; Zhang, D.; Huang, F.; Fang, X.; Zhao, X.; Zhang, X.; Chen, A.; Chen, T. Novel Target Capture DNA Library Preparation Method Using CircLigase-Mediated Hook Ligation. New Biotechnol. 2020, 59, 44–50. [Google Scholar] [CrossRef]
- Berglund, E.C.; Lindqvist, C.M.; Hayat, S.; Övernäs, E.; Henriksson, N.; Nordlund, J.; Wahlberg, P.; Forestier, E.; Lönnerholm, G.; Syvänen, A.-C. Accurate Detection of Subclonal Single Nucleotide Variants in Whole Genome Amplified and Pooled Cancer Samples Using HaloPlex Target Enrichment. BMC Genom. 2013, 14, 856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moens, L.N.; Falk-Sörqvist, E.; Ljungström, V.; Mattsson, J.; Sundström, M.; La Fleur, L.; Mathot, L.; Micke, P.; Nilsson, M.; Botling, J. HaloPlex Targeted Resequencing for Mutation Detection in Clinical Formalin-Fixed, Paraffin-Embedded Tumor Samples. J. Mol. Diagn. 2015, 17, 729–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, J.F.; Kohl, T.A.; Kotrová, M.; Rönsch, K.; Paprotka, T.; Mohr, V.; Hutzenlaub, T.; Brüggemann, M.; Zengerle, R.; Niemann, S. Library Preparation for next Generation Sequencing: A Review of Automation Strategies. Biotechnol. Adv. 2020, 41, 107537. [Google Scholar] [CrossRef]
- Mora-Castilla, S.; To, C.; Vaezeslami, S.; Morey, R.; Srinivasan, S.; Dumdie, J.N.; Cook-Andersen, H.; Jenkins, J.; Laurent, L.C. Miniaturization Technologies for Efficient Single-Cell Library Preparation for next-Generation Sequencing. J. Lab. Autom. 2016, 21, 557–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; De Jonghe, J.; Kulesa, A.B.; Feldman, D.; Vatanen, T.; Bhattacharyya, R.P.; Berdy, B.; Gomez, J.; Nolan, J.; Epstein, S. High-Throughput Automated Microfluidic Sample Preparation for Accurate Microbial Genomics. Nat. Commun. 2017, 8, 13919. [Google Scholar] [CrossRef]
