
Metabolism and Vascular Retinopathies: Current Perspectives and Future Directions

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
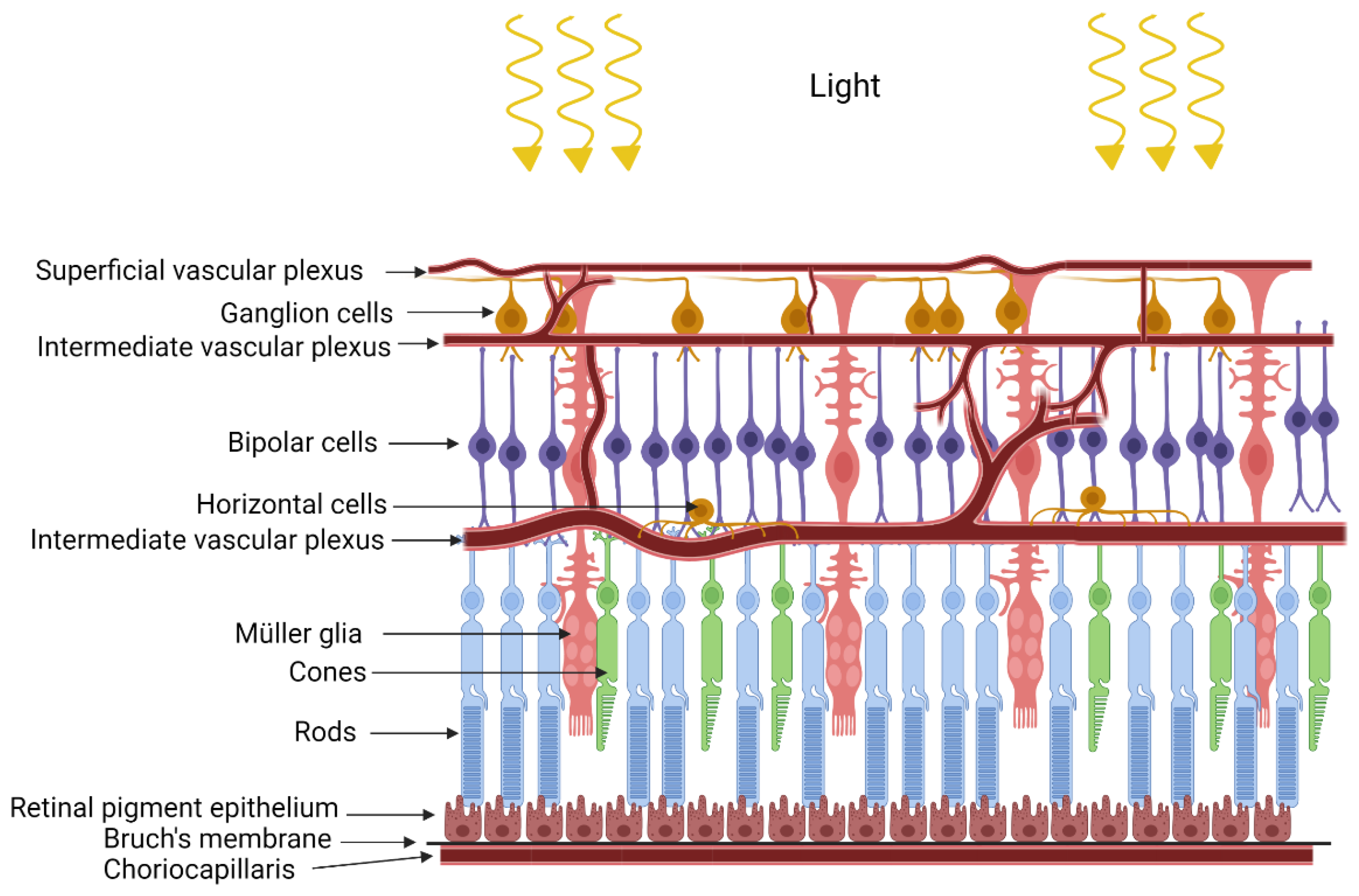
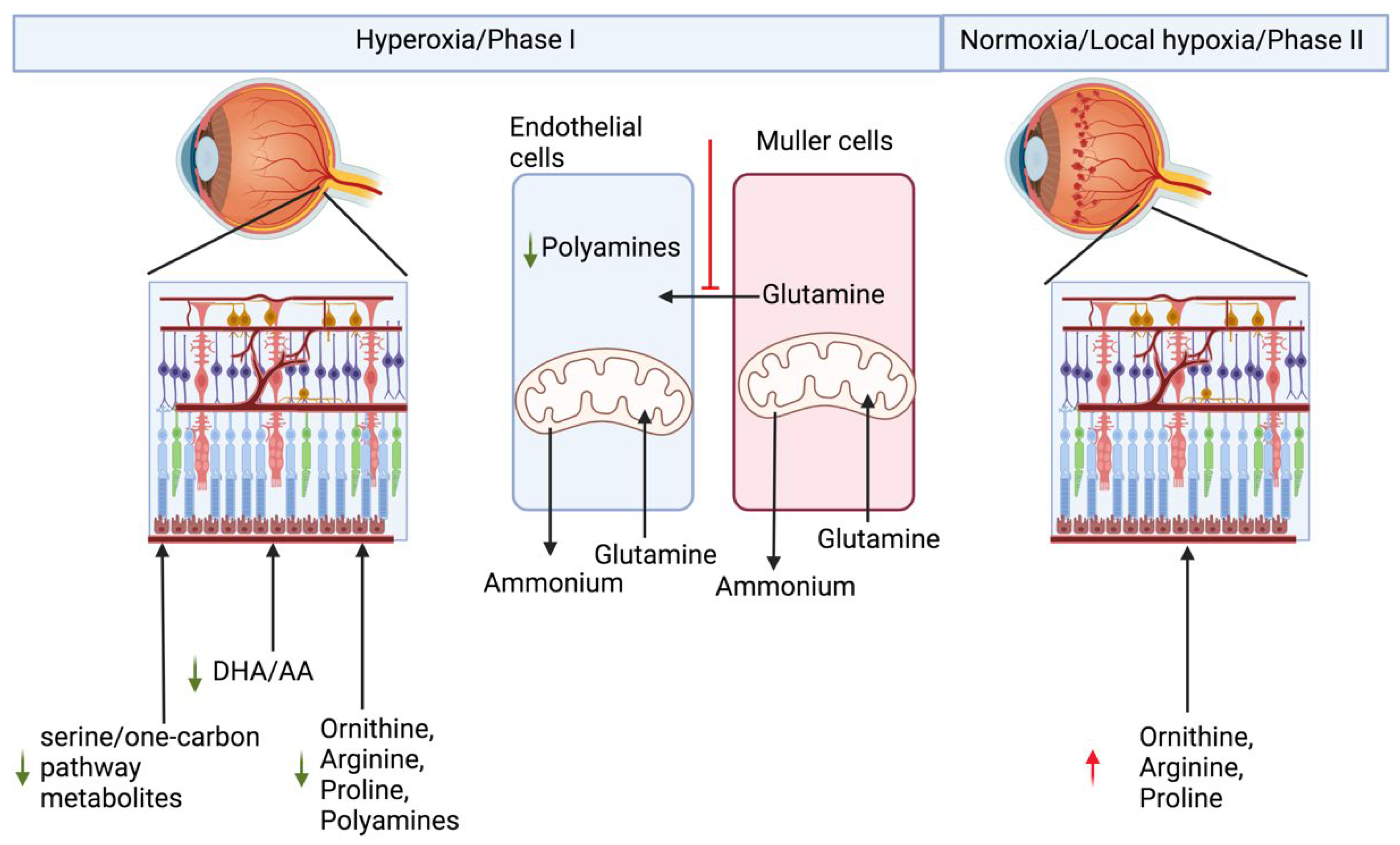
2. Retinopathy of Prematurity
3. Age-Related Macular Degeneration (AMD)
4. Diabetic Retinopathy
5. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- De Schaepdrijver, L.; Simoens, P.; Lauwers, H.; De Geest, J.P. Retinal vascular patterns in domestic animals. Res. Vet. Sci. 1989, 47, 34–42. [Google Scholar] [CrossRef]
- Gardner, W.T.; Antonetti, D.A.; Barber, A.J.; LaNoue, K.F.; Levison, S.W. Diabetic Retinopathy: More Than Meets the Eye. Surv. Ophthalmol. 2002, 47 (Suppl. S2), S253–S262. [Google Scholar] [CrossRef]
- Erickson, K.K.; Sundstrom, J.M.; Antonetti, D.A. Vascular permeability in ocular disease and the role of tight junctions. Angiogenesis 2007, 10, 103. [Google Scholar] [CrossRef] [PubMed]
- Goldman, D. Müller glial cell reprogramming and retina regeneration. Nat. Rev. Neurosci. 2014, 15, 431–442. [Google Scholar] [CrossRef]
- Kumar, A.; Pandey, R.K.; Miller, L.J.; Singh, P.K.; Kanwar, M. Muller glia in retinal innate immunity: A perspective on their roles in endophthalmitis. Crit. Rev. Immunol. 2013, 33, 119–135. [Google Scholar] [CrossRef]
- Bringmann, A.; Wiedemann, P. Müller Glial Cells in Retinal Disease. Ophthalmologica 2012, 227, 1–19. [Google Scholar] [CrossRef]
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.N.; Osborne, N.N.; Reichenbach, A. Müller cells in the healthy and diseased retina. Prog. Retin. Eye Res. 2006, 25, 397–424. [Google Scholar] [CrossRef]
- Bringmann, A.; Reichenbach, A. Role of Muller cells in retinal degenerations. Front. Biosci. Landmark 2001, 6, 72–92. [Google Scholar] [CrossRef]
- Germer, A.; Biedermann, B.; Wolburg, H.; Schuck, J.; Grosche, J.; Kuhrt, H.; Reichelt, W.; Schousboe, A.; Paasche, G.; Mack, A.F.; et al. Distribution of mitochondria within Müller cells—I. Correlation with retinal vascularization in different mammalian species. J. Neurocytol. 1998, 27, 329–345. [Google Scholar] [CrossRef]
- Hurley, J.B. Retina Metabolism and Metabolism in the Pigmented Epithelium: A Busy Intersection. Annu. Rev. Vis. Sci. 2021, 7, 665–692. [Google Scholar] [CrossRef]
- Du, J.; Zhu, S.; Lim, R.R.; Chao, J.R. Proline metabolism and transport in retinal health and disease. Amino Acids 2021, 53, 1789–1806. [Google Scholar] [CrossRef]
