
POEMS Syndrome: Presented as Idiopathic Multicentric Castleman Disease of Plasma Cell Variant for Eight Years and Dramatic Treatment with Siltuximab Followed by Autologous Peripheral Blood Stem Cell Transplantation



Abstract
:1. Introduction
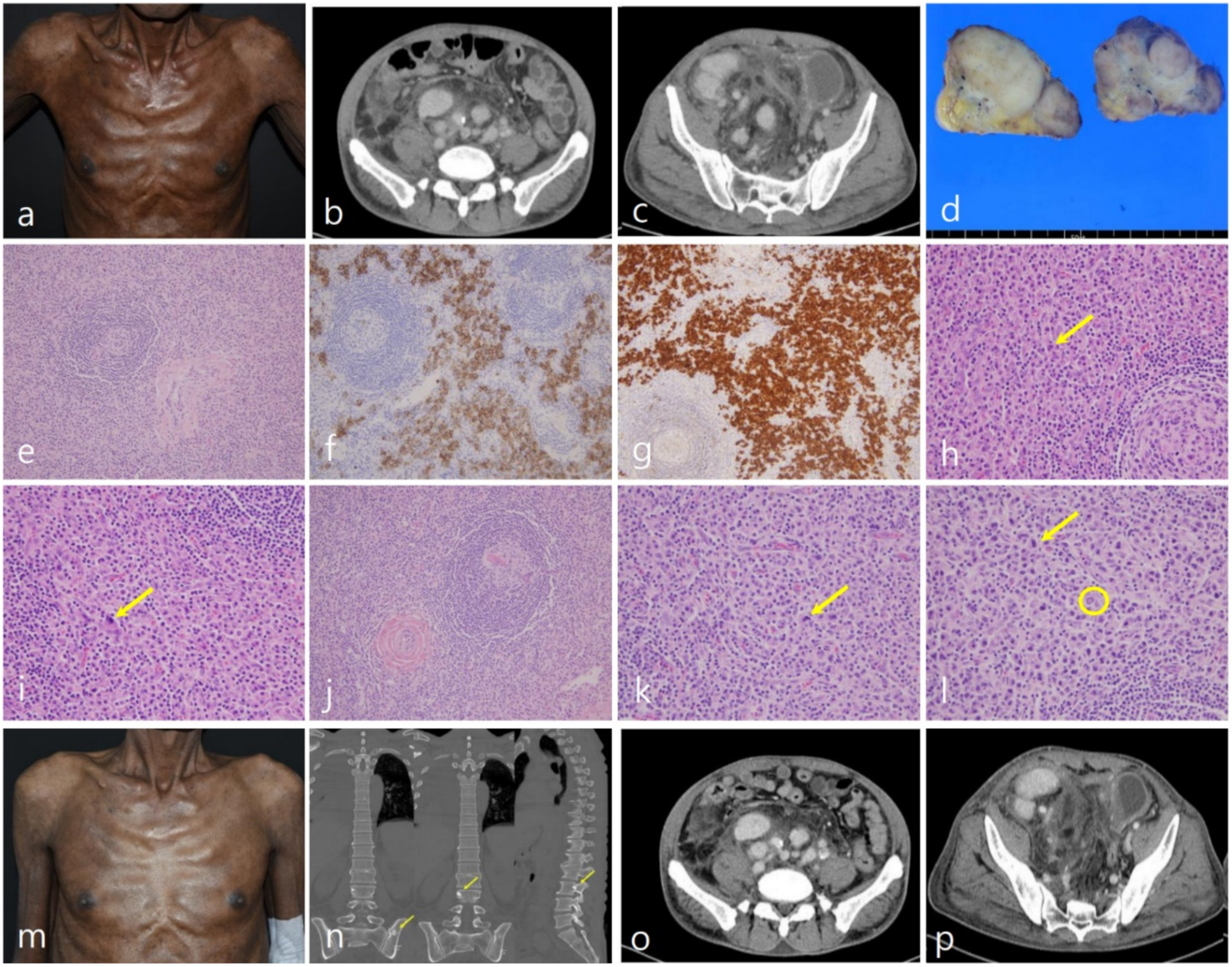
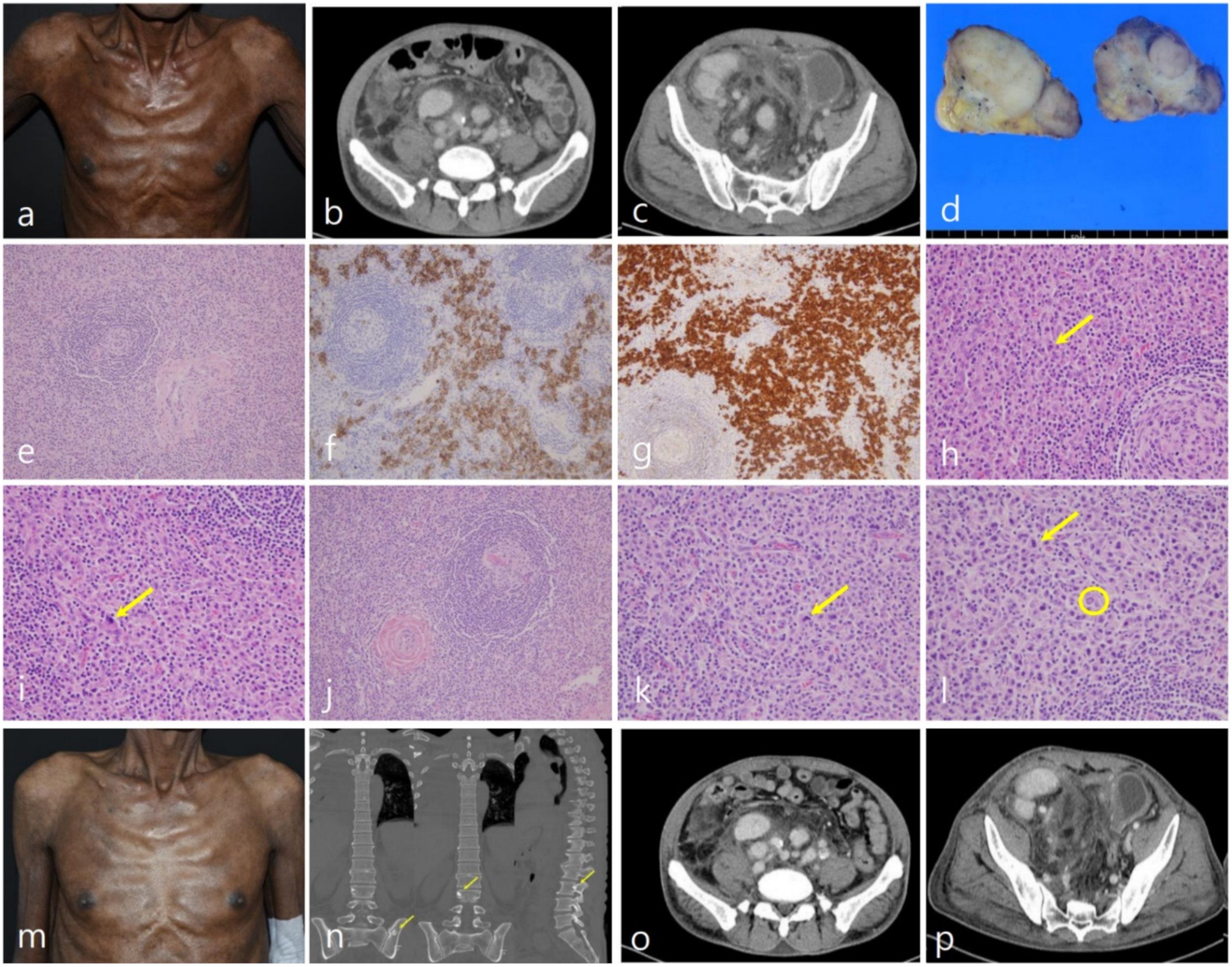
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Dispenzieri, A. POEMS Syndrome: 2019 Update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2019, 94, 812–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanada, I.; Takatsuki, K. Plasma Cell Dyscrasia with Polyneuropathy and Endocrine Disorder: Clinical and Laboratory Features of 109 Reported Cases. Jpn. J. Clin. Oncol. 1983, 13, 543–556. [Google Scholar] [CrossRef]
- Nakanishi, T.; Sobue, I.; Toyokura, Y.; Nishitani, H.; Kuroiwa, Y.; Satoyoshi, E.; Tsubaki, T.; Igata, A.; Ozaki, Y. The Crow-Fukase syndrome: A study of 102 cases in Japan. Neurology 1984, 34, 712. [Google Scholar] [CrossRef] [PubMed]
- Nasu, S.; Misawa, S.; Sekiguchi, Y.; Shibuya, K.; Kanai, K.; Fujimaki, Y.; Ohmori, S.; Mitsuma, S.; Koga, S.; Kuwabara, S. Different neurological and physiological profiles in POEMS syndrome and chronic inflammatory demyelinating polyneuropathy. J. Neurol. Neurosurg. Psychiatry 2012, 83, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Frizzera, G.; Peterson, B.A.; Bayrd, E.D.; Goldman, A. A systemic lymphoproliferative disorder with morphologic features of Castleman’s disease: Clinical findings and clinicopathologic correlations in 15 patients. J. Clin. Oncol. 1985, 3, 1202–1216. [Google Scholar] [CrossRef] [PubMed]