- Burghel, G.J.; Hurst, C.D.; Watson, C.M.; Chambers, P.A.; Dickinson, H.; Roberts, P.; Knowles, M.A. Towards a Next-Generation Sequencing Diagnostic Service for Tumour Genotyping: A Comparison of Panels and Platforms. BioMed Res. Int. 2015, 2015, 478017. [Google Scholar] [CrossRef] [Green Version]
- Samorodnitsky, E.; Datta, J.; Jewell, B.M.; Hagopian, R.; Miya, J.; Wing, M.R.; Damodaran, S.; Lippus, J.M.; Reeser, J.W.; Bhatt, D. Comparison of Custom Capture for Targeted Next-Generation DNA Sequencing. J. Mol. Diagn. 2015, 17, 64–75. [Google Scholar] [CrossRef] [Green Version]
- Samorodnitsky, E.; Jewell, B.M.; Hagopian, R.; Miya, J.; Wing, M.R.; Lyon, E.; Damodaran, S.; Bhatt, D.; Reeser, J.W.; Datta, J. Evaluation of Hybridization Capture versus Amplicon-Based Methods for Whole-Exome Sequencing. Hum. Mutat. 2015, 36, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Zakrzewski, F.; Gieldon, L.; Rump, A.; Seifert, M.; Grützmann, K.; Krüger, A.; Loos, S.; Zeugner, S.; Hackmann, K.; Porrmann, J. Targeted Capture-Based NGS Is Superior to Multiplex PCR-Based NGS for Hereditary BRCA1 and BRCA2 Gene Analysis in FFPE Tumor Samples. BMC Cancer 2019, 19, 396. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Hoon, D.S.; Pantel, K. Cell-Free Nucleic Acids as Biomarkers in Cancer Patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Lam, S.N.; Zhou, Y.C.; Chan, Y.M.; Foo, C.M.; Lee, P.Y.; Mok, W.Y.; Wong, W.S.; Fung, Y.Y.; Wong, K.Y.; Huang, J.Y. Comparison of Target Enrichment Platforms for Circulating Tumor DNA Detection. Sci. Rep. 2020, 10, 4124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
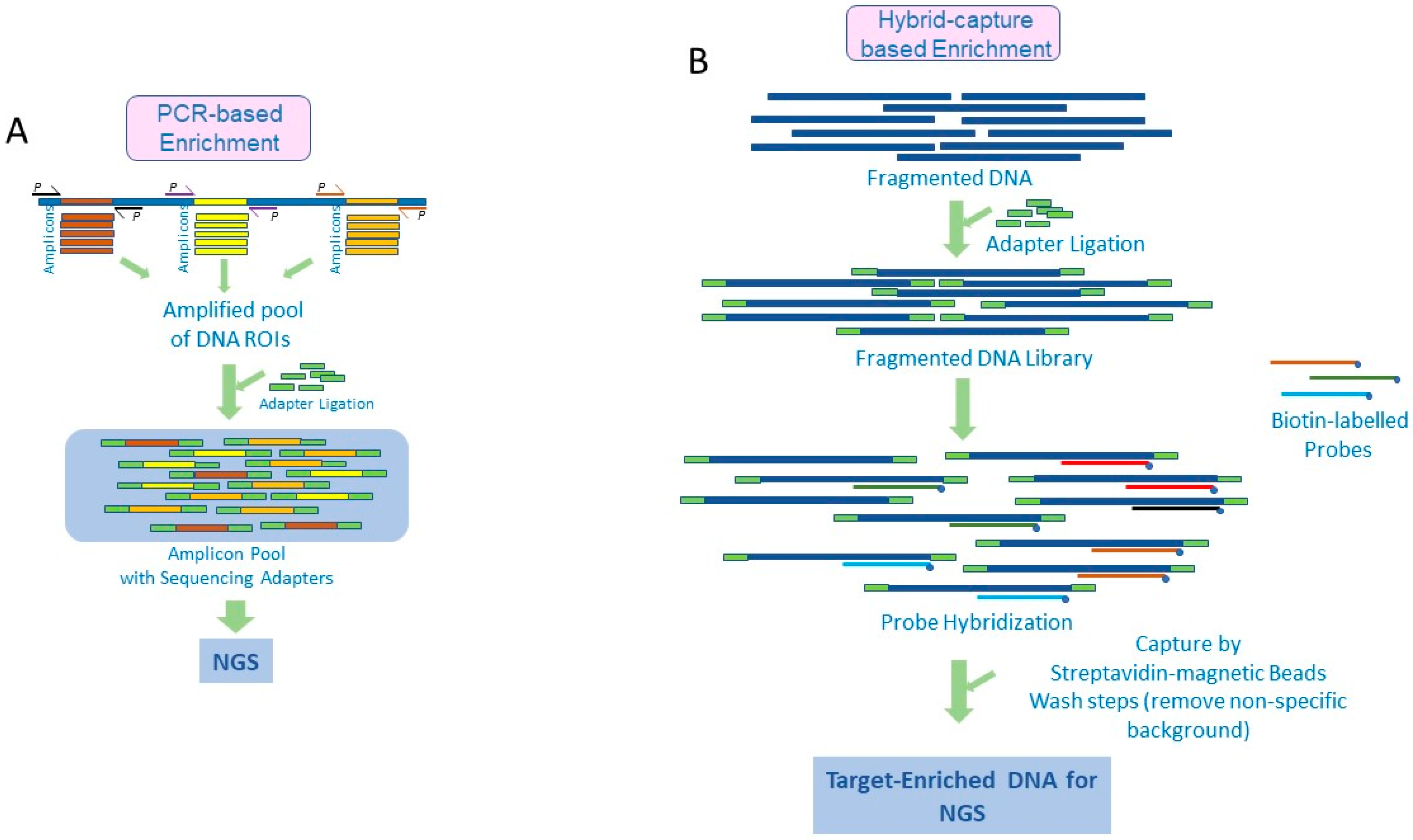

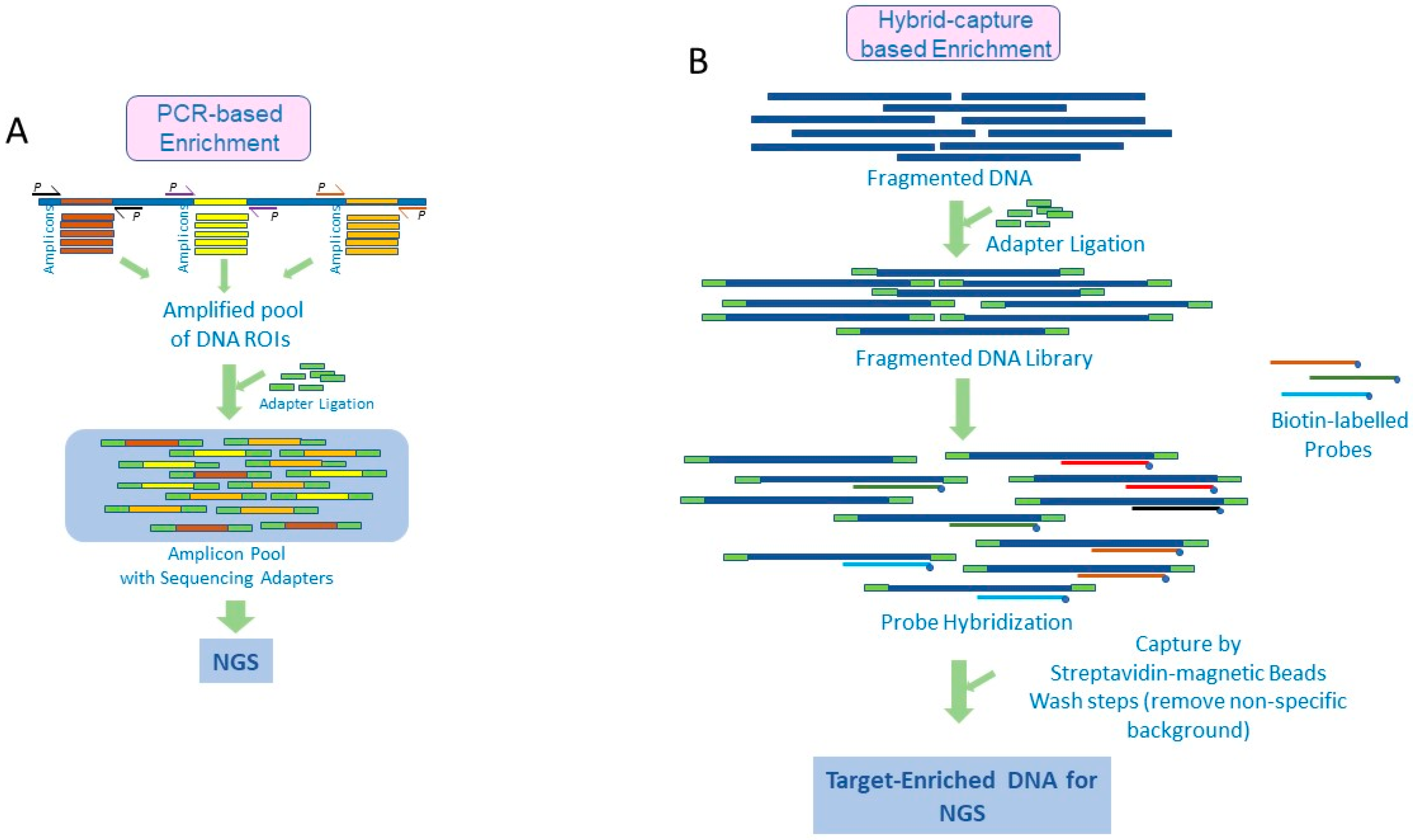{kind=link}
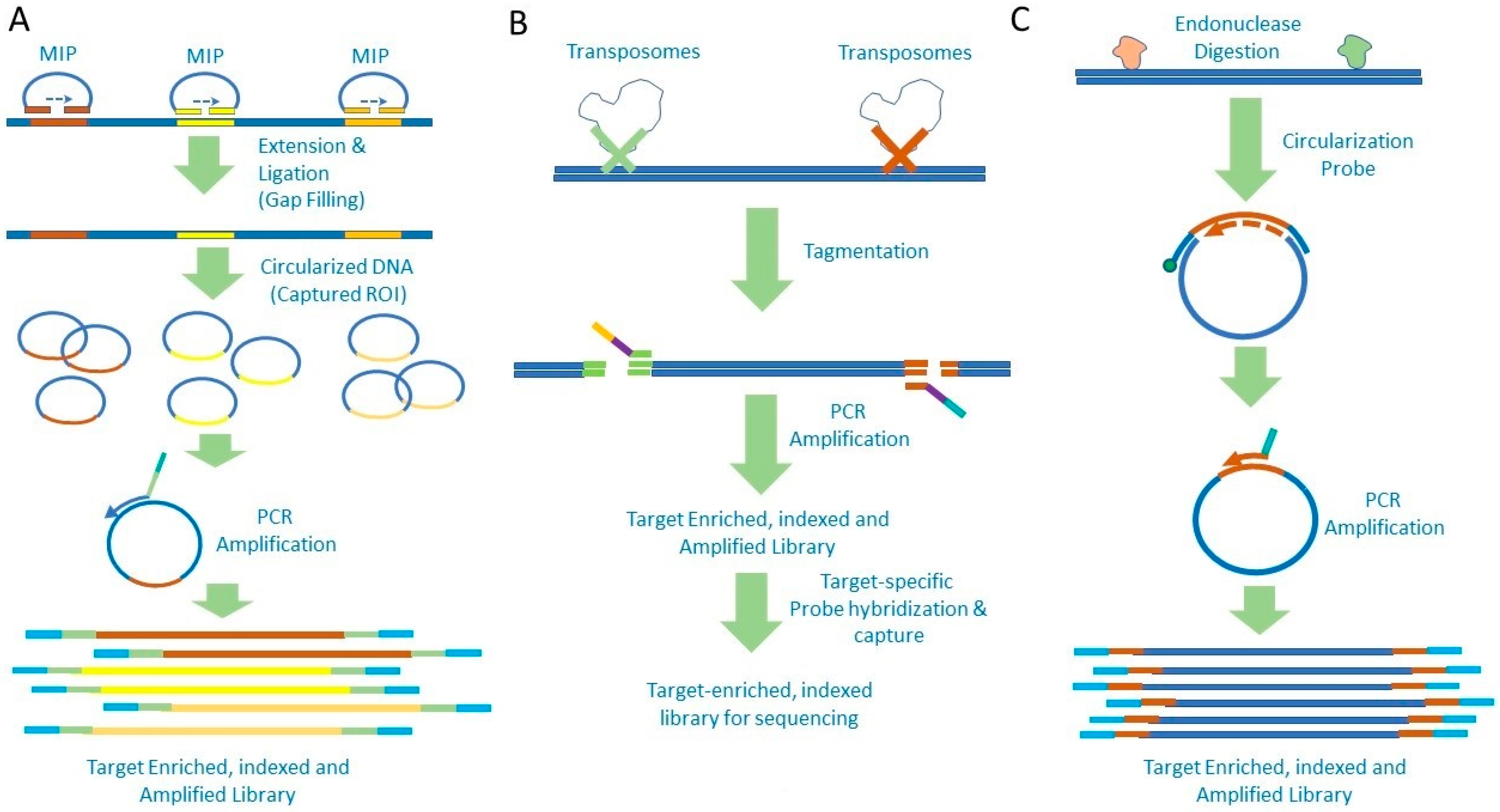{kind=link}
| Hybrid Capture-Based Enrichment | PCR Amplicon-Based Enrichment | |