- Swarup, A.; Samuels, I.S.; Bell, B.A.; Han, J.Y.S.; Du, J.; Massenzio, E.; Abel, E.D.; Boesze-Battaglia, K.; Peachey, N.S.; Philp, N.J. Modulating GLUT1 expression in retinal pigment epithelium decreases glucose levels in the retina: Impact on photoreceptors and Müller glial cells. Am. J. Physiol. Cell Physiol. 2019, 316, C121–C133. [Google Scholar] [CrossRef] [PubMed]
- Yam, M.; Engel, A.L.; Wang, Y.; Zhu, S.; Hauer, A.; Zhang, R.; Lohner, D.; Huang, J.; Dinterman, M.; Zhao, C.; et al. Proline mediates metabolic communication between retinal pigment epithelial cells and the retina. J. Biol. Chem. 2019, 294, 10278–10289. [Google Scholar] [CrossRef] [PubMed]
- Chinchore, Y.; Begaj, T.; Wu, D.; Drokhlyansky, E.; Cepko, C.L. Glycolytic reliance promotes anabolism in photoreceptors. Elife 2017, 6, e25946. [Google Scholar] [CrossRef] [PubMed]
- Winkler, B.S. Glycolytic and oxidative metabolism in relation to retinal function. J. Gen. Physiol. 1981, 77, 667–692. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.B.; Lindsay, K.J.; Du, J. Glucose, lactate, and shuttling of metabolites in vertebrate retinas. J. Neurosci. Res. 2015, 93, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.H.; Noell, W.K. Glucose catabolism of rabbit retina before and after development of visual function. J. Neurochem. 1960, 5, 253–276. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Lowry, O.H.; Roberts, N.R.; Schulz, D.W.; Clow, J.E.; Clark, J.R. Quantitative Histochemistry of Retina: II. Enzymes of Glucose Metabolism. J. Biol. Chem. 1961, 236, 2813–2820. [Google Scholar] [CrossRef]
- Lowry, O.H.; Roberts, N.R.; Lewis, C. The quantitative histochemistry of the retina. J. Biol. Chem. 1956, 220, 879–892. [Google Scholar] [CrossRef]
- Joyal, J.S.; Sun, Y.; Gantner, M.L.; Shao, Z.; Evans, L.P.; Saba, N.; Fredrick, T.; Burnim, S.; Kim, J.S.; Patel, G.; et al. Retinal lipid and glucose metabolism dictates angiogenesis through the lipid sensor Ffar1. Nat. Med. 2016, 22, 439–445. [Google Scholar] [CrossRef]
- Atsuzawa, K.; Nakazawa, A.; Mizutani, K.; Fukasawa, M.; Yamamoto, N.; Hashimoto, T.; Usuda, N. Immunohistochemical localization of mitochondrial fatty acid β-oxidation enzymes in Müller cells of the retina. Histochem. Cell Biol. 2010, 134, 565–579. [Google Scholar] [CrossRef]