- Lachant, N.A.; Sun, N.C.; Leong, L.A.; Oseas, R.S.; Prince, H.E. Multicentric angiofollicular lymph node hyperplasia (Castleman’s disease) followed by Kaposi’s sarcoma in two homosexual males with the acquired immunodeficiency syndrome (AIDS). Am. J. Clin. Pathol. 1985, 83, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dispenzieri, A.; Kyle, R.A.; Lacy, M.Q.; Rajkumar, S.V.; Therneau, T.M.; Larson, D.R.; Greipp, P.R.; Witzig, T.E.; Basu, R.; Suarez, G.A.; et al. POEMS syndrome: Definitions and long-term outcome. Blood 2003, 101, 2496–2506. [Google Scholar] [CrossRef] [PubMed]
- Van Rhee, F.; Voorhees, P.; Dispenzieri, A.; Dispenzeri, A.; Fossa, A.; Srkalovic, G.; Ide, M.; Munshi, N.; Schey, S.; Streetly, M.; et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood 2018, 132, 2115–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, G.-J.; Jeon, Y.-W.; Park, S.-S.; Park, S.; Shin, S.-H.; Yahng, S.-A.; Yoon, J.-H.; Lee, S.-E.; Cho, B.-S.; Eom, K.-S.; et al. The clinical, laboratory, and radiologic improvement due to siltuximab treatment in idiopathic multicentric Castleman’s disease. Korean J. Intern. Med. 2021, 36, 424–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauermann, M.L. The Peripheral Neuropathies of POEMS Syndrome and Castleman Disease. Hematol. Clin. N. Am. 2018, 32, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Inada, K.; Hamazaki, M. Localized mediastinal lymph-node hyperplasia resembling thymoma; a case report. Ann. Surg. 1958, 147, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Nanki, T.; Tomiyama, J.; Arai, S. Mixed connective tissue disease associated with multicentric Castleman’s disease. Scand. J. Rheumatol. 1994, 23, 215–217. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Mandatory Major Criteria | Other Major Criteria (One Required) | Minor Criteria |