|---|---|---|
| Enrichment Principle | Hybridization capture-based enrichment using single-stranded DNA or RNA probes complementary to the genomic regions of interest | PCR-based amplification using sequence-specific primers flanking genomic regions of interest |
| Nucleic Acid Input | Requires relatively high quantity of nucleic acid input | Compatible with low quantity of nucleic acid input |
| Nucleic Acid Quality | Compatible with challenging sample types; however, success depends on obtaining sufficient yield | Compatible with challenging sample types (e.g., FFPE and decalcified samples) |
| Fragmentation of Nucleic Acid Input | Required. Nucleic acids need to be enzymatic-digested oracoustically sheared prior to hybrid capture | Not required |
| Workflow Time | Relatively long due to the hybrid capture step | Significantly shorter workflow |
| Workflow Complexity | High complexity workflow with multiple steps | Relatively simple workflow |
| Gene Targets per Panel | No limitations and can include any number of gene targets. Preferred methodology for large panels and whole-exome sequencing. | Generally suited for smaller number of gene targets. Limited by the multiplexing capability of the primers |
| Uniformity of Sequence Enrichment | Higher uniformity of target enrichment and lower rates of sequencing failures in regions of interest | Relatively low target enrichment uniformity and higher sequencing failures |
| Off-Target Sequencing Rate | Relatively high. Has more possibility of off-target sequences captured and sequenced | Lower off-target sequencing rate |