- Heckel, E.; Cagnone, G.; Agnihotri, T.; Cakir, B.; Das, A.; Kim, J.S.; Kim, N.; Lavoie, G.; Situ, A.; Pundir, S.; et al. Triglyceride-derived fatty acids reduce autophagy in a model of retinal angiomatous proliferation. JCI Insight 2022. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.W.; Marsch, E.; Treps, L.; Baes, M.; Carmeliet, P. Endothelial cell metabolism in health and disease: Impact of hypoxia. EMBO J. 2017, 36, 2187–2203. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.; Hoppe, G.; Tran, V.; McCollum, L.; Bolok, Y.; Song, W.; Sharma, A.; Brunengraber, H.; Sears, J.E. Serine and 1-carbon metabolism are required for HIF-mediated protection against retinopathy of prematurity. JCI Insight 2019, 4, e129398. [Google Scholar] [CrossRef] [PubMed]
- Hartnett, M.E.; Lane, R.H. Effects of oxygen on the development and severity of retinopathy of prematurity. J. Aapos 2013, 17, 229–234. [Google Scholar] [CrossRef]
- Smith, L.E. Pathogenesis of retinopathy of prematurity. Semin. Neonatol. 2003, 8, 469–473. [Google Scholar] [CrossRef]
- Smith, L.E. Through the eyes of a child: Understanding retinopathy through ROP the Friedenwald lecture. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5177–5182. [Google Scholar] [CrossRef] [PubMed]
- Hartnett, M.E.; Penn, J.S. Mechanisms and Management of Retinopathy of Prematurity. N. Engl. J. Med. 2012, 367, 2515–2526. [Google Scholar] [CrossRef] [PubMed]
- Madan, A.; Penn, J.S. Animal models of oxygen-induced retinopathy. Front. Biosci. 2003, 8, 1030–1043. [Google Scholar]
- Smith, L.E.; Wesolowski, E.; McLellan, A.; Kostyk, S.K.; D’Amato, R.; Sullivan, R.; D’Amore, P.A. Oxygen-induced retinopathy in the mouse. Investig. Ophthalmol. Vis. Sci. 1994, 35, 101–111. [Google Scholar]
- Kim, B.; Li, J.; Jang, C.; Arany, Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017, 36, 2321–2333. [Google Scholar] [CrossRef] [PubMed]
- De Bock, K.; Georgiadou, M.; Carmeliet, P. Role of endothelial cell metabolism in vessel sprouting. Cell Metab. 2013, 18, 634–647. [Google Scholar] [CrossRef]
- Newman, E.; Reichenbach, A. The Müller cell: A functional element of the retina. Trends Neurosci. 1996, 19, 307–312. [Google Scholar] [CrossRef]
- Joyal, J.-S.; Gantner, M.L.; Smith, L.E.H. Retinal energy demands control vascular supply of the retina in development and disease: The role of neuronal lipid and glucose metabolism. Prog. Retin. Eye Res. 2018, 64, 131–156. [Google Scholar] [CrossRef] [PubMed]
- Tojo, Y.; Sekine, H.; Hirano, I.; Pan, X.; Souma, T.; Tsujita, T.; Kawaguchi, S.-I.; Takeda, N.; Takeda, K.; Fong, G.-H.; et al. Hypoxia Signaling Cascade for Erythropoietin Production in Hepatocytes. Mol. Cell. Biol. 2015, 35, 2658–2672. [Google Scholar] [CrossRef] [PubMed]