|---|---|---|
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.-M.; Choi, Y.S.; Kim, J.-M. POEMS Syndrome: Presented as Idiopathic Multicentric Castleman Disease of Plasma Cell Variant for Eight Years and Dramatic Treatment with Siltuximab Followed by Autologous Peripheral Blood Stem Cell Transplantation. Diagnostics 2022, 12, 998. https://doi.org/10.3390/diagnostics12040998
Lee Y-M, Choi YS, Kim J-M. POEMS Syndrome: Presented as Idiopathic Multicentric Castleman Disease of Plasma Cell Variant for Eight Years and Dramatic Treatment with Siltuximab Followed by Autologous Peripheral Blood Stem Cell Transplantation. Diagnostics. 2022; 12(4):998. https://doi.org/10.3390/diagnostics12040998
Chicago/Turabian StyleLee, Yong-Moon, Yoon Seok Choi, and Jin-Man Kim. 2022. "POEMS Syndrome: Presented as Idiopathic Multicentric Castleman Disease of Plasma Cell Variant for Eight Years and Dramatic Treatment with Siltuximab Followed by Autologous Peripheral Blood Stem Cell Transplantation" Diagnostics 12, no. 4: 998. https://doi.org/10.3390/diagnostics12040998
APA StyleLee, Y.-M., Choi, Y. S., & Kim, J.-M. (2022). POEMS Syndrome: Presented as Idiopathic Multicentric Castleman Disease of Plasma Cell Variant for Eight Years and Dramatic Treatment with Siltuximab Followed by Autologous Peripheral Blood Stem Cell Transplantation. Diagnostics, 12(4), 998. https://doi.org/10.3390/diagnostics12040998