| Commercial Options | SureSelect (Agilent Technologies) Haloplex (Agilent Technologies) XGen NGS Hybrid capture (Integrated DNA Technologies) TruSight Hybris capture (Illumina Inc.) Swift Hybrid Capture (Swift Biosciences, Ann Arbor, MI, USA) | AmpliSeq (ThermoFisher Scientific) AccesArray (Fluidigm Corporation, South San Francisco, CA, USA ) GeneRead (Qiagen) RainStorm (RainDance Technologies, Lexington, MA, USA) TruSeq (Illumina Inc.) HEAT-Seq (Roche) XGen NGS Amplicon Sequencing (Integrated DNA Technologies) Accel-Amplicon (Swift Biosciences) |
| Commercial Enrichment Methods Compared | Enrichment Approach | Genome Targets and Sample Types | Findings | Reference |
|---|---|---|---|---|
| Fluidigm Access array (Fluidigm) | Microfluidic PCR | DNA from 8 human bladder cancer cell lines (both fresh and formalin-fixed samples). 24 mutations in 6 genes (BRAF, FGFR3, KRAS, NRAS, PIK3CA, and TP53) Fresh and formalin-fixed and paraffin-embedded DNA samples. | Complete concordance of results for fresh DNA SureSeq panel performed the best followed by Fluidigm and IonAmpliseq | [46] |
| SureSeq panel | ||||
| (Oxfore Gene Technology) | Hybrid-capture | |||
| Ion AmpliSeq | ||||
| (ThermoFisher Scientific) | PCR amplicon-based | |||
| SureSelect (Agilent Technologies) | Hybrid capture | Single nucleotide variants (SNVs) and copy number variations (CNVs) in a panel of 257 cancer-related genes. Cancer cell lines and tumor samples (breast, melanoma, lung, and colon cancer) | Comparable cost of workflow across the methods High level of concordance observed for SNV and CNV detection across methods SureSelect and SeqCap showed better library complexity and overall sequencing uniformity | [47] |
| Haloplex (Agilent Technologies) | Hybrid capture | |||
| Nextera (Illumina Inc) | Hybrid capture | |||
| SeqCap EZ (Roche Nimblegen) | Hybrid capture | |||
| SureSelect (Agilent Technologies) | Hybrid capture | Whole-Exome Sequencing | Hybrid capture methods provided better library complexity, uniformity of coverage, analytical sensitivity, and specificity | [48] |