- Akita, T.; Murohara, T.; Ikeda, H.; Sasaki, K.; Shimada, T.; Egami, K.; Imaizumi, T. Hypoxic preconditioning augments efficacy of human endothelial progenitor cells for therapeutic neovascularization. Lab. Investig. 2003, 83, 65–73. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gidday, J.M.; Fitzgibbons, J.C.; Shah, A.R.; Park, T.S. Neuroprotection from ischemic brain injury by hypoxic preconditioning in the neonatal rat. Neurosci. Lett. 1994, 168, 221–224. [Google Scholar] [CrossRef]
- Grimm, C.; Wenzel, A.; Groszer, M.; Mayser, H.; Seeliger, M.; Samardzija, M.; Bauer, C.; Gassmann, M.; Remé, C.E. HIF-1-induced erythropoietin in the hypoxic retina protects against light-induced retinal degeneration. Nat. Med. 2002, 8, 718–724. [Google Scholar] [CrossRef]
- Eckle, T.; Köhler, D.; Lehmann, R.; Kasmi, K.C.E.; Eltzschig, H.K. Hypoxia-Inducible Factor-1 Is Central to Cardioprotection. Circulation 2008, 118, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Sears, J.E.; Hoppe, G.; Ebrahem, Q.; Anand-Apte, B. Prolyl hydroxylase inhibition during hyperoxia prevents oxygen-induced retinopathy. Proc. Natl. Acad. Sci. USA 2008, 105, 19898–19903. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, G.; Yoon, S.; Gopalan, B.; Savage, A.R.; Brown, R.; Case, K.; Vasanji, A.; Chan, E.R.; Silver, R.B.; Sears, J.E. Comparative systems pharmacology of HIF stabilization in the prevention of retinopathy of prematurity. Proc. Natl. Acad. Sci. USA 2016, 113, E2516–E2525. [Google Scholar] [CrossRef]
- Singh, C.; Sharma, A.; Hoppe, G.; Song, W.; Bolok, Y.; Sears, J.E. 3-Hydroxypyruvate Destabilizes Hypoxia Inducible Factor and Induces Angiostasis. Investig. Ophthalmol. Vis. Sci. 2018, 59, 3440–3448. [Google Scholar] [CrossRef]
- Paris, L.P.; Johnson, C.H.; Aguilar, E.; Usui, Y.; Cho, K.; Hoang, L.T.; Feitelberg, D.; Benton, H.P.; Westenskow, P.D.; Kurihara, T.; et al. Global metabolomics reveals metabolic dysregulation in ischemic retinopathy. Metabolomics 2015, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Cagnone, G.; Fu, Z.; Cakir, B.; Kotoda, Y.; Asakage, M.; Wakabayashi, Y.; Hellström, A.; Joyal, J.-S.; Talukdar, S.; et al. Vitreous metabolomics profiling of proliferative diabetic retinopathy. Diabetologia 2021, 64, 70–82. [Google Scholar] [CrossRef]
- Zhou, Y.; Tan, W.; Zou, J.; Cao, J.; Huang, Q.; Jiang, B.; Yoshida, S.; Li, Y. Metabolomics Analyses of Mouse Retinas in Oxygen-Induced Retinopathy. Investig. Ophthalmol. Vis. Sci. 2021, 62, 9. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, Y.; Zhang, X.; Huang, Q.; Tan, W.; Yang, Y.; He, X.; Yoshida, S.; Zhao, P.; Li, Y. Plasma levels of amino acids and derivatives in retinopathy of prematurity. Int. J. Med. Sci. 2021, 18, 3581–3587. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.P.; Xu, Z.; Putluri, N.; Sreekumar, A.; Lemtalsi, T.; Caldwell, R.W.; Caldwell, R.B. Arginase 2 deficiency reduces hyperoxia-mediated retinal neurodegeneration through the regulation of polyamine metabolism. Cell Death Dis. 2014, 5, e1075. [Google Scholar] [CrossRef]
- Singh, C.; Benos, A.; Grenell, A.; Rao, S.; Anand-Apte, B.; Sears, J.E. Hyperoxia Inhibits Proliferation of Retinal Endothelial Cells in a Myc-Dependent Manner. Life 2021, 11, 614. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Nishimura, K.; Saiki, R.; Okudaira, H.; Tome, M.; Higashi, K.; Nakamura, M.; Terui, Y.; Fujiwara, K.; Kashiwagi, K.; et al. Role of polyamines at the G1/S boundary and G2/M phase of the cell cycle. Int. J. Biochem. Cell Biol. 2013, 45, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.; Tran, V.; McCollum, L.; Bolok, Y.; Allan, K.; Yuan, A.; Hoppe, G.; Brunengraber, H.; Sears, J.E. Hyperoxia induces glutamine-fuelled anaplerosis in retinal Müller cells. Nat. Commun. 2020, 11, 1277. [Google Scholar] [CrossRef] [PubMed]
- Hellström, A.; Nilsson, A.K.; Wackernagel, D.; Pivodic, A.; Vanpee, M.; Sjöbom, U.; Hellgren, G.; Hallberg, B.; Domellöf, M.; Klevebro, S.; et al. Effect of Enteral Lipid Supplement on Severe Retinopathy of Prematurity: A Randomized Clinical Trial. JAMA Pediatr. 2021, 175, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Löfqvist, C.A.; Najm, S.; Hellgren, G.; Engström, E.; Sävman, K.; Nilsson, A.K.; Andersson, M.X.; Hård, A.L.; Smith, L.E.H.; Hellström, A. Association of Retinopathy of Prematurity with Low Levels of Arachidonic Acid: A Secondary Analysis of a Randomized Clinical Trial. JAMA Ophthalmol. 2018, 136, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Chen, C.T.; Cagnone, G.; Heckel, E.; Sun, Y.; Cakir, B.; Tomita, Y.; Huang, S.; Li, Q.; Britton, W.; et al. Dyslipidemia in retinal metabolic disorders. EMBO Mol. Med. 2019, 11, e10473. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Usui-Ouchi, A.; Nilsson, A.K.; Yang, J.; Ko, M.; Hellström, A.; Fu, Z. Metabolism in Retinopathy of Prematurity. Life 2021, 11, 1119. [Google Scholar] [CrossRef] [PubMed]