| Haloplex (Agilent Technologies) | Hybrid capture | |||
| SeqCap EZ (Roche Nimblegen) | Hybrid capture | |||
| Ion AmpliSeq | ||||
| (ThermoFisher Scientific) | PCR amplicon-based | |||
| TruSight (Illumina Inc) | Hybrid capture | BRCA1 and BRCA2 sequencing FFPE tumor samples | TruSight performed better with regards to uniformity of sequencing, analytical specificity, and detection of mutations and CNVs | [49] |
| TruSeq custom amplicon | ||||
| (Illumina Inc) | PCR amplicon-based | |||
| Avenio CtDNA panel (Roche) | Hybrid capture | Screening circulating cell-free tumor DNA (ctDNA) for cancer-related markers | QiaSeq had shorter workflow (1 day) in comparison to Avenio (3 days) Avenio fared better in enriching targets of interest (99% vs. 85% for QIASeq). Analytical sensitivity for Avenio (92.3%) was better than QiaSeq (86.4%). Detection sensitivity for low-level variants (≤ 5%) was better for Avenio (75%) vs. QiaSeq (53.8%) | [51] |
| QIAseq Human Comprehensive Cancer | ||||
| panel (Qiagen) | PCR amplicon-based |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, R.R. Target Enrichment Approaches for Next-Generation Sequencing Applications in Oncology. Diagnostics 2022, 12, 1539. https://doi.org/10.3390/diagnostics12071539
Singh RR. Target Enrichment Approaches for Next-Generation Sequencing Applications in Oncology. Diagnostics. 2022; 12(7):1539. https://doi.org/10.3390/diagnostics12071539
Chicago/Turabian StyleSingh, Rajesh R. 2022. "Target Enrichment Approaches for Next-Generation Sequencing Applications in Oncology" Diagnostics 12, no. 7: 1539. https://doi.org/10.3390/diagnostics12071539
APA StyleSingh, R. R. (2022). Target Enrichment Approaches for Next-Generation Sequencing Applications in Oncology. Diagnostics, 12(7), 1539. https://doi.org/10.3390/diagnostics12071539