- Connor, K.M.; SanGiovanni, J.P.; Lofqvist, C.; Aderman, C.M.; Chen, J.; Higuchi, A.; Hong, S.; Pravda, E.A.; Majchrzak, S.; Carper, D.; et al. Increased dietary intake of omega-3-polyunsaturated fatty acids reduces pathological retinal angiogenesis. Nat. Med. 2007, 13, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Lofqvist, C.A.; Shao, Z.; Sun, Y.; Joyal, J.S.; Hurst, C.G.; Cui, R.Z.; Evans, L.P.; Tian, K.; SanGiovanni, J.P.; et al. Dietary ω-3 polyunsaturated fatty acids decrease retinal neovascularization by adipose-endoplasmic reticulum stress reduction to increase adiponectin. Am. J. Clin. Nutr. 2015, 101, 879–888. [Google Scholar] [CrossRef]
- Miller, J.W. Age-related macular degeneration revisited—Piecing the puzzle: The LXIX Edward Jackson memorial lecture. Am. J. Ophthalmol. 2013, 155, 1–35.e13. [Google Scholar] [CrossRef]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 14682–14687. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.H.; Bell, B.; Singh, R.; Batoki, J.; Wolk, A.; Cutler, A.; Prayson, N.; Ali, M.; Stoehr, H.; Anand-Apte, B. Sorsby Fundus Dystrophy Mutation in Tissue Inhibitor of Metalloproteinase 3 (TIMP3) promotes Choroidal Neovascularization via a Fibroblast Growth Factor-dependent Mechanism. Sci. Rep. 2019, 9, 17429. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, J.R. Juvenile neuronal ceroid lipofuscinosis (Batten disease): Current insights. Degener. Neurol. Neuromuscul. Dis. 2016, 6, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.N.; Fearnley, I.M.; Walker, J.E.; Hall, N.A.; Lake, B.D.; Wolfe, L.S.; Haltia, M.; Martinus, R.D.; Jolly, R.D. Mitochondrial ATP synthase subunit c storage in the ceroid-lipofuscinoses (Batten disease). Am. J. Med. Genet. 1992, 42, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.R.; Knight, K.; Engel, A.L.; Jankowski, C.; Wang, Y.; Manson, M.A.; Gu, H.; Djukovic, D.; Raftery, D.; Hurley, J.B.; et al. Human retinal pigment epithelial cells prefer proline as a nutrient and transport metabolic intermediates to the retinal side. J. Biol. Chem. 2017, 292, 12895–12905. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Yanagida, A.; Knight, K.; Engel, A.L.; Vo, A.H.; Jankowski, C.; Sadilek, M.; Tran, V.T.; Manson, M.A.; Ramakrishnan, A.; et al. Reductive carboxylation is a major metabolic pathway in the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2016, 113, 14710–14715. [Google Scholar] [CrossRef]
- Laíns, I.; Chung, W.; Kelly, R.S.; Gil, J.; Marques, M.; Barreto, P.; Murta, J.N.; Kim, I.K.; Vavvas, D.G.; Miller, J.B.; et al. Human Plasma Metabolomics in Age-Related Macular Degeneration: Meta-Analysis of Two Cohorts. Metabolites 2019, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.L.; Ma, C.; Scott, W.K.; Agarwal, A.; Pericak-Vance, M.A.; Haines, J.L.; Jones, D.P.; Uppal, K.; Brantley, M.A., Jr. Plasma Metabolomics of Intermediate and Neovascular Age-Related Macular Degeneration Patients. Cells 2021, 10, 3141. [Google Scholar] [CrossRef]
- Acar, İ.E.; Lores-Motta, L.; Colijn, J.M.; Meester-Smoor, M.A.; Verzijden, T.; Cougnard-Gregoire, A.; Ajana, S.; Merle, B.M.J.; de Breuk, A.; Heesterbeek, T.J.; et al. Integrating Metabolomics, Genomics, and Disease Pathways in Age-Related Macular Degeneration: The EYE-RISK Consortium. Ophthalmology 2020, 127, 1693–1709. [Google Scholar] [CrossRef]
- Duncan, J.L. Mouse models may provide new insight into the relation between cholesterol and age related macular degeneration. Br. J. Ophthalmol. 2005, 89, 1549–1551. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Holoman, N.C.; Aiello, J.J.; Trobenter, T.D.; Tarchick, M.J.; Kozlowski, M.R.; Makowski, E.R.; De Vivo, D.C.; Singh, C.; Sears, J.E.; Samuels, I.S. Reduction of Glut1 in the Neural Retina But Not the RPE Alleviates Polyol Accumulation and Normalizes Early Characteristics of Diabetic Retinopathy. J. Neurosci. 2021, 41, 3275–3299. [Google Scholar] [CrossRef] [PubMed]
- Hammes, H.P.; Federoff, H.J.; Brownlee, M. Nerve growth factor prevents both neuroretinal programmed cell death and capillary pathology in experimental diabetes. Mol. Med. 1995, 1, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.H.; Kim, J.M.; Jeon, H.J.; Oh, T.; Choi, H.J.; Kim, B.J. Metabolomics profiles associated with diabetic retinopathy in type 2 diabetes patients. PLoS ONE 2020, 15, e0241365. [Google Scholar] [CrossRef]
- Zhu, X.R.; Yang, F.Y.; Lu, J.; Zhang, H.R.; Sun, R.; Zhou, J.B.; Yang, J.K. Plasma metabolomic profiling of proliferative diabetic retinopathy. Nutr. Metab. 2019, 16, 37. [Google Scholar] [CrossRef]
- Sumarriva, K.; Uppal, K.; Ma, C.; Herren, D.J.; Wang, Y.; Chocron, I.M.; Warden, C.; Mitchell, S.L.; Burgess, L.G.; Goodale, M.P.; et al. Arginine and Carnitine Metabolites Are Altered in Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3119–3126. [Google Scholar] [CrossRef]
- Peters, K.S.; Rivera, E.; Warden, C.; Harlow, P.A.; Mitchell, S.L.; Calcutt, M.W.; Samuels, D.C.; Brantley, M.A., Jr. Plasma Arginine and Citrulline are Elevated in Diabetic Retinopathy. Am. J. Ophthalmol. 2021, 235, 154–162. [Google Scholar] [CrossRef]
- Sun, Y.; Zou, H.; Li, X.; Xu, S.; Liu, C. Plasma Metabolomics Reveals Metabolic Profiling for Diabetic Retinopathy and Disease Progression. Front. Endocrinol. 2021, 12, 757088. [Google Scholar] [CrossRef]
- Mora-Ortiz, M.; Nuñez Ramos, P.; Oregioni, A.; Claus, S.P. NMR metabolomics identifies over 60 biomarkers associated with Type II Diabetes impairment in db/db mice. Metabolomics 2019, 15, 89. [Google Scholar] [CrossRef]
- Patrick, A.T.; He, W.; Madu, J.; Sripathi, S.R.; Choi, S.; Lee, K.; Samson, F.P.; Powell, F.L.; Bartoli, M.; Jee, D.; et al. Mechanistic dissection of diabetic retinopathy using the protein-metabolite interactome. J. Diabetes Metab. Disord. 2020, 19, 829–848. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Peng, Y.-R.; van Zyl, T.; Regev, A.; Shekhar, K.; Juric, D.; Sanes, J.R. Cell Atlas of The Human Fovea and Peripheral Retina. Sci. Rep. 2020, 10, 9802. [Google Scholar] [CrossRef] [PubMed]
- Fligor, C.M.; Langer, K.B.; Sridhar, A.; Ren, Y.; Shields, P.K.; Edler, M.C.; Ohlemacher, S.K.; Sluch, V.M.; Zack, D.J.; Zhang, C.; et al. Three-Dimensional Retinal Organoids Facilitate the Investigation of Retinal Ganglion Cell Development, Organization and Neurite Outgrowth from Human Pluripotent Stem Cells. Sci. Rep. 2018, 8, 14520. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lowe, A.; Dharmat, R.; Lee, S.; Owen, L.A.; Wang, J.; Shakoor, A.; Li, Y.; Morgan, D.J.; Hejazi, A.A.; et al. Generation, transcriptome profiling, and functional validation of cone-rich human retinal organoids. Proc. Natl. Acad. Sci. USA 2019, 116, 10824–10833. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, C. Metabolism and Vascular Retinopathies: Current Perspectives and Future Directions. Diagnostics 2022, 12, 903. https://doi.org/10.3390/diagnostics12040903
Singh C. Metabolism and Vascular Retinopathies: Current Perspectives and Future Directions. Diagnostics. 2022; 12(4):903. https://doi.org/10.3390/diagnostics12040903
Chicago/Turabian StyleSingh, Charandeep. 2022. "Metabolism and Vascular Retinopathies: Current Perspectives and Future Directions" Diagnostics 12, no. 4: 903. https://doi.org/10.3390/diagnostics12040903
APA StyleSingh, C. (2022). Metabolism and Vascular Retinopathies: Current Perspectives and Future Directions. Diagnostics, 12(4), 903. https://doi.org/10.3390/diagnostics12